ANCOM-BC clean - cells with >= 1000 features only
Will Macnair
Institute for Molecular Life Sciences, University of Zurich, SwitzerlandSwiss Institute of Bioinformatics (SIB), University of Zurich, SwitzerlandJune 04, 2021
Last updated: 2021-06-04
Checks: 4 3
Knit directory: MS_lesions/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
The global environment had objects present when the code in the R Markdown file was run. These objects can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment. Use wflow_publish or wflow_build to ensure that the code is always run in an empty environment.
The following objects were defined in the global environment when these results were created:
| Name | Class | Size |
|---|---|---|
| q | function | 1008 bytes |
The command set.seed(20210118) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
- calc_ancombc
- calc_clr_pcas
- calc_counts_wide
- calc_join_conos_meta
- calc_props_dt
- calc_qc
- calc_scCODA
- load_conos
- load_meta
- plot_ancom_cis_signif
- plot_clr_heatmap
- plot_clr_pca
- plot_neuro_prop
- plot_sample_splits_clrs
- plot_sccoda_cis_signif
- setup_input
- setup_outputs
To ensure reproducibility of the results, delete the cache directory ms09_ancombc_clean_1e3_cache and re-run the analysis. To have workflowr automatically delete the cache directory prior to building the file, set delete_cache = TRUE when running wflow_build() or wflow_publish().
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 5ab29c3. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rprofile
Ignored: .Rproj.user/
Ignored: ._.DS_Store
Ignored: ._MS_lesions.sublime-project
Ignored: .log/
Ignored: MS_lesions.sublime-project
Ignored: MS_lesions.sublime-workspace
Ignored: analysis/.__site.yml
Ignored: analysis/figure/
Ignored: analysis/ms02_doublet_id_cache/
Ignored: analysis/ms03_SampleQC_cache/
Ignored: analysis/ms04_conos_cache/
Ignored: analysis/ms05_splitting_cache/
Ignored: analysis/ms06_sccaf_cache/
Ignored: analysis/ms07_soup_cache/
Ignored: analysis/ms08_modules_cache/
Ignored: analysis/ms09_ancombc_cache/
Ignored: analysis/ms09_ancombc_clean_1e3_cache/
Ignored: analysis/ms10_muscat_run01_cache/
Ignored: analysis/ms10_muscat_run02_cache/
Ignored: analysis/ms10_muscat_template_cache/
Ignored: analysis/ms11_paga_cache/
Ignored: analysis/ms12_markers_cache/
Ignored: analysis/ms13_labelling_cache/
Ignored: analysis/supp06_sccaf_cache/
Ignored: analysis/supp10_muscat_cache/
Ignored: code/muscat_plan.txt
Ignored: data/
Ignored: figures/
Ignored: output/
Untracked files:
Untracked: Rplots.pdf
Unstaged changes:
Modified: analysis/ms09_ancombc_clean_1e3.Rmd
Modified: analysis/ms09_ancombc_clean_2e3.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/ms09_ancombc_clean_1e3.Rmd) and HTML (docs/ms09_ancombc_clean_1e3.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 9d1ab6c | Macnair | 2021-06-03 | Updated ACNOM-BC with type_fine |
| Rmd | 129c53d | Macnair | 2021-04-16 | Renamed a lot of things to add ms07_soup |
Notes
I’ve done a range of runs of ANCOM-BC with slightly different parameters, to see which works best. As usual, there’s not so much difference between them (which is good). Here’s a quick description of the runs:
- lesions_WM_mad: Testing lesions vs healthy, WM only, samples with unusually high neuronal propn excluded.
- lesions_WM_big_mad: Testing lesions vs healthy, WM only, samples with unusually high neuronal propn excluded, celltypes with very few cells excluded.
- lesions_GM_mad: Testing lesions vs healthy, GM only, samples with unusually low neuronal propn excluded (actually only excludes one sample).
- lesions_GM_big_mad: Testing lesions vs healthy, GM only, samples with unusually low neuronal propn excluded, celltypes with very few cells excluded.
- lesions_WM_no_neuro: Testing lesions vs healthy, WM only, samples with unusually high neuronal propn excluded, restricted to non-neuronal celltypes only.
- lesions_GM_neuro: Testing lesions vs healthy, GM only, samples with unusually low neuronal propn excluded, restricted to ONLY neuronal celltypes.
- lesions_WM_all: Testing lesions vs healthy, WM only, all samples included.
- lesions_GM_all: Testing lesions vs healthy, GM only, all samples included.
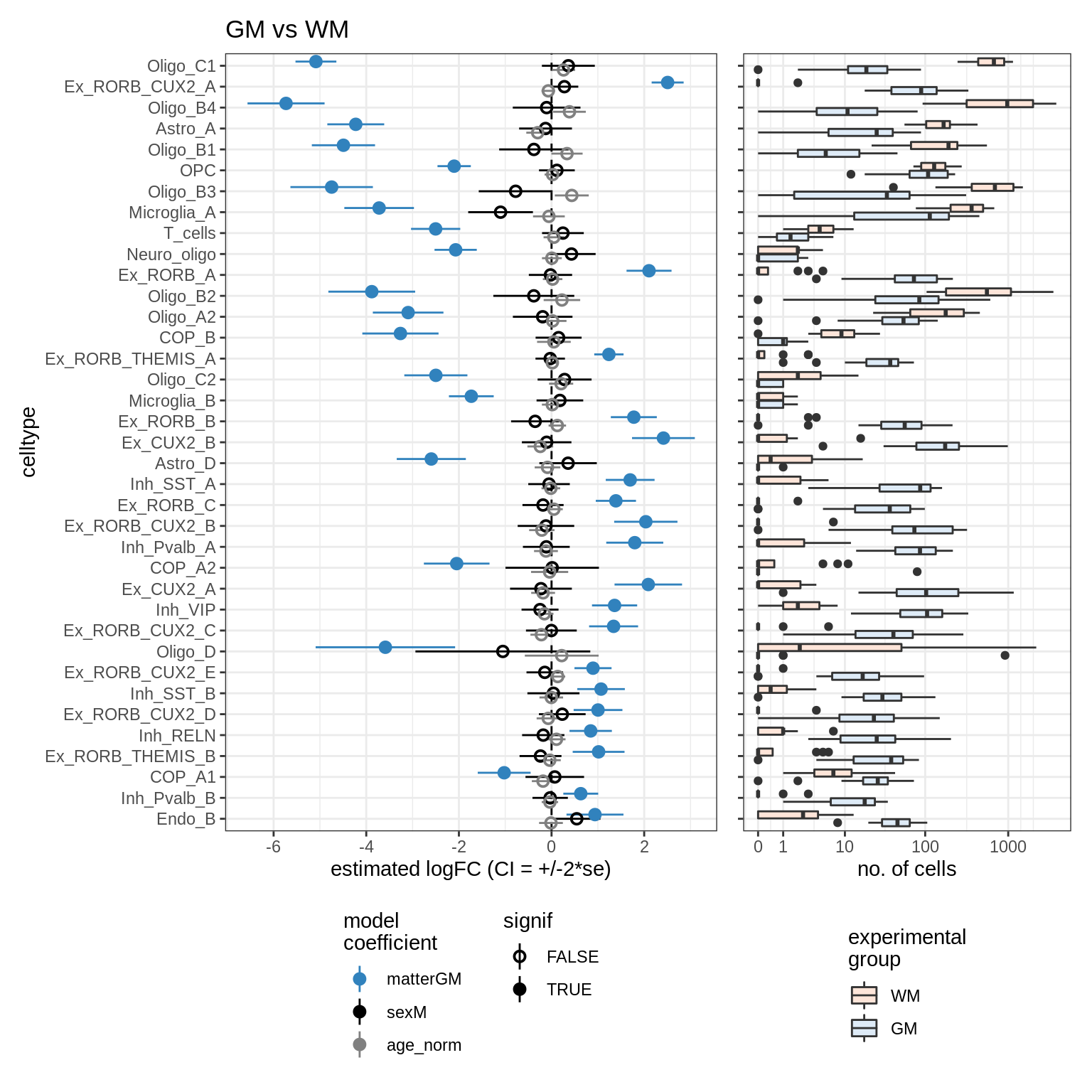
- GM_vs_WM: Testing healthy GM vs healthy WM. This doesn’t work so well with
ANCOM-BC, as it looks for a number of unchanging celltypes to use as a reference, and in this comparison it’s only OPCs that could plausibly not change between them. - lesions_WM_oligos: Testing lesions vs healthy, WM only, oligos + OPCs only, samples with unusually high neuronal propn excluded.
- lesions_GM_oligos: Testing lesions vs healthy, GM only, oligos + OPCs only, samples with unusually high neuronal propn excluded.
- GM_vs_WM_oligos: Testing healthy GM vs healthy WM, WM only, oligos + OPCs only.
The same samples + celltypes are used in running scCODA, but in scCODA you have to manually specify a reference celltype that you assume doesn’t change. For this, I’ve used one broad celltype for each run, as follows:
- lesions_WM_mad: Excitatory neurons
- lesions_WM_big_mad: Excitatory neurons
- lesions_GM_mad: Excitatory neurons
- lesions_GM_big_mad: Excitatory neurons
- lesions_WM_no_neuro: Astrocytes
- lesions_GM_neuro: Excitatory neurons
- lesions_WM_all: Excitatory neurons
- lesions_GM_all: Excitatory neurons
- GM_vs_WM: OPCs / COPs
Setup / definitions
Libraries
Helper functions
source('code/ms00_utils.R')
source('code/ms04_conos.R')
source('code/ms09_ancombc.R')
use_condaenv('sccoda', required=TRUE)
source_python('code/ms09_scCODA_fns.py')Inputs
# define run
labels_f = 'data/byhand_markers/validation_markers_2021-05-31.csv'
labelled_f = 'output/ms13_labelling/conos_labelled_2021-05-31.txt.gz'
meta_f = 'data/metadata/metadata_updated_20201127.txt'
byhand_f = paste0('data/byhand_markers/Copy of Copy of ',
'Marker_selection_for_validation_MS_snucseq_30102020 ',
'Ediinburgh.xlsx - final markers for Cartana panel.csv')
qc_dir = 'output/ms03_SampleQC'
