Gene Ontology Enrichment Analysis
Che-Wei Chang
2021-10-25
Last updated: 2022-01-18
Checks: 7 0
Knit directory: Barley1kGBS_Proj/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it's best to always run the code in an empty environment.
The command set.seed(20210831) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version da73ff9. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/GenomeEnvAssociation_cache/
Ignored: analysis/GenomicVarPartitioning_cache/
Ignored: analysis/PreprocessEnvData_cache/
Untracked files:
Untracked: VennDiagram2022-01-18_16-08-05.log
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/GOanalysis.Rmd) and HTML (public/GOanalysis.html) files. If you've configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | 5f53139 | Che-Wei Chang | 2022-01-18 | Build site. |
| Rmd | 597a627 | Che-Wei Chang | 2021-11-30 | update analyses. unPC, ResistanceGA and GO enrichment |
| html | 597a627 | Che-Wei Chang | 2021-11-30 | update analyses. unPC, ResistanceGA and GO enrichment |
Introduction
To investigate biological functions related to putatively adaptive loci, we conducted gene ontology (GO) enrichment analysis with gene annotations of the barley 'Morex v2' genome. We searched over-represented GO terms for genes within 500 bp adjacent intervals of candidate SNPs detected by the genome scans. This analysis was done using the R package SNP2GO (Szkiba et al. 2014) with a significant level of FDR < 0.05.
Searching genes within 500 bp intervals
We first looked for genes within 500 bp intervals adjacent to significant SNPs identified by genome scans. To do this, we used a customized function search_gene_anno.
Reading required data
rm(list = ls())
source("./code/Rfunctions.R")
library(SNP2GO)
library(vcfR)
library(vegan)
library(qvalue)
library(robust)
library(IRanges)
library(GenomicRanges)
library(RCurl)
# to save computational time, we load the results of genome scans done previously
## load RDA results
rda.simple <- readRDS("./data/2020-04-10-simple_rda_191B1K+53KS_12envs_27147SNPs_maf0.05.RDS")
rda.ancescoff <- readRDS("./data/2020-04-10-partial_rda_ALStructure_K4_ancestry_coeff_191B1K+53KS_12envs_27147SNPs_maf0.05.RDS")
## load LFMM results
lfmm.p <- read.delim("./data/2020-04-18-191B1K+53KS_LFMM_K4_27147SNPs_maf0.05_12envs_adj_pvalues.tsv")
lfmm.q <- read.delim("./data/2020-04-18-191B1K+53KS_LFMM_K4_27147SNPs_maf0.05_12envs_adj_qvalues.tsv")
## load BAYPASS results
XtX <- read.table("./data/baypass_output_191B1K+53KS_in4pops_summary_pi_xtx.out", stringsAsFactors = F, header = T)
POD <- read.table("./data/wildbarley_POD_summary_pi_xtx.out", header = T)
pod.thres <- quantile(POD$M_XtX, 0.995)
# load gene annotations
gen.anno <- read.delim("./data/Barley_Morex_V2_gene_annotation_PGSB.all.descriptions.txt", header = T, stringsAsFactors = F)
# load VCF
geno <- read.vcfR("./data/GBS_244accessions_27147SNP_Beagle5Imp.vcf.gz", verbose = FALSE)
all.snp <- as.data.frame(geno@fix[,1:3])
CHR <- paste0("chr",as.numeric(geno@fix[,"CHROM"]), "H")
POS <- as.numeric(geno@fix[,"POS"])
# check the neighbour genes of all SNPs
pseudo.sig <- data.frame(chr = CHR, pos = POS)
gene.data <- data.frame(gene = gen.anno$gene, chr = gen.anno$agp_chr, start = gen.anno$start, end = gen.anno$end)Identification of genes nearby significant SNPs
Simple RDA and partial RDA
k = 4
qcut <- 0.05 # FDR threshold
rda.simple.pq <- rdadapt(rda.simple, K = k)
rda.ancescoff.pq <- rdadapt(rda.ancescoff, K = k)
# search genes within a 500 bp interval
## Simple RDA
gene.data <- data.frame(gene = gen.anno$gene, chr = gen.anno$agp_chr, start = gen.anno$start, end = gen.anno$end)
rda.simple.sig <- data.frame(chr = CHR[rda.simple.pq$q.values < qcut], pos = POS[rda.simple.pq$q.values < qcut])
rda.simple.gene <-
gen.anno[gen.anno$gene %in% unique(unname(unlist(sapply(search_gene_anno(gene.pos = gene.data,snp.pos = rda.simple.sig,search.bp = 500),
function(x){strsplit(x, split = ";")})))),]
## Partial RDA
rda.ancescoeff.sig <- data.frame(chr = CHR[rda.ancescoff.pq$q.values < qcut], pos = POS[rda.ancescoff.pq$q.values < qcut])
rda.ancescoeff.gene <-
gen.anno[gen.anno$gene %in% unique(unname(unlist(sapply(search_gene_anno(gene.pos = gene.data,snp.pos = rda.ancescoeff.sig, search.bp = 500),
function(x){strsplit(x, split = ";")})))),]
str(rda.simple.gene)'data.frame': 159 obs. of 12 variables:
$ genotype : chr "Morex" "Morex" "Morex" "Morex" ...
$ gene : chr "HORVU.MOREX.r2.1HG0004200" "HORVU.MOREX.r2.1HG0039290" "HORVU.MOREX.r2.1HG0041990" "HORVU.MOREX.r2.1HG0042300" ...
$ agp_chr : chr "chr1H" "chr1H" "chr1H" "chr1H" ...
$ chr : int 1 1 1 1 1 1 1 1 2 2 ...
$ start : int 10397928 336523412 359228317 361583171 378787645 385282025 503288645 508003287 4715619 43627318 ...
$ end : int 10404246 336534989 359230673 361585578 378790491 385286453 503291401 508003934 4718433 43628617 ...
$ orientation: chr "+" "+" "+" "+" ...
$ confidence : chr "HC" "HC" "HC" "HC" ...
$ confidence2: chr "HC1" "HC1" "HC1" "HC1" ...
$ gene_length: int 6319 11578 2357 2408 2847 4429 2757 648 2815 1300 ...
$ ntranscript: int 1 1 1 1 1 1 1 1 1 1 ...
$ annotation : chr "Mitochondrial import inner membrane translocase subunit tim23" "Aldo/keto reductase family protein, putative, expressed" "Xyloglucan endotransglucosylase/hydrolase" "Urea-proton symporter DUR3" ...str(rda.ancescoeff.gene)'data.frame': 194 obs. of 12 variables:
$ genotype : chr "Morex" "Morex" "Morex" "Morex" ...
$ gene : chr "HORVU.MOREX.r2.1HG0004380" "HORVU.MOREX.r2.1HG0007840" "HORVU.MOREX.r2.1HG0017670" "HORVU.MOREX.r2.1HG0027260" ...
$ agp_chr : chr "chr1H" "chr1H" "chr1H" "chr1H" ...
$ chr : int 1 1 1 1 1 1 1 1 1 1 ...
$ start : int 10810978 21790779 80671317 197534363 359228317 370914136 417218200 448576690 503288645 516201537 ...
$ end : int 10812240 21792317 80675966 197534759 359230673 370916310 417219711 448578184 503291401 516205231 ...
$ orientation: chr "+" "+" "+" "+" ...
$ confidence : chr "LC" "HC" "HC" "LC" ...
$ confidence2: chr "LC2" "HC1" "HC2" "LC2" ...
$ gene_length: int 1263 1539 4650 397 2357 2175 1512 1495 2757 3695 ...
$ ntranscript: int 1 1 1 1 1 1 1 1 1 1 ...
$ annotation : chr "alanine-tRNA ligase, putative (DUF760)" "Tryptophan decarboxylase" "Disease resistance protein (NBS-LRR class) family" "RNA polymerase II transcription mediator" ...BAYPASS
## BAYPASS
baypass.sig <- data.frame(chr = CHR[XtX$M_XtX > pod.thres], pos = POS[XtX$M_XtX > pod.thres])
baypass.gene <-
gen.anno[gen.anno$gene %in% unique(unname(unlist(sapply(search_gene_anno(gene.pos = gene.data,snp.pos = baypass.sig, search.bp = 500),
function(x){strsplit(x, split = ";")})))),]
str(baypass.gene)'data.frame': 125 obs. of 12 variables:
$ genotype : chr "Morex" "Morex" "Morex" "Morex" ...
$ gene : chr "HORVU.MOREX.r2.1HG0040730" "HORVU.MOREX.r2.1HG0042510" "HORVU.MOREX.r2.1HG0042550" "HORVU.MOREX.r2.1HG0043040" ...
$ agp_chr : chr "chr1H" "chr1H" "chr1H" "chr1H" ...
$ chr : int 1 1 1 1 1 2 2 2 2 2 ...