qc_f = file.path(qc_dir, 'ms_qc_dt.txt')Outputs
# where to save?
save_dir = 'output/ms09_ancombc'
date_tag = '2021-06-03'
if (!dir.exists(save_dir))
dir.create(save_dir)
# strange samples
sample_vars = c('sample_id', 'matter', 'lesion_type',
'neuro_ok', 'neuro_prop', 'source', 'patient_id',
'sex', 'age_norm', 'pmi_cat')
mad_cut = 2
feat_cut = 1e3
# define models
subset_list = list(
lesions_WM_mad = list(
subset_spec = list(matter = 'WM', neuro_ok = TRUE)
),
lesions_WM_big_mad = list(
subset_spec = list(matter = 'WM', neuro_ok = TRUE),
size_spec = list(min_count = 10, min_prop = 0.1)),
lesions_GM_mad = list(
subset_spec = list(matter = 'GM', neuro_ok = TRUE)
),
lesions_GM_big_mad = list(
subset_spec = list(matter = 'GM', neuro_ok = TRUE),
size_spec = list(min_count = 10, min_prop = 0.1)),
lesions_WM_no_neuro = list(
subset_spec = list(matter = 'WM', neuro_ok = TRUE),
type_spec = setdiff(broad_ord,
c('Excitatory neurons', 'Inhibitory neurons'))
),
lesions_GM_neuro = list(
subset_spec = list(matter = 'GM', neuro_ok = TRUE),
type_spec = c('Excitatory neurons', 'Inhibitory neurons')
),
lesions_WM_all = list(