$ start : int 347323332 363097440 363407332 367308627 520540660 89939523 92732808 117960598 381092014 455012235 ...
$ end : int 347323640 363097799 363407963 367309736 520551806 89940710 92740831 117968687 381099563 455019398 ...
$ orientation: chr "+" "+" "+" "+" ...
$ confidence : chr "LC" "HC" "HC" "HC" ...
$ confidence2: chr "LC2" "HC2" "HC2" "HC2" ...
$ gene_length: int 309 360 632 1110 11147 1188 8024 8090 7550 7164 ...
$ ntranscript: int 1 1 1 1 1 1 1 1 1 1 ...
$ annotation : chr "D-tagatose-1,6-bisphosphate aldolase subunit GatY" "Expansin-like protein" "MADS-box transcription factor" "F-box protein SKIP22" ...LFMM
## LFMM
lfmm.sig.tag <- apply(lfmm.q, 1, function(x){any(x < qcut)})
lfmm.sig <- data.frame(chr = CHR[lfmm.sig.tag], pos = POS[lfmm.sig.tag])
lfmm.gene <-
gen.anno[gen.anno$gene %in% unique(unname(unlist(sapply(search_gene_anno(gene.pos = gene.data,snp.pos = lfmm.sig, search.bp = 500),
function(x){strsplit(x, split = ";")})))),]
str(lfmm.gene)'data.frame': 166 obs. of 12 variables:
$ genotype : chr "Morex" "Morex" "Morex" "Morex" ...
$ gene : chr "HORVU.MOREX.r2.1HG0023750" "HORVU.MOREX.r2.1HG0059900" "HORVU.MOREX.r2.1HG0071770" "HORVU.MOREX.r2.1HG0074820" ...
$ agp_chr : chr "chr1H" "chr1H" "chr1H" "chr1H" ...
$ chr : int 1 1 1 1 2 2 2 2 2 3 ...
$ start : int 146233302 469696741 504404498 511691892 16831745 614556615 619314636 629639994 649885232 5233815 ...
$ end : int 146234762 469700484 504404758 511694152 16835037 614556956 619316254 629646699 649885783 5239725 ...
$ orientation: chr "+" "+" "+" "+" ...
$ confidence : chr "HC" "HC" "HC" "HC" ...
$ confidence2: chr "HC2" "HC1" "HC2" "HC2" ...
$ gene_length: int 1461 3744 261 2261 3293 342 1619 6706 552 5911 ...
$ ntranscript: int 1 1 1 1 1 1 1 1 1 1 ...
$ annotation : chr "DUF641 family protein" "Phosphatidylinositol:ceramide inositolphosphotransferase" "Clavata3/ESR (CLE) gene family member" "UDP-N-acetylglucosamine 1-carboxyvinyltransferase" ...Genes identified by multiple approaches
# intersect of simple RDA and partial RDA
nrow(rda.simple.gene[rda.simple.gene$gene %in% rda.ancescoeff.gene$gene,])[1] 32# intersect of simple RDA and Baypass
nrow(rda.simple.gene[rda.simple.gene$gene %in% baypass.gene$gene,])[1] 51# intersect of simple RDA and LFMM