subset_spec = list(matter = 'WM')
),
lesions_GM_all = list(
subset_spec = list(matter = 'GM')
),
GM_vs_WM = list(
subset_spec = list(lesion_type = c('GM', 'WM'), neuro_ok = TRUE)
),
lesions_WM_oligos = list(
subset_spec = list(matter = 'WM', neuro_ok = TRUE),
type_spec = c('Oligodendrocytes', 'OPCs / COPs')
),
lesions_GM_oligos = list(
subset_spec = list(matter = 'GM', neuro_ok = TRUE),
type_spec = c('Oligodendrocytes', 'OPCs / COPs')
),
GM_vs_WM_oligos = list(
subset_spec = list(lesion_type = c('GM', 'WM'), neuro_ok = TRUE),
type_spec = c('Oligodendrocytes', 'OPCs / COPs')
)
)
model_names = names(subset_list)
formulae = list(
'~ lesion_type + sex + age_norm',
'~ lesion_type + sex + age_norm',
'~ lesion_type + sex + age_norm',
'~ lesion_type + sex + age_norm',
'~ lesion_type + sex + age_norm',
'~ lesion_type + sex + age_norm',
'~ lesion_type + sex + age_norm',
'~ lesion_type + sex + age_norm',
'~ matter + sex + age_norm',
'~ lesion_type + sex + age_norm',
'~ lesion_type + sex + age_norm',
'~ matter + sex + age_norm'
) %>% setNames(model_names)
names_list = list(
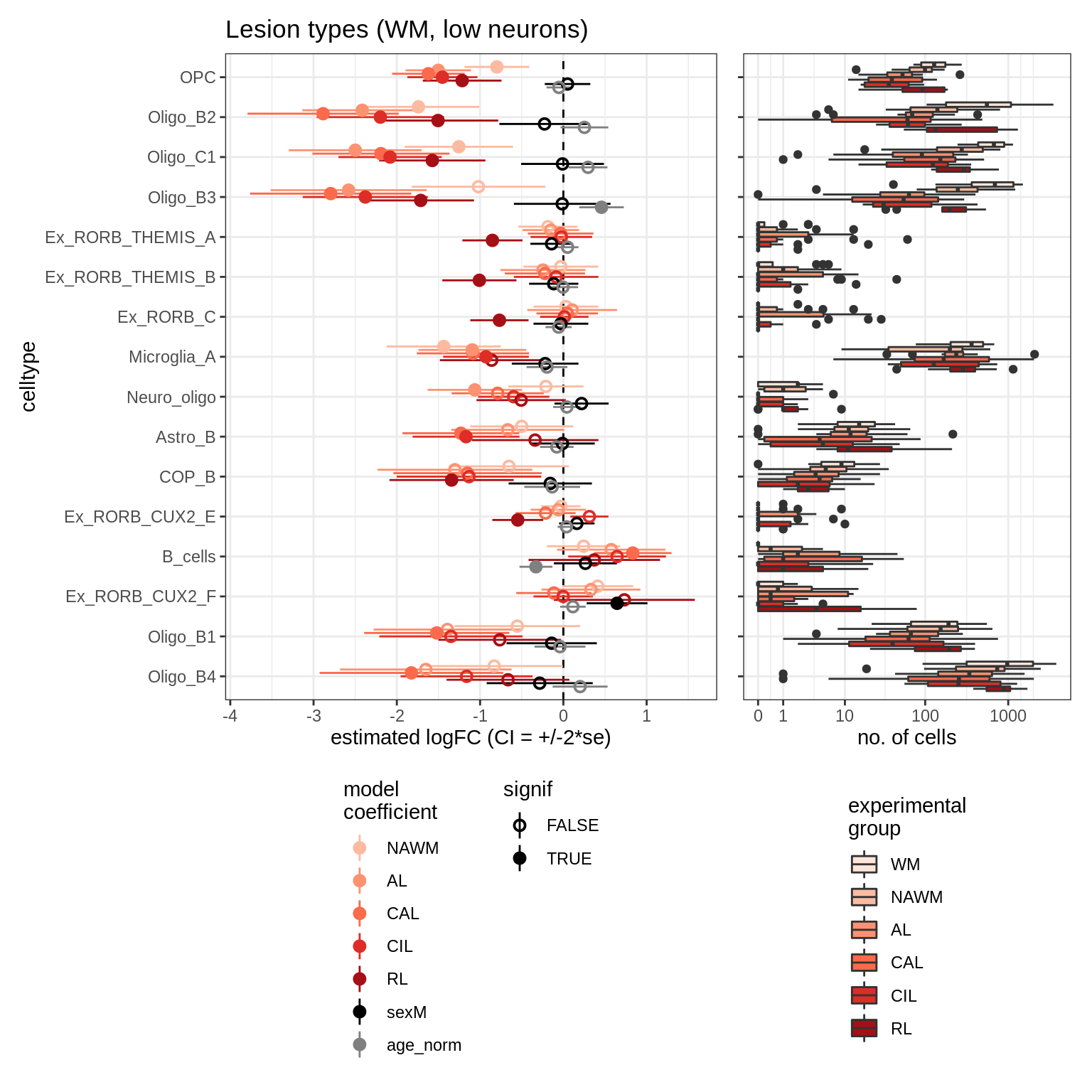
'Lesion types (WM, low neurons)',
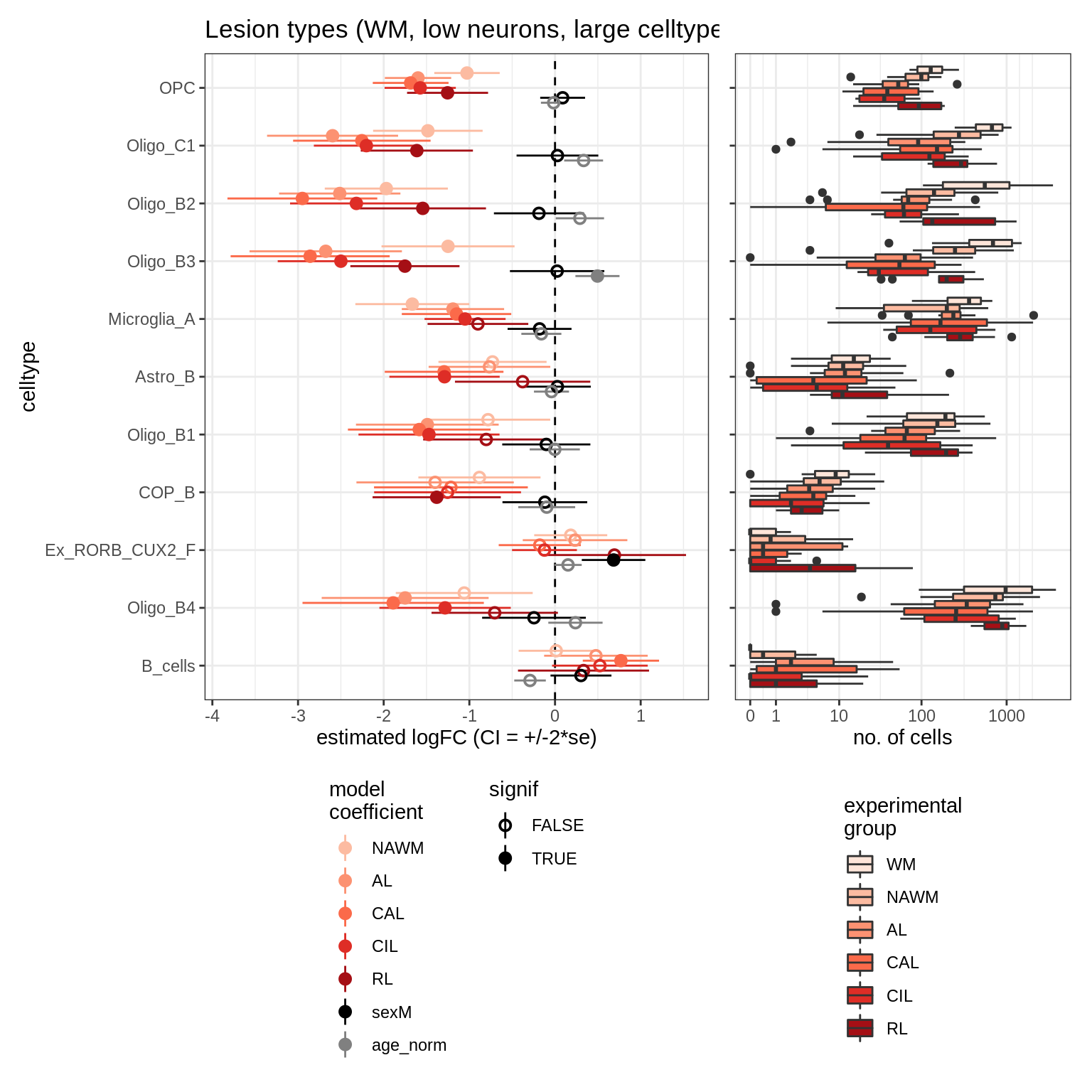
'Lesion types (WM, low neurons, large celltypes)',
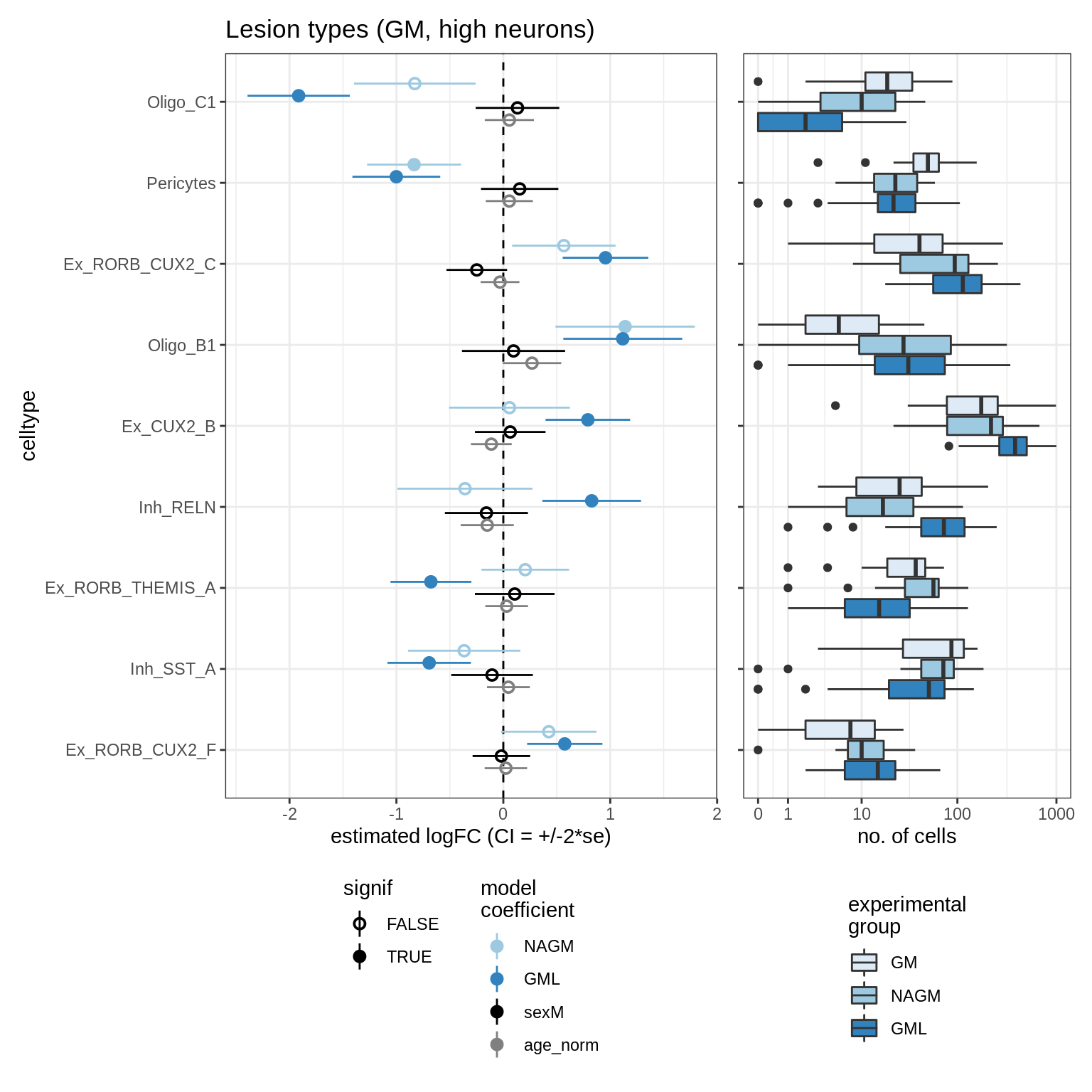
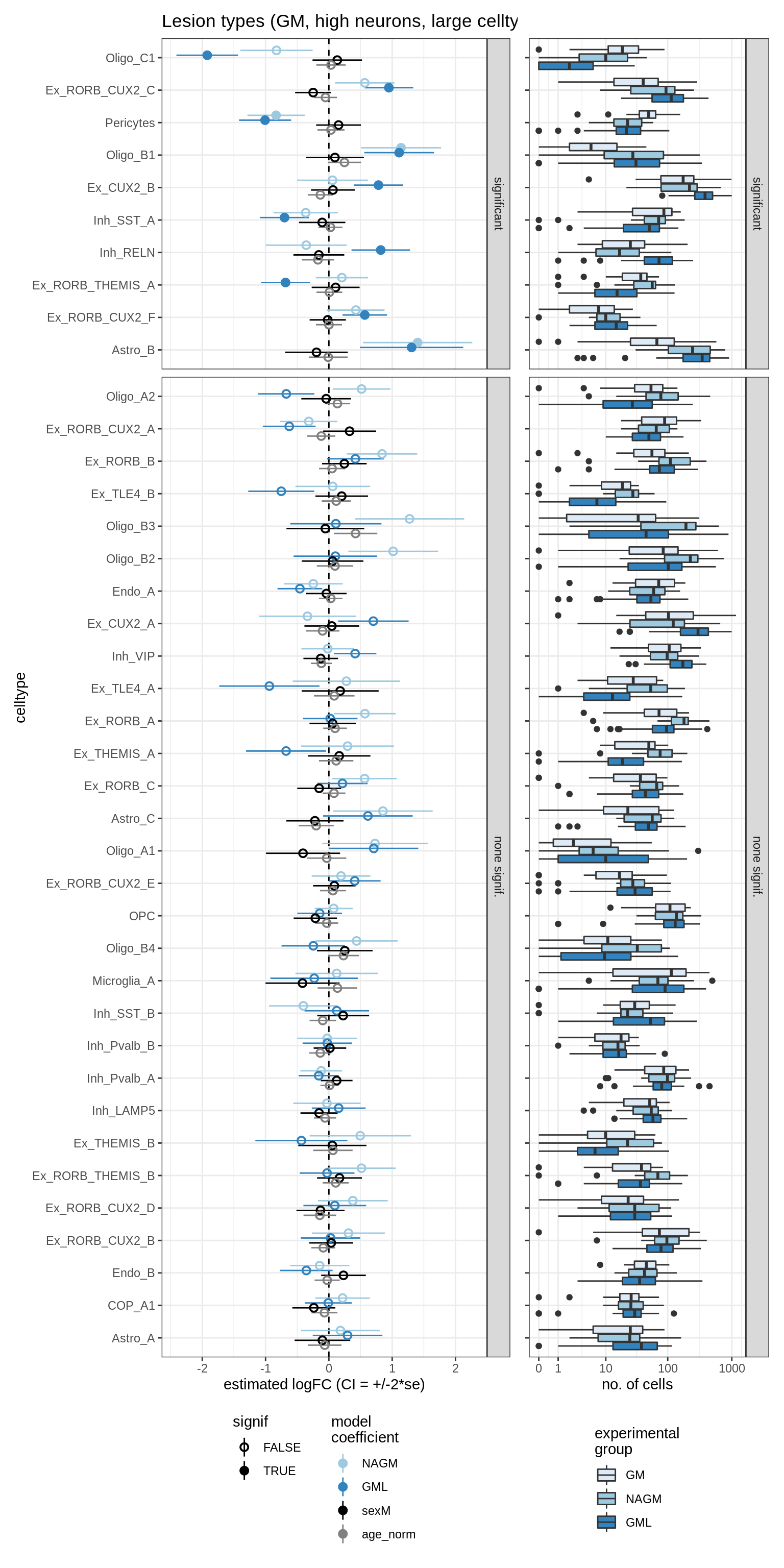
'Lesion types (GM, high neurons)',
'Lesion types (GM, high neurons, large celltypes)',
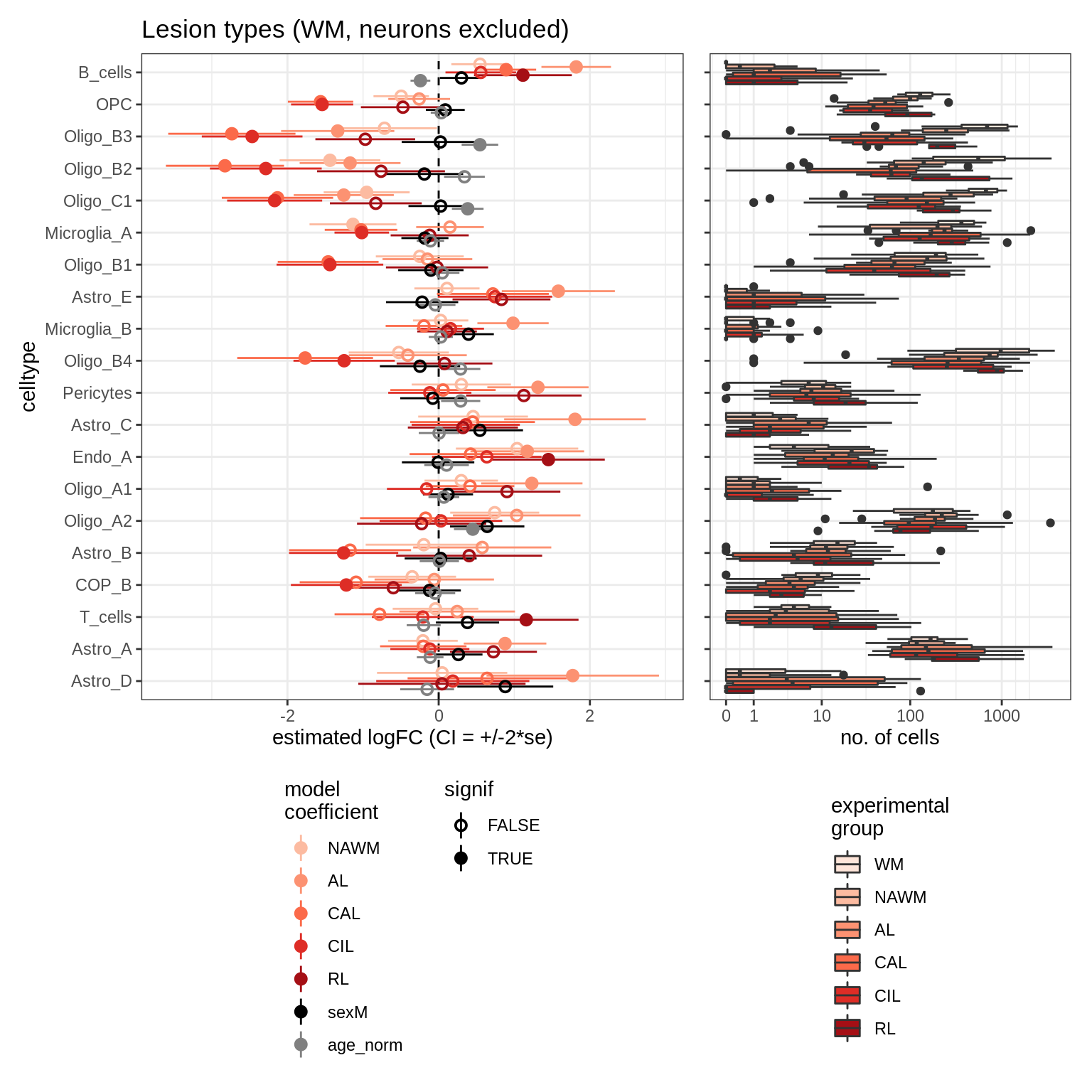
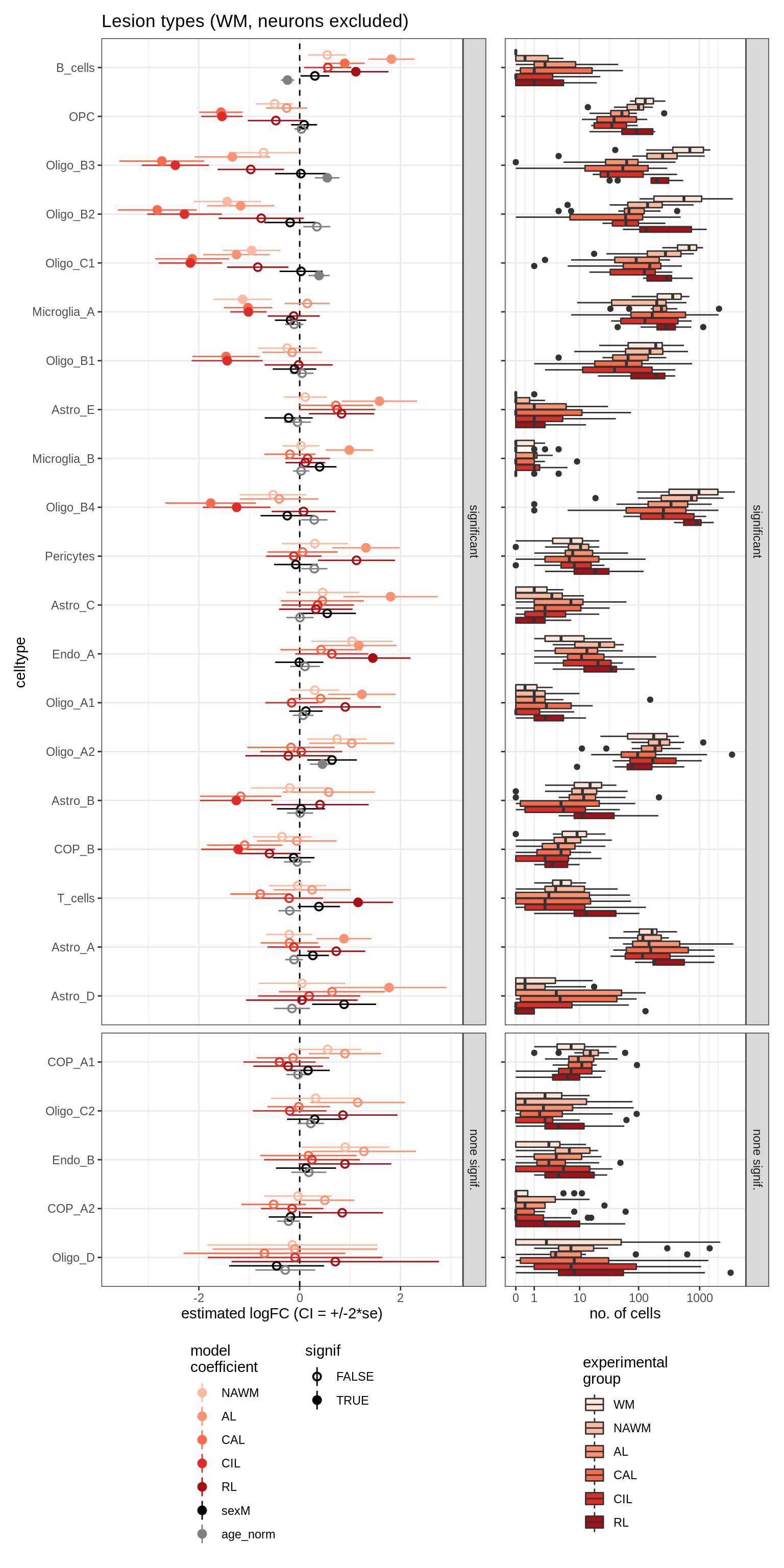
'Lesion types (WM, neurons excluded)',
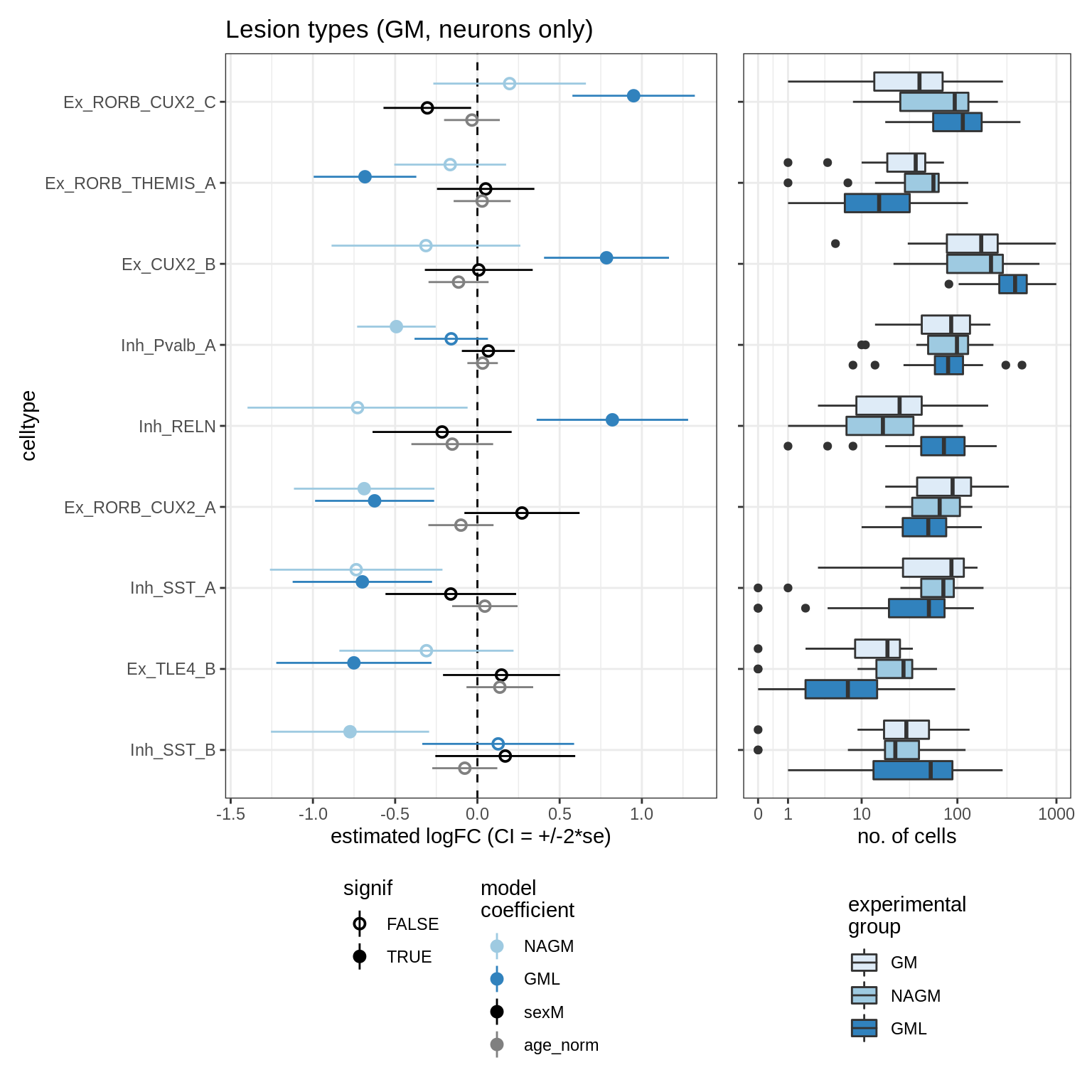
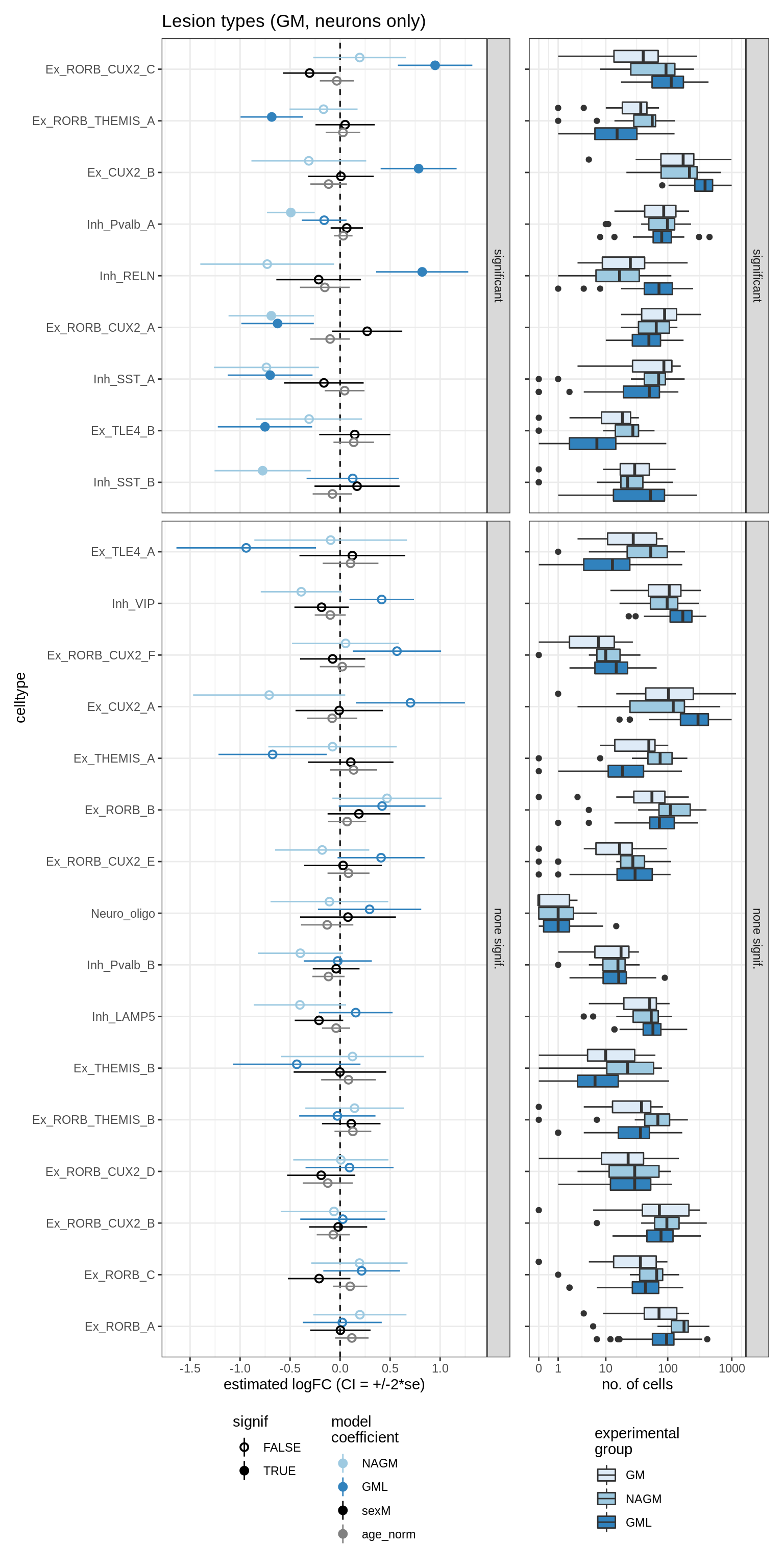
'Lesion types (GM, neurons only)',
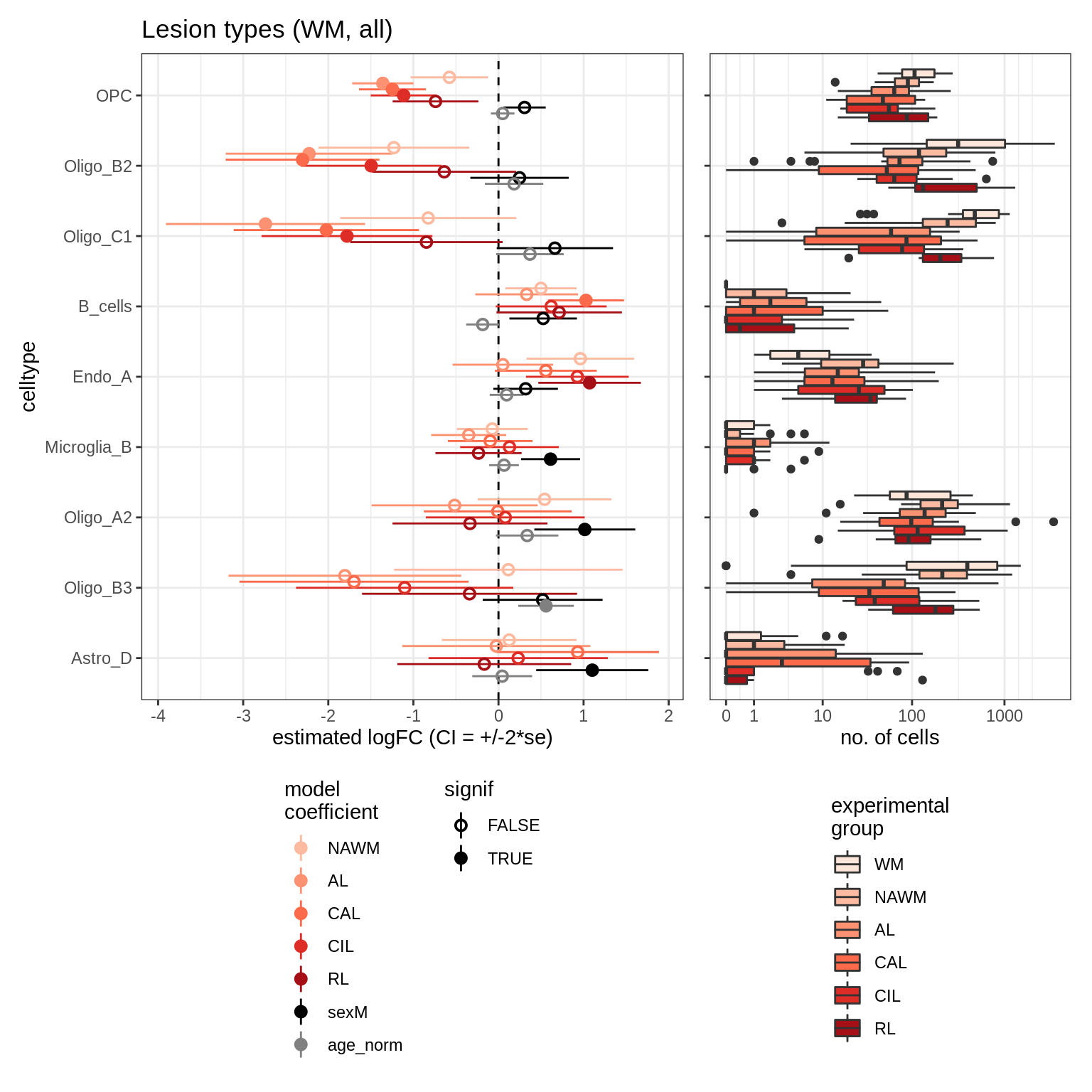
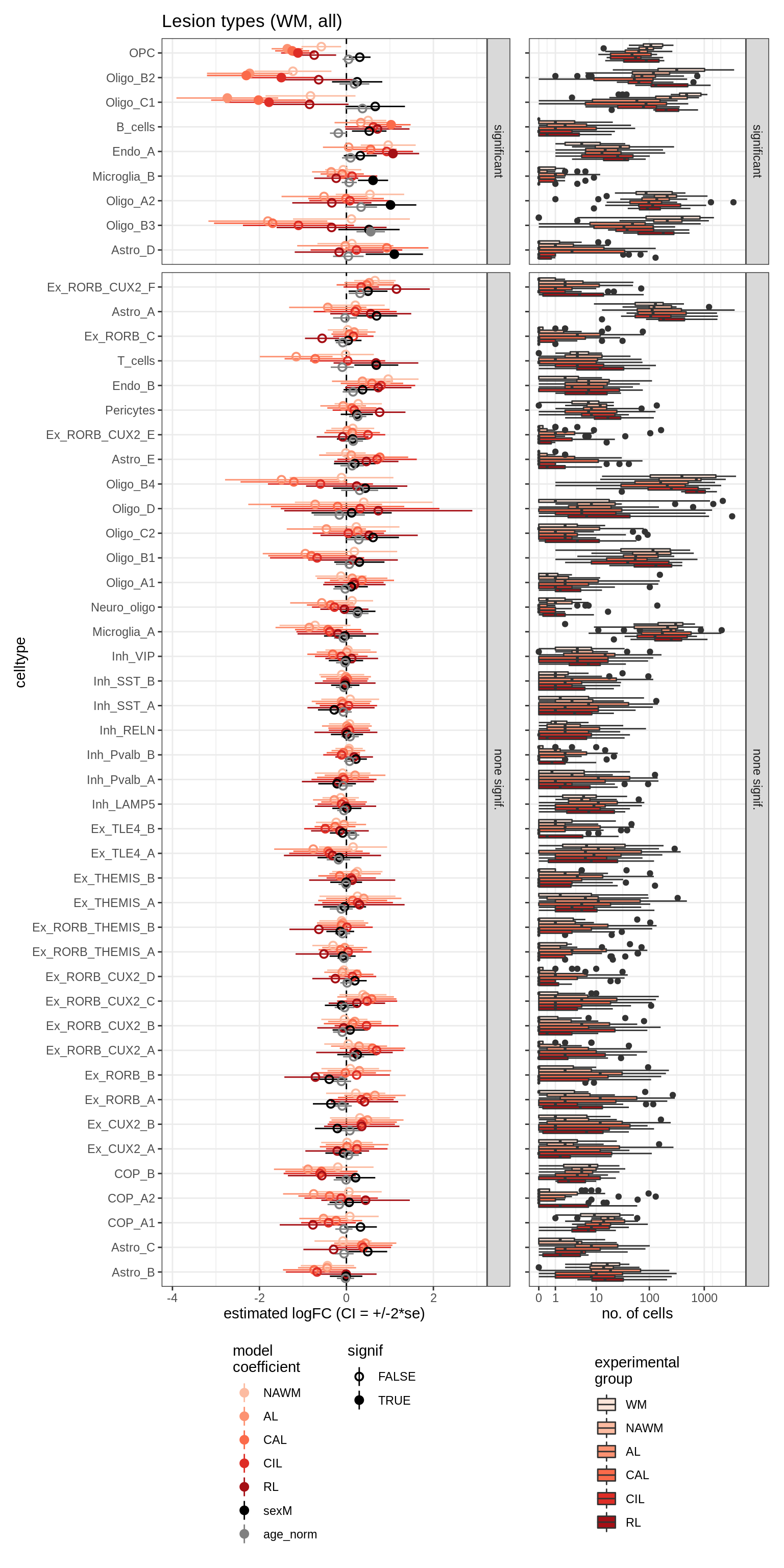
'Lesion types (WM, all)',
'Lesion types (GM, all)',
'GM vs WM',
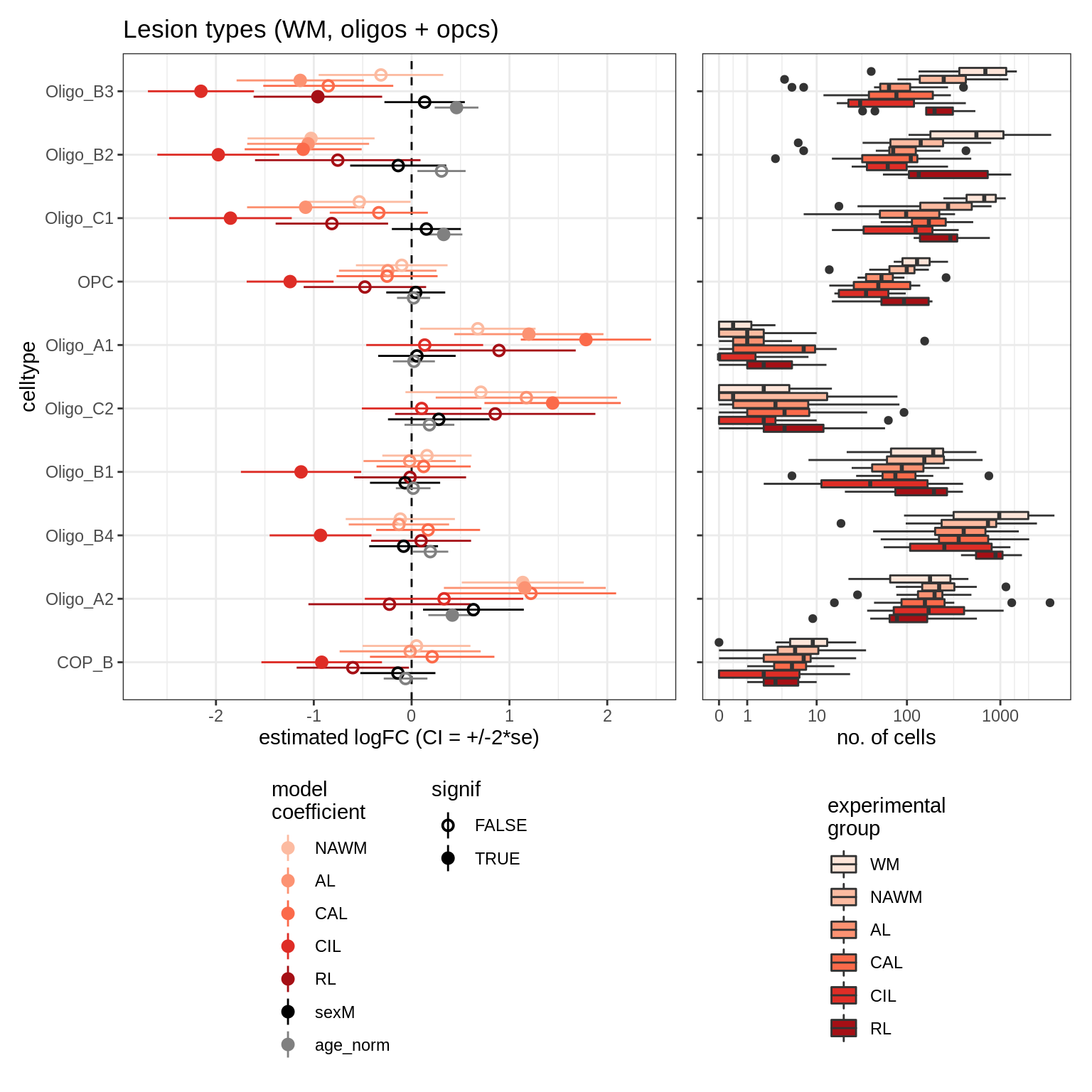
'Lesion types (WM, oligos + opcs)',
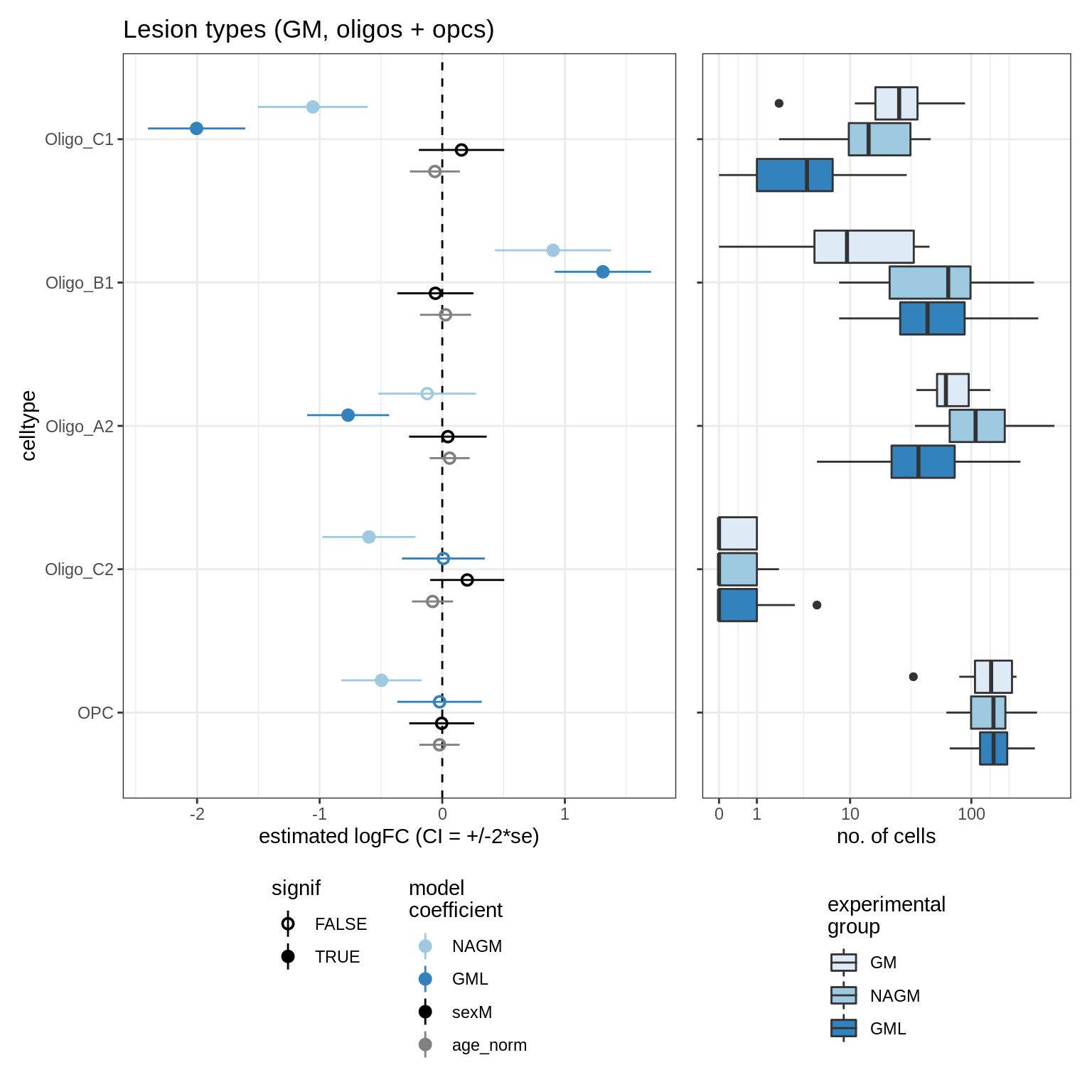
'Lesion types (GM, oligos + opcs)',
'GM vs WM (oligos + opcs)'
) %>% setNames(model_names)
whatplot_list = list(
'lesion_type',
'lesion_type',
'lesion_type',
'lesion_type',
'lesion_type',
'lesion_type',
'lesion_type',
'lesion_type',
'matter',
'lesion_type',
'lesion_type',
'matter'
) %>% setNames(model_names)