nrow(rda.simple.gene[rda.simple.gene$gene %in% lfmm.gene$gene,])[1] 7# intersect of partial RDA and Baypass
nrow(rda.ancescoeff.gene[rda.ancescoeff.gene$gene %in% baypass.gene$gene,])[1] 9# intersect of partial RDA and LFMM
nrow(rda.ancescoeff.gene[rda.ancescoeff.gene$gene %in% lfmm.gene$gene,])[1] 20# intersect of Baypass and LFMM
nrow(baypass.gene[baypass.gene$gene %in% lfmm.gene$gene,])[1] 3# intersect of all methods
overlap.gene <- rda.ancescoeff.gene[rda.ancescoeff.gene$gene %in% lfmm.gene$gene & rda.ancescoeff.gene$gene %in% baypass.gene$gene & rda.ancescoeff.gene$gene %in% rda.simple.gene$gene,]
overlap.gene genotype gene agp_chr chr start end
47345 Morex HORVU.MOREX.r2.4HG0308420 chr4H 4 312111636 312189673
47635 Morex HORVU.MOREX.r2.4HG0314300 chr4H 4 392019895 392026683
orientation confidence confidence2 gene_length ntranscript
47345 - HC HC1 78038 1
47635 - HC HC1 6789 1
annotation
47345 ATP-dependent RNA helicase
47635 Nucleolar GTP-binding protein 2Venn diagram
# make Venn diagram
uniq.gene <- unique(c(as.character(rda.simple.gene$gene),
as.character(rda.ancescoeff.gene$gene),
as.character(baypass.gene$gene), as.character(lfmm.gene$gene)))
venn.input <- list("Simple RDA" = which(uniq.gene %in% as.character(rda.simple.gene$gene)),
"Partial RDA" = which(uniq.gene %in% as.character(rda.ancescoeff.gene$gene)),
"LFMM" = which(uniq.gene %in% as.character(lfmm.gene$gene)),
"BAYPASS" = which(uniq.gene %in% as.character(baypass.gene$gene))
)
library(VennDiagram)Loading required package: gridLoading required package: futile.loggervenn <-
venn.diagram(venn.input,fill = "white"
, cex = 1.5,cat.fontface = 4,lty =2, imagetype = "png",
filename = NULL,
cat.pos = c(0), resolution = 400, cat.cex = 2
)
grid.draw(venn)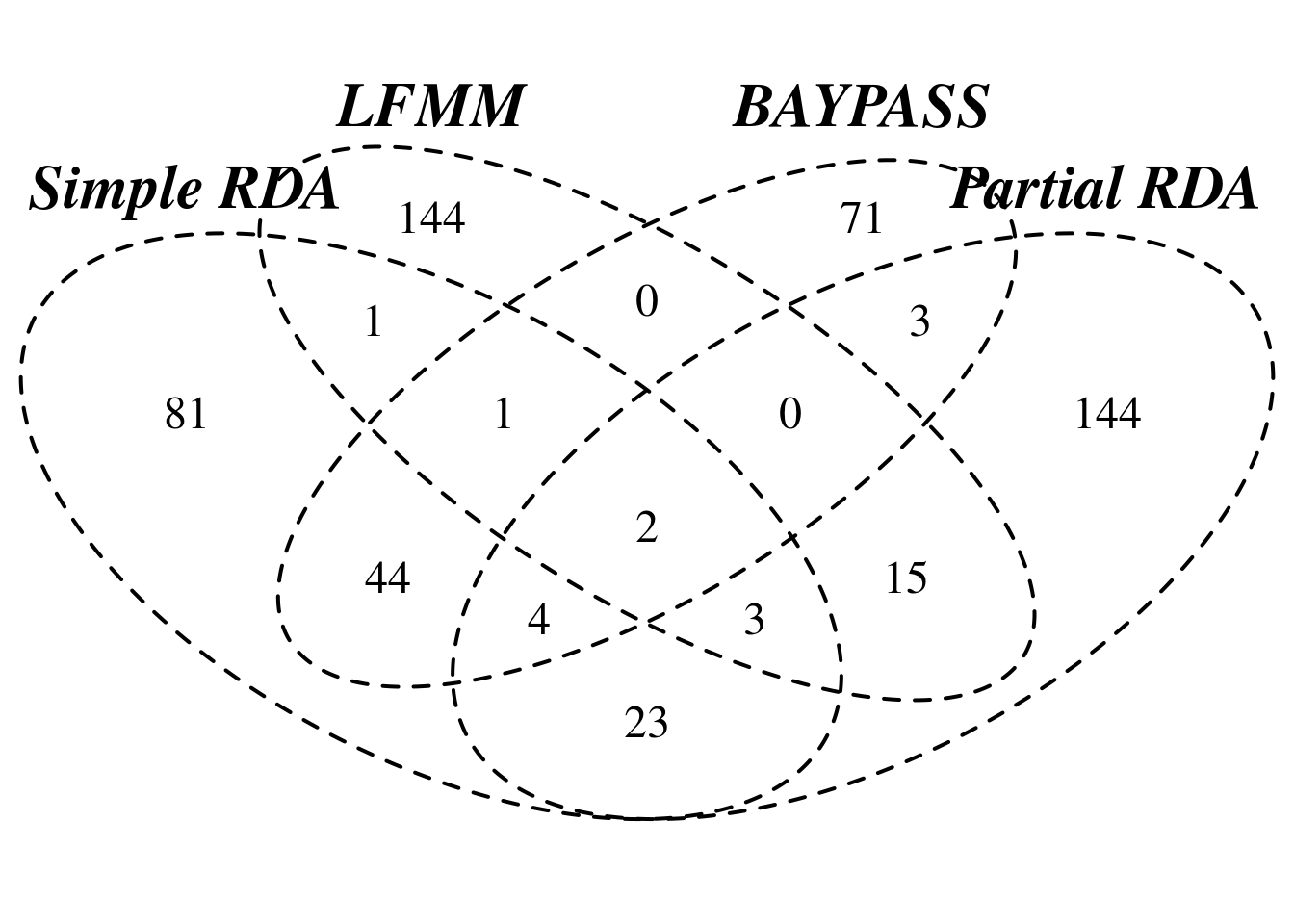
| Version | Author | Date |
|---|---|---|
| 597a627 | Che-Wei Chang | 2021-11-30 |
GO term enrichment analysis
Data preparation
all.snp[,2] <- as.numeric(as.character(all.snp[,2]))
all.snp[,1] <- paste("chr",all.snp[,1], "H", sep = "") # make the coding of chromosome is identical to the `gff3` file
all.snp.G<- GRanges(seqnames=all.snp[,1],ranges=IRanges(all.snp[,2],all.snp[,2]))
# select candidates according to FDR
cand.rda.simple <- all.snp.G[which(rda.simple.pq[,2] < qcut)]
notcand.rda.simple <- all.snp.G[which(!rda.simple.pq[,2] < qcut)]