ref_type_list = list(
c(`Excitatory neurons` = 'excit-ref'),
c(`Excitatory neurons` = 'excit-ref'),
c(`Excitatory neurons` = 'excit-ref'),
c(`Excitatory neurons` = 'excit-ref'),
c(`Astrocytes` = 'astro-ref'),
c(`Excitatory neurons` = 'excit-ref'),
c(`Excitatory neurons` = 'excit-ref'),
c(`Excitatory neurons` = 'excit-ref'),
c(`OPCs / COPs` = 'opc_cop-ref'),
c(`OPCs / COPs` = 'opc_cop-ref'),
c(`OPCs / COPs` = 'opc_cop-ref'),
c(`OPCs / COPs` = 'opc_cop-ref')
) %>% setNames(model_names)
p_cut = 0.05
# define filenames
ancom_pat = sprintf('%s/ancom_obj_clean_1e3_%s_%s.rds', save_dir, date_tag, '%s')
# define sccoda setup
n_results = 20000L
n_burnin = 5000L
coda_pat = sprintf('%s/sccoda_obj_clean_1e3_%s_%s.p', save_dir, date_tag, '%s')Load inputs
labels_dt = load_names_dt(labels_f) %>%
.[, cluster_id := type_fine]
conos_dt = load_labelled_dt(labelled_f, labels_f)meta_dt = load_meta_dt(meta_f)Processing / calculations
conos_dt = merge(conos_dt, meta_dt, by = 'sample_id') %>%
add_neuro_props(mad_cut = mad_cut)qc_dt = qc_f %>% fread %>%
.[, .(cell_id, log_feats = log10(all_feats),
log_counts = log10(all_counts))] %>%
.[, feat_ok := log_feats >= log10(feat_cut)]
feat_ok_ids = qc_dt[feat_ok == TRUE]$cell_id
conos_dt = conos_dt[cell_id %in% feat_ok_ids]props_dt = calc_props_dt(conos_dt, sample_vars)counts_wide = calc_counts_wide(props_dt, sample_vars)pca_list = calc_pca(props_dt, by_what = 'sample')
pca_wm = calc_pca(props_dt[matter == 'WM' & neuro_ok == TRUE], by_what = 'sample')
pca_gm = calc_pca(props_dt[matter == 'GM' & neuro_ok == TRUE], by_what = 'sample')# do fitting
n_models = length(model_names)
bpparam = MulticoreParam(workers = n_models)
ancom_list = bplapply(seq.int(n_models),
function(i) {
nn = model_names[[i]]
if (!is.null(subset_list[[nn]]$type_spec)) {
conos_tmp = conos_dt[type_broad %in% subset_list[[nn]]$type_spec]
} else {
conos_tmp = copy(conos_dt)
}
props_tmp = calc_props_dt(conos_tmp, sample_vars)
wide_tmp = calc_counts_wide(props_tmp, sample_vars)
fit_ancombc_fn(ancom_pat, nn, wide_tmp, subset_list[[nn]]$subset_spec,
subset_list[[nn]]$size_spec, formulae[[nn]], sample_vars, seed = i)
}, BPPARAM = bpparam) %>% setNames(model_names)
bpstop()# do fitting
coda_list = lapply(seq.int(n_models),