cand.rda.ancestry <- all.snp.G[which(rda.ancescoff.pq[,2] < qcut)]
notcand.rda.ancestry <- all.snp.G[which(!rda.ancescoff.pq[,2] < qcut)]
# 'snp2go' only search the lines of 'gene' for GO terms in the gff file
# but in our gff3 file the GO term is described in the lines of 'mRNA'
# so, I have to modify the our gff file to add GO terms to the 'gene'
gff_file <- "./data/Barley_Morex_V2_gene_annotation_PGSB.all.gff3"
gff <- readLines(gff_file)
gene <- gff[grepl(pattern = "\tgene\t", gff, perl = T)] #
mRNA <- gff[grepl(pattern = "\tmRNA\t", gff, perl = T)] #
new.gene <- paste(gene, sapply(strsplit(mRNA, split = ";"), function(x){paste(x[-1], collapse = ";")}), sep = ";")
gff[grepl(pattern = "\tgene\t", gff, perl = T)] <- new.gene
fileout<-file("./output/Barley_Morex_V2_gene_annotation_PGSB_append_GO.all.gff")
writeLines(gff, fileout)
close(fileout)Run SNP2GO
gff_file <- "./output/Barley_Morex_V2_gene_annotation_PGSB_append_GO.all.gff"
ext = 500 # setting the searching range as 500 bp
GO.simpleRDA <- snp2go(gff=gff_file,
candidateSNPs=cand.rda.simple,
noncandidateSNPs=notcand.rda.simple,
extension = ext,FDR = 0.05, runs = 10000)
GO.partialRDA <- snp2go(gff=gff_file,
candidateSNPs=cand.rda.ancestry,
noncandidateSNPs=notcand.rda.ancestry,
extension = ext, FDR = 0.05, runs = 10000) # use 'FDR = 0.05' to keep only the significant results
# GO enrichment using LFMM result
qcut <- 0.05
ext = 500
for(i in 1:ncol(lfmm.q)){
if(length(which(lfmm.q[i] < qcut) == 0)){next()} # skip if there is no candidate
cand.lfmm <- all.snp.G[which(lfmm.q[i] < qcut)] # select candidate
notcand.lfmm <- all.snp.G[which(!lfmm.q[i] < qcut)]
env <- colnames(lfmm.q)[i]
GO.lfmm <- snp2go(gff=gff_file,
candidateSNPs=cand.lfmm,
noncandidateSNPs=notcand.lfmm,
extension = ext,FDR = 0.05, runs = 10000)
saveRDS(GO.lfmm, paste("./output/", env,"-LFMM_FDR",qcut,"_SNP2GO_",ext,"bp_GOenrichFDR0.05.RDS", sep = ""))
}
# GO enrivhment using XtX result
ext = 500
cand.xtx <- all.snp.G[which(XtX$M_XtX > pod.thres)] # select candidate
notcand.xtx <- all.snp.G[which(!XtX$M_XtX > pod.thres)]
GO.xtx <- snp2go(gff=gff_file,
candidateSNPs=cand.xtx,
noncandidateSNPs=notcand.xtx,
extension = ext,FDR = 0.05, runs = 10000)Check the number of enriched GO terms
We identified no enriched GO term based on the SNPs detected by LFMM.