function(i) {
nn = model_names[[i]]
if (!is.null(subset_list[[nn]]$type_spec)) {
conos_tmp = conos_dt[type_broad %in% subset_list[[nn]]$type_spec]
} else {
conos_tmp = copy(conos_dt)
}
# fit coda
coda_obj = run_sccoda(coda_pat, nn,
conos_tmp, sample_vars, ref_type_list[[nn]],
subset_list[[nn]]$subset_spec, subset_list[[nn]]$size_spec,
formulae[[nn]], num_results = n_results, num_burnin = n_burnin)
}) %>% setNames(model_names)Analysis
Check for outliers
Proportion of neurons by sample
(plot_neuro_prop(props_dt, mad_cut))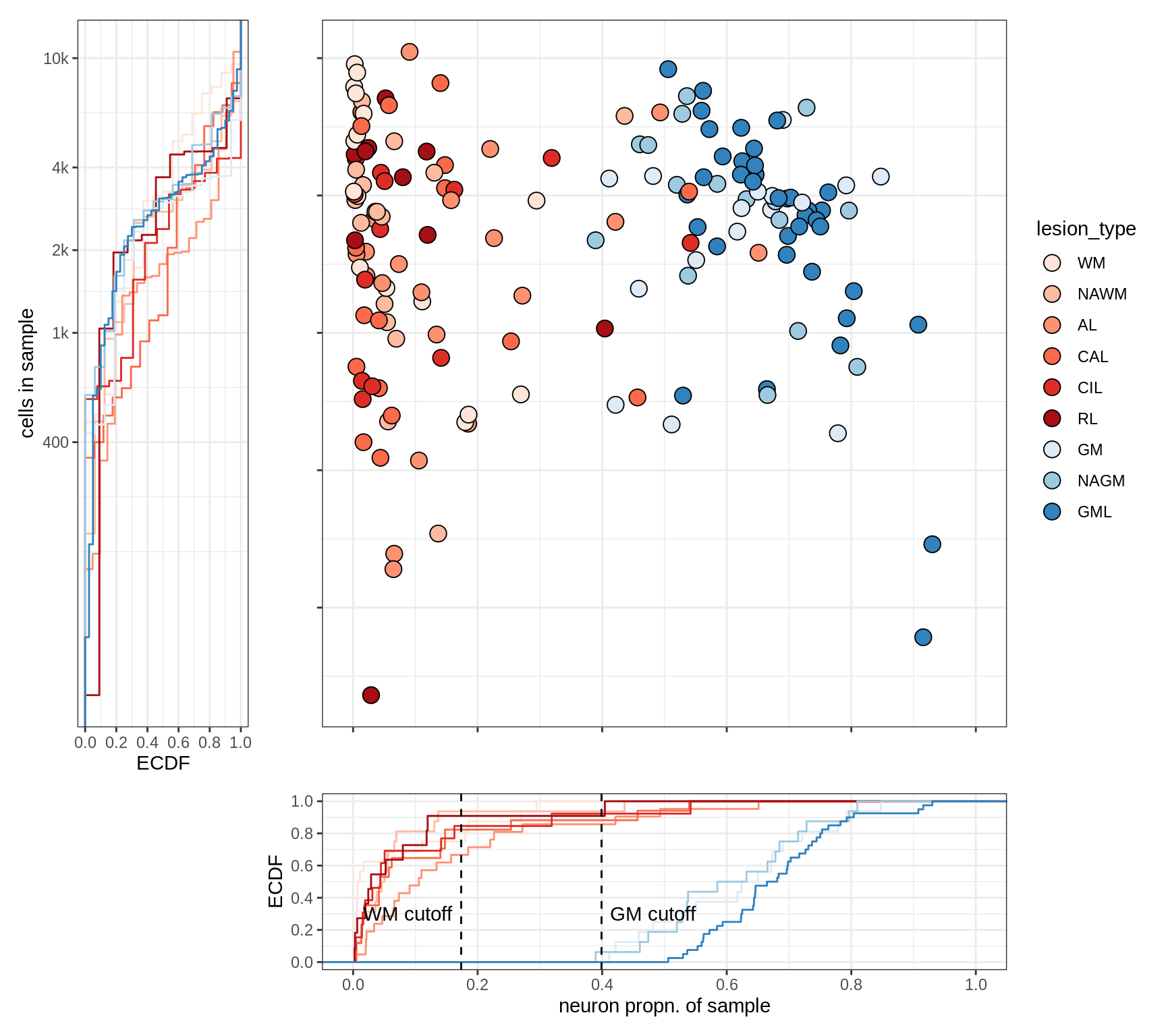
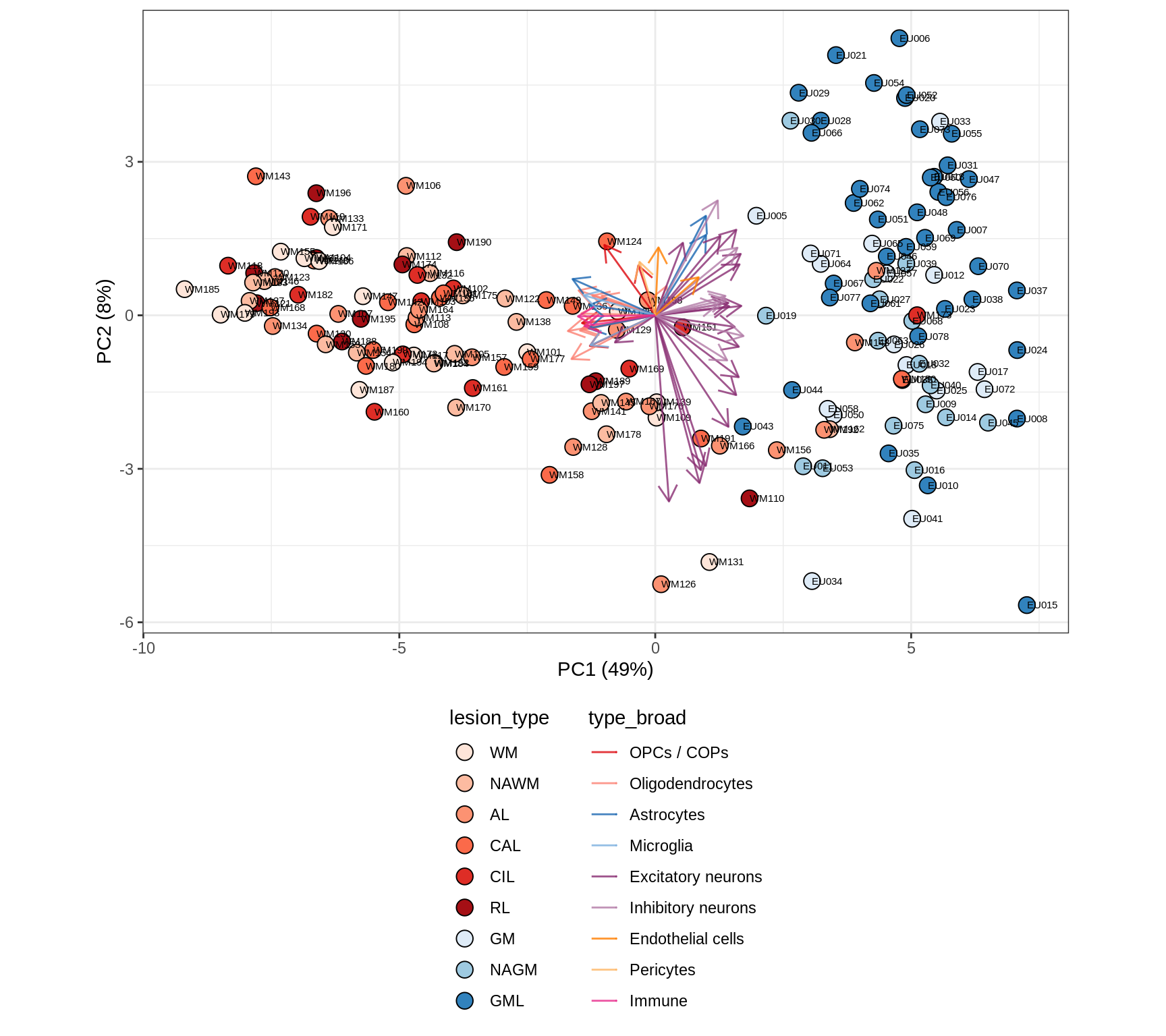
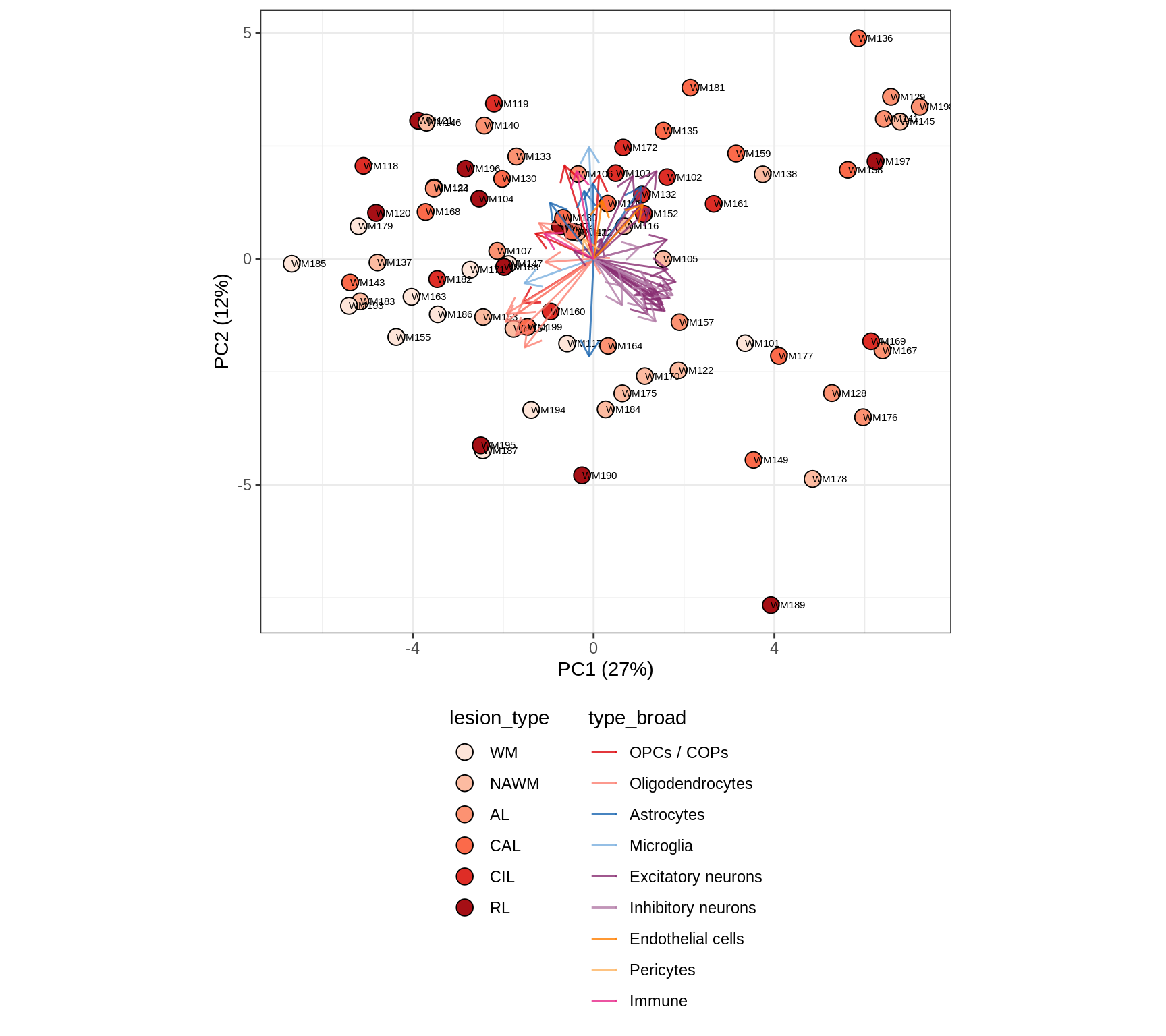
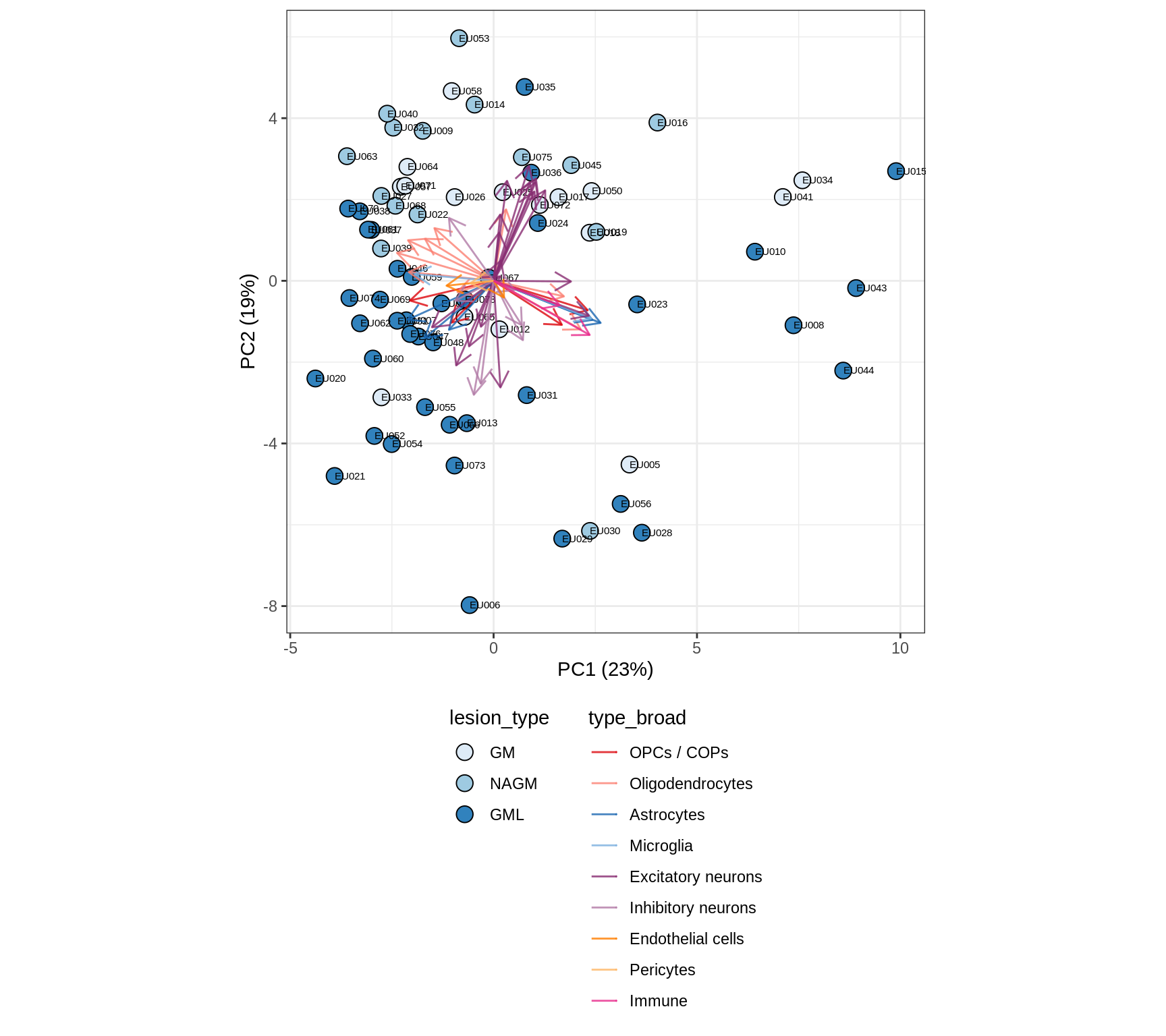
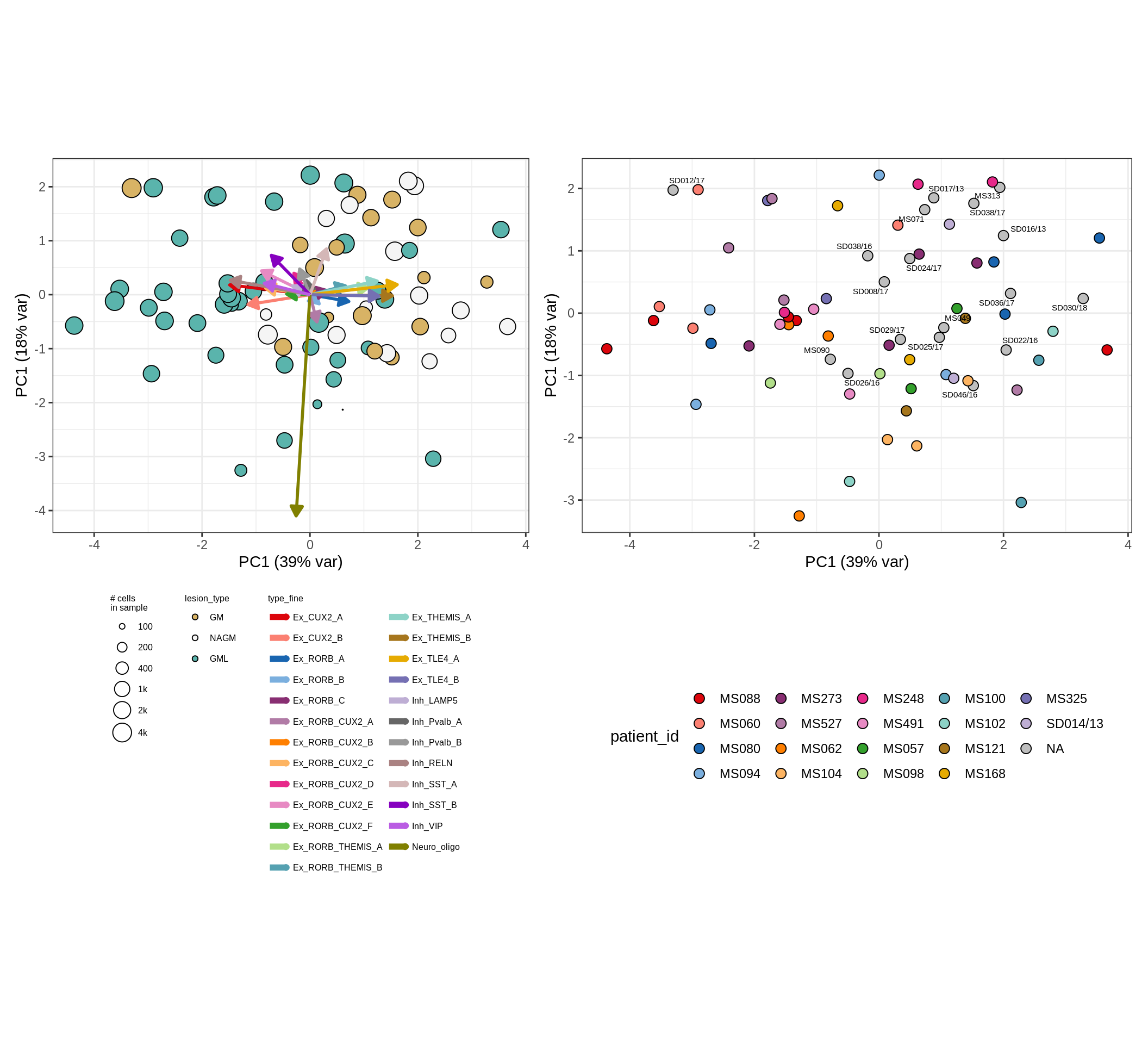
PCA of celltype proportions
for (what in c('all', 'WM', 'GM')) {
cat('#### ', what, '\n')
if (what == 'all') {
print(plot_pca(pca_list))
} else if (what == 'WM') {
print(plot_pca(pca_wm))
} else if (what == 'GM') {
print(plot_pca(pca_gm))
}
cat('\n\n')
}all
WM
GM
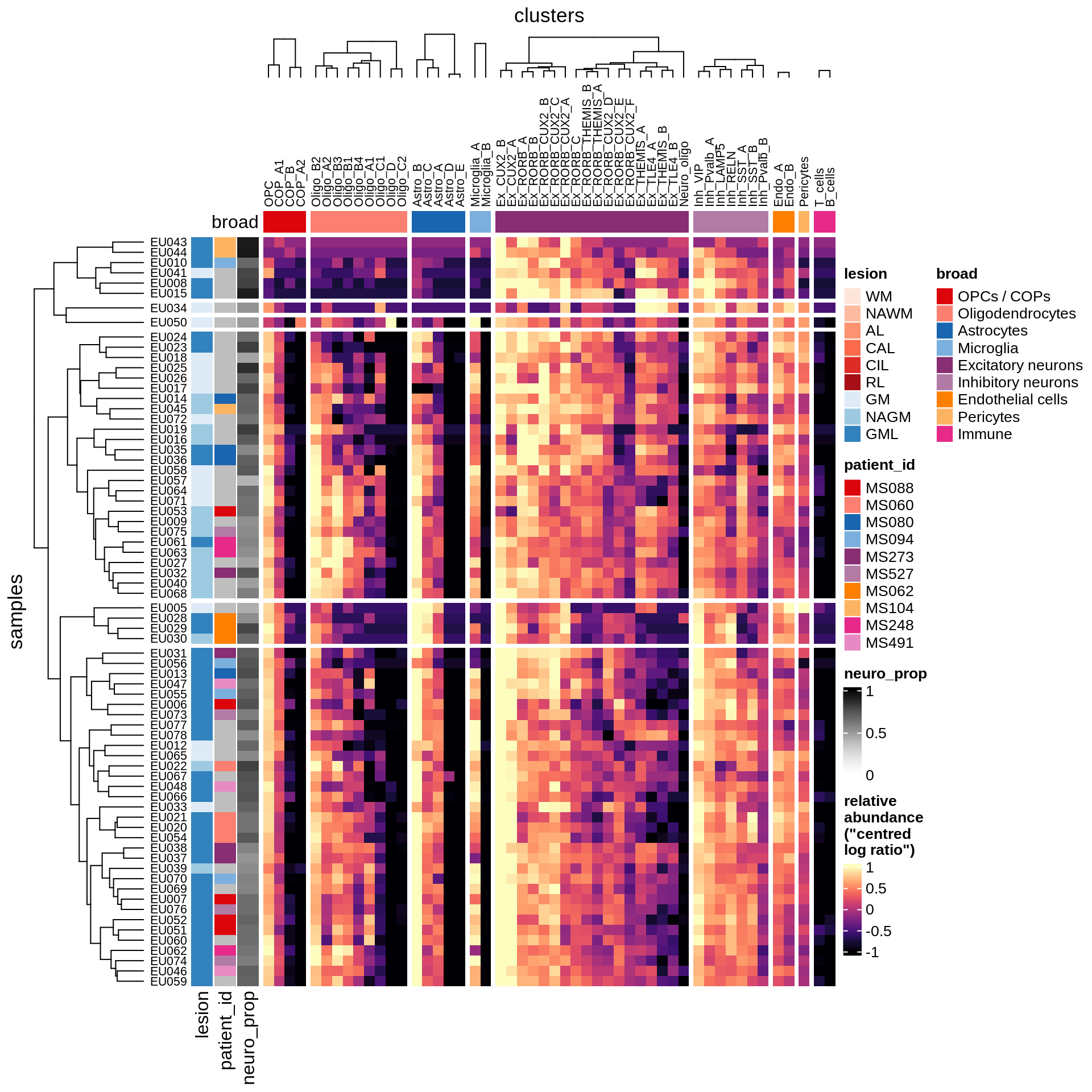
Cluster samples by celltype proportions
for (what in c('all', 'WM', 'GM')) {