sapply(list.files("./output/",pattern = "*LFMM_FDR*", full.names = TRUE), function(x){
nrow(readRDS(x)$enriched)
}) # no enriched GO term for LFMM results ./output//Aspect-LFMM_FDR0.05_SNP2GO_500bp_GOenrichFDR0.05.RDS
0
./output//BD_sl12-LFMM_FDR0.05_SNP2GO_500bp_GOenrichFDR0.05.RDS
0
./output//OCC_sl4-LFMM_FDR0.05_SNP2GO_500bp_GOenrichFDR0.05.RDS
0
./output//Slope-LFMM_FDR0.05_SNP2GO_500bp_GOenrichFDR0.05.RDS
0
./output//SLT_sl1234-LFMM_FDR0.05_SNP2GO_500bp_GOenrichFDR0.05.RDS
0 Ten and two GO terms enriched based on the SNPs detected by XtX and the simple RDA, respectively. However, no GO term was enriched based on the SNPs detected by the partial RDA.
nrow(GO.xtx$enriched) # 10 enriched GO terms[1] 10nrow(GO.simpleRDA$enriched) # 2 enriched GO terms[1] 2nrow(GO.partialRDA$enriched) # no enriched GO term[1] 0
sessionInfo()R version 3.6.0 (2019-04-26)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 16.04.6 LTS
Matrix products: default
BLAS: /usr/lib/libblas/libblas.so.3.6.0
LAPACK: /usr/lib/lapack/liblapack.so.3.6.0
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] grid stats4 parallel stats graphics grDevices utils
[8] datasets methods base
other attached packages:
[1] VennDiagram_1.6.20 futile.logger_1.4.3 RCurl_1.98-1.1
[4] robust_0.4-18.2 fit.models_0.5-14 qvalue_2.16.0
[7] vegan_2.5-6 lattice_0.20-38 permute_0.9-5
[10] vcfR_1.9.0 SNP2GO_1.0.6 GenomicRanges_1.36.1
[13] GenomeInfoDb_1.20.0 goProfiles_1.46.0 stringr_1.4.0
[16] CompQuadForm_1.4.3 GO.db_3.8.2 AnnotationDbi_1.46.1
[19] IRanges_2.18.3 S4Vectors_0.22.1 Biobase_2.44.0
[22] BiocGenerics_0.30.0 workflowr_1.6.2
loaded via a namespace (and not attached):
[1] nlme_3.1-139 bitops_1.0-6 fs_1.5.0
[4] bit64_0.9-7 rprojroot_1.3-2 tools_3.6.0
[7] backports_1.1.5 utf8_1.2.2 R6_2.5.1
[10] DBI_1.1.1 mgcv_1.8-28 colorspace_2.0-2
[13] tidyselect_1.1.1 bit_4.0.4 compiler_3.6.0
[16] git2r_0.26.1 formatR_1.7 scales_1.1.1
[19] DEoptimR_1.0-8 mvtnorm_1.0-12 robustbase_0.93-5
[22] digest_0.6.28 rmarkdown_2.3 XVector_0.24.0
[25] rrcov_1.5-2 pkgconfig_2.0.3 htmltools_0.5.2
[28] highr_0.9 fastmap_1.1.0 rlang_0.4.12
[31] RSQLite_2.2.0 generics_0.1.0 dplyr_1.0.7
[34] magrittr_2.0.1 GenomeInfoDbData_1.2.1 Matrix_1.2-18
[37] Rcpp_1.0.7 munsell_0.5.0 fansi_0.5.0
[40] ape_5.3 lifecycle_1.0.1 stringi_1.7.5
[43] whisker_0.4 yaml_2.2.1 MASS_7.3-51.1
[46] zlibbioc_1.30.0 plyr_1.8.6 pinfsc50_1.1.0
[49] blob_1.2.1 promises_1.2.0.1 crayon_1.4.1
[52] splines_3.6.0 knitr_1.36 pillar_1.6.4
[55] reshape2_1.4.3 futile.options_1.0.1 glue_1.4.2
[58] evaluate_0.14 lambda.r_1.2.4 memuse_4.0-0
[61] vctrs_0.3.8 httpuv_1.6.3 gtable_0.3.0
[64] purrr_0.3.4 assertthat_0.2.1 cachem_1.0.6
[67] ggplot2_3.3.5 xfun_0.27 later_1.3.0
[70] pcaPP_1.9-73 viridisLite_0.4.0 tibble_3.1.5
[73] memoise_2.0.0 cluster_2.0.8 ellipsis_0.3.2