cat('### ', what, '\n')
if (what == 'all') {
draw(plot_clr_heatmap(props_dt), row_dend_width = unit(1, "in"))
} else if (what == 'WM') {
draw(plot_clr_heatmap(props_dt[matter == 'WM' & neuro_ok == TRUE]),
row_dend_width = unit(1, "in"))
} else if (what == 'GM') {
draw(plot_clr_heatmap(props_dt[matter == 'GM' & neuro_ok == TRUE]),
row_dend_width = unit(1, "in"))
}
cat('\n\n')
}all
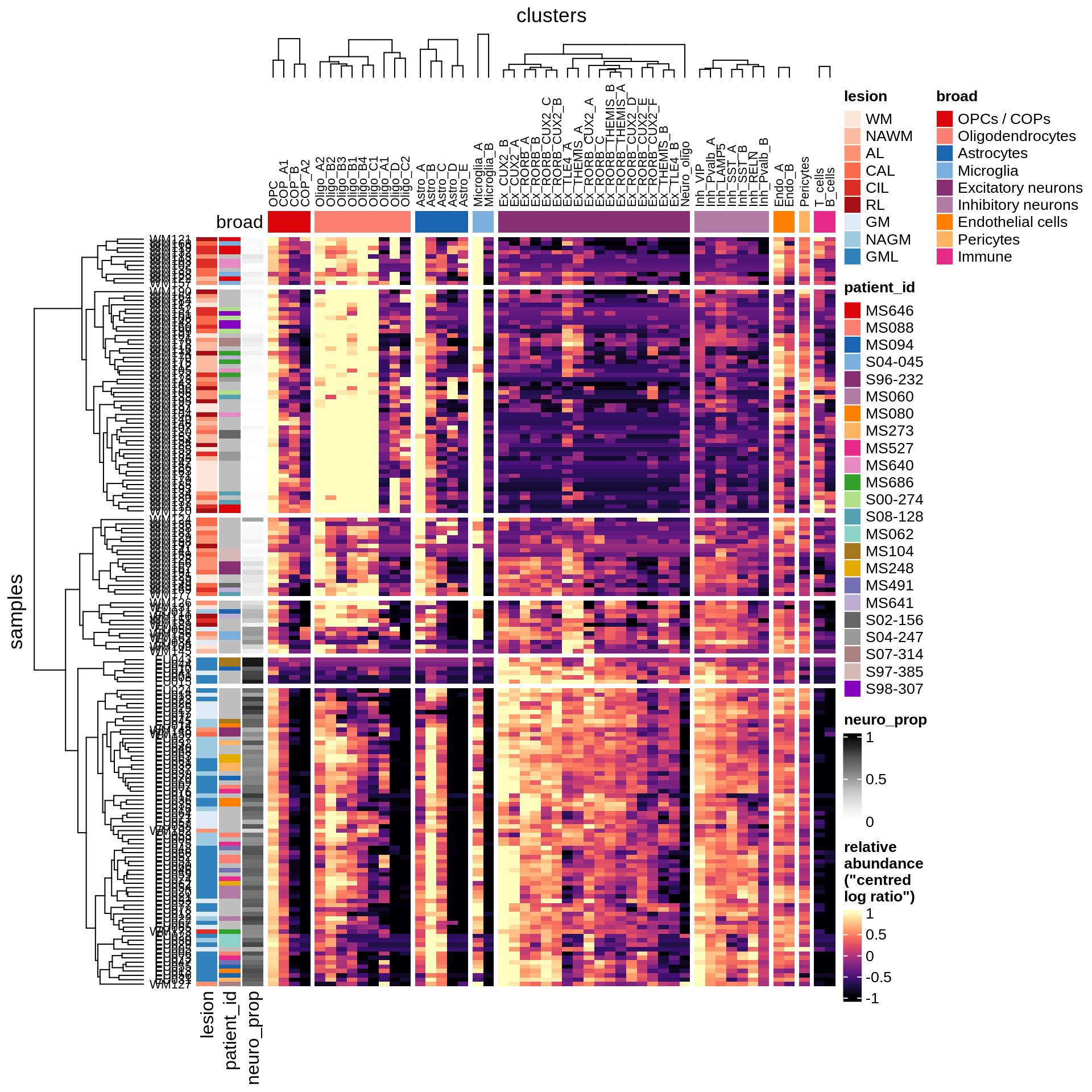
WM
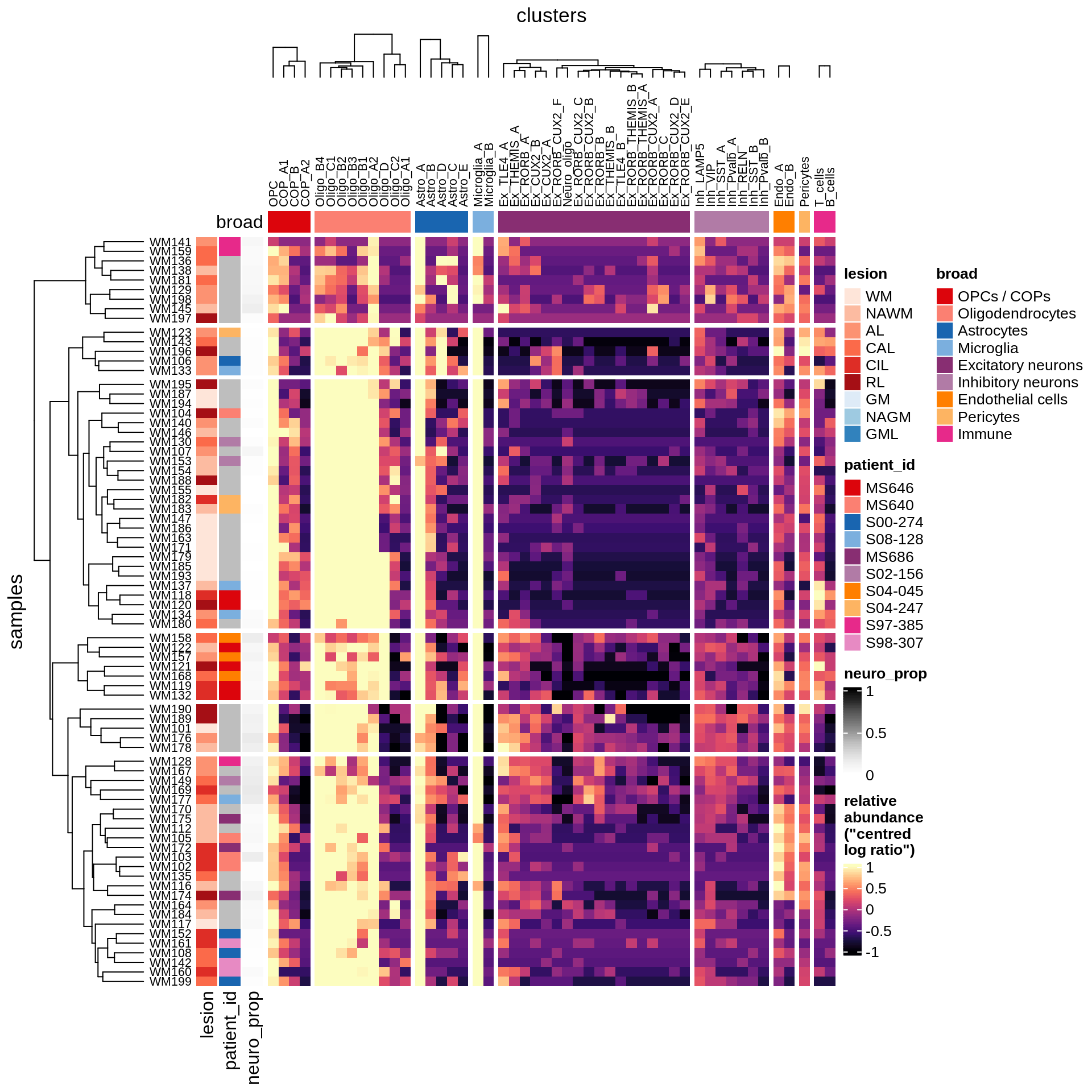
GM
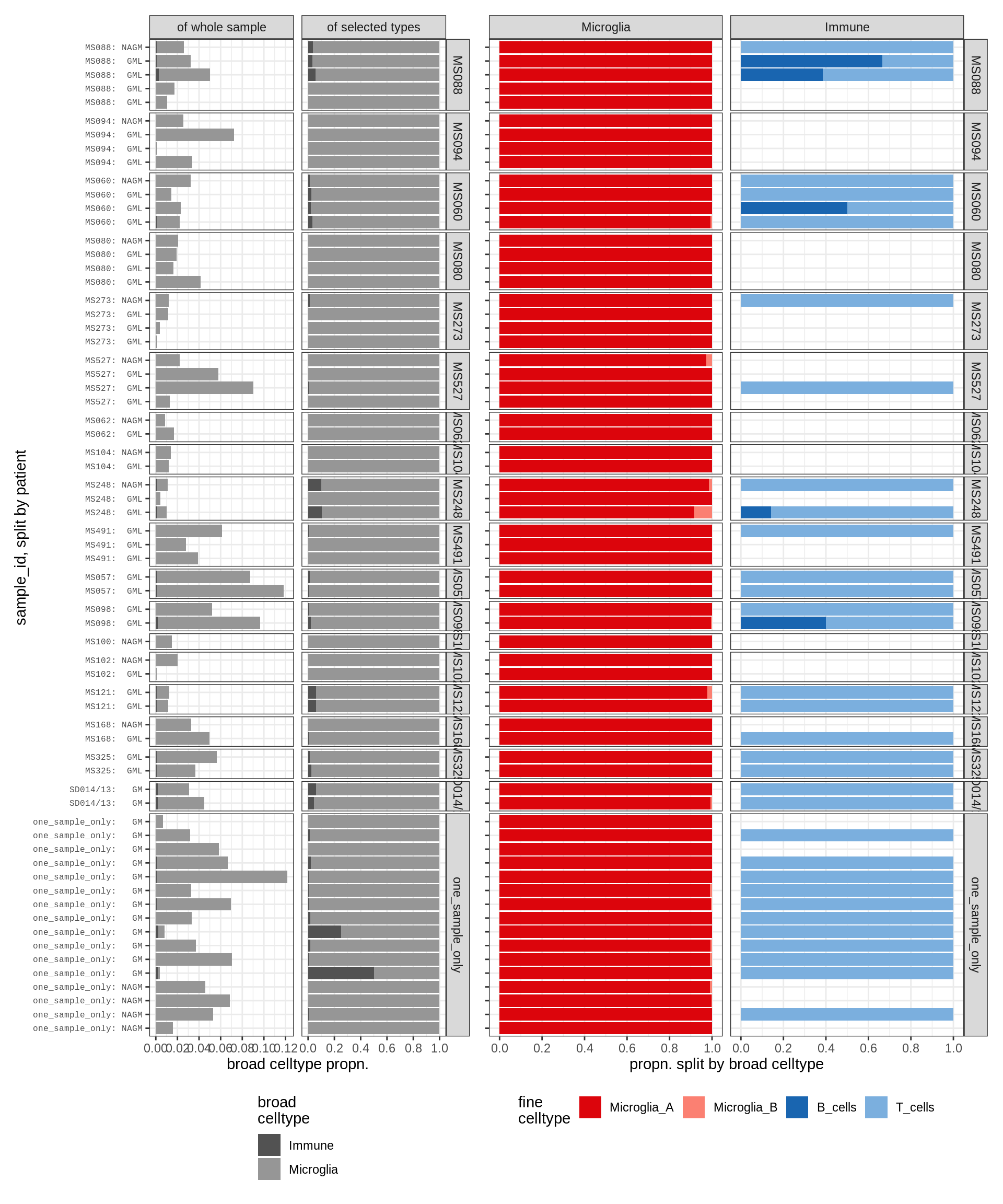
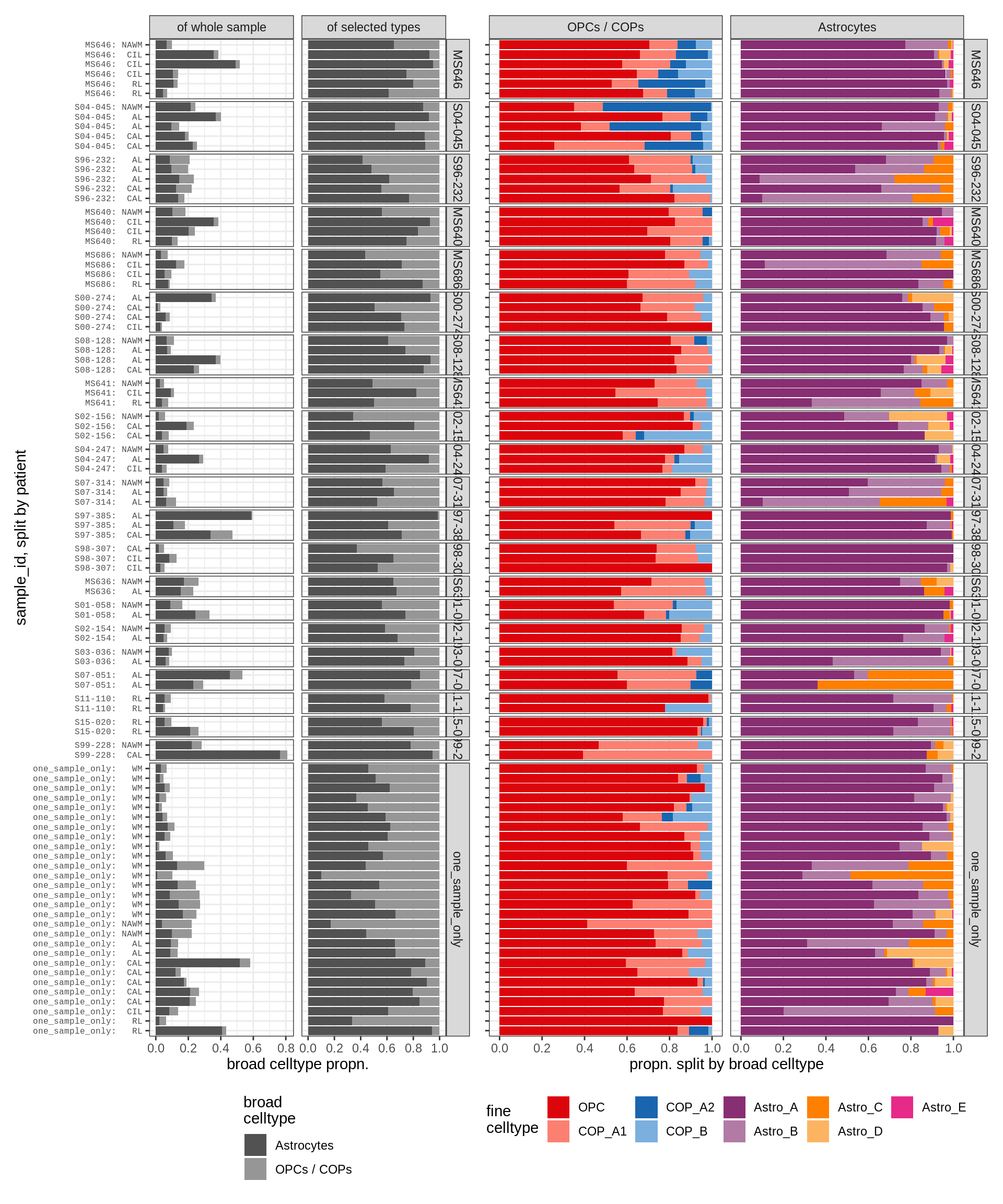
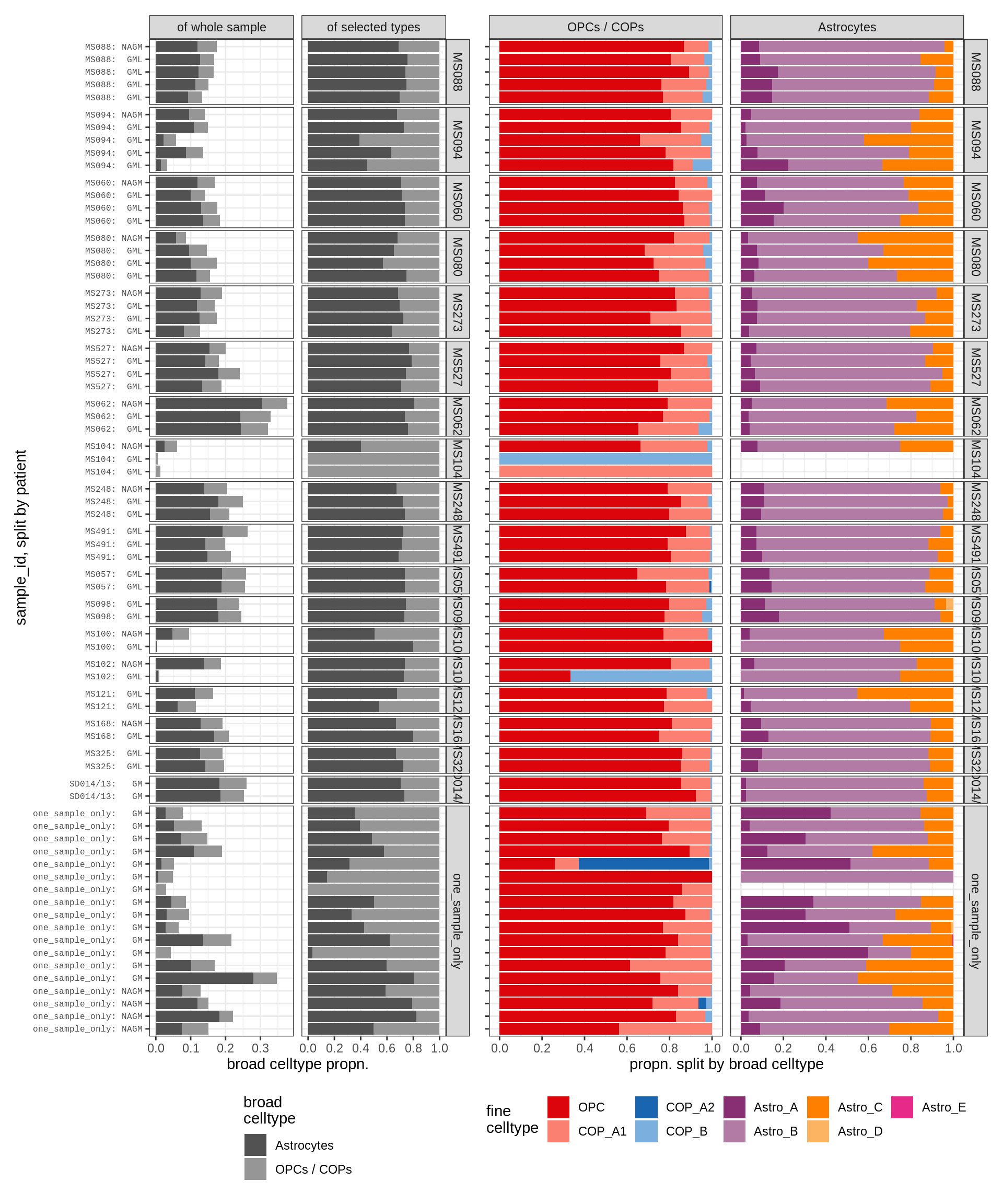
Barplot of celltype proportions split by sample
types_list = list(
oligo_opc = c('OPCs / COPs', 'Oligodendrocytes'),
neurons = c('Excitatory neurons', 'Inhibitory neurons'),
micro_immune = c('Microglia', 'Immune'),
astro_opc = c('OPCs / COPs', 'Astrocytes'),
endo_peri = c('Endothelial cells', 'Pericytes')
)
for (t in names(types_list)) {
for (m in c('WM', 'GM')) {
cat('### ', t, ', ', m, '\n')
types = types_list[[t]]
print(plot_sample_splits(conos_dt[matter == m], types = types))
cat('\n\n')
}
}oligo_opc , WM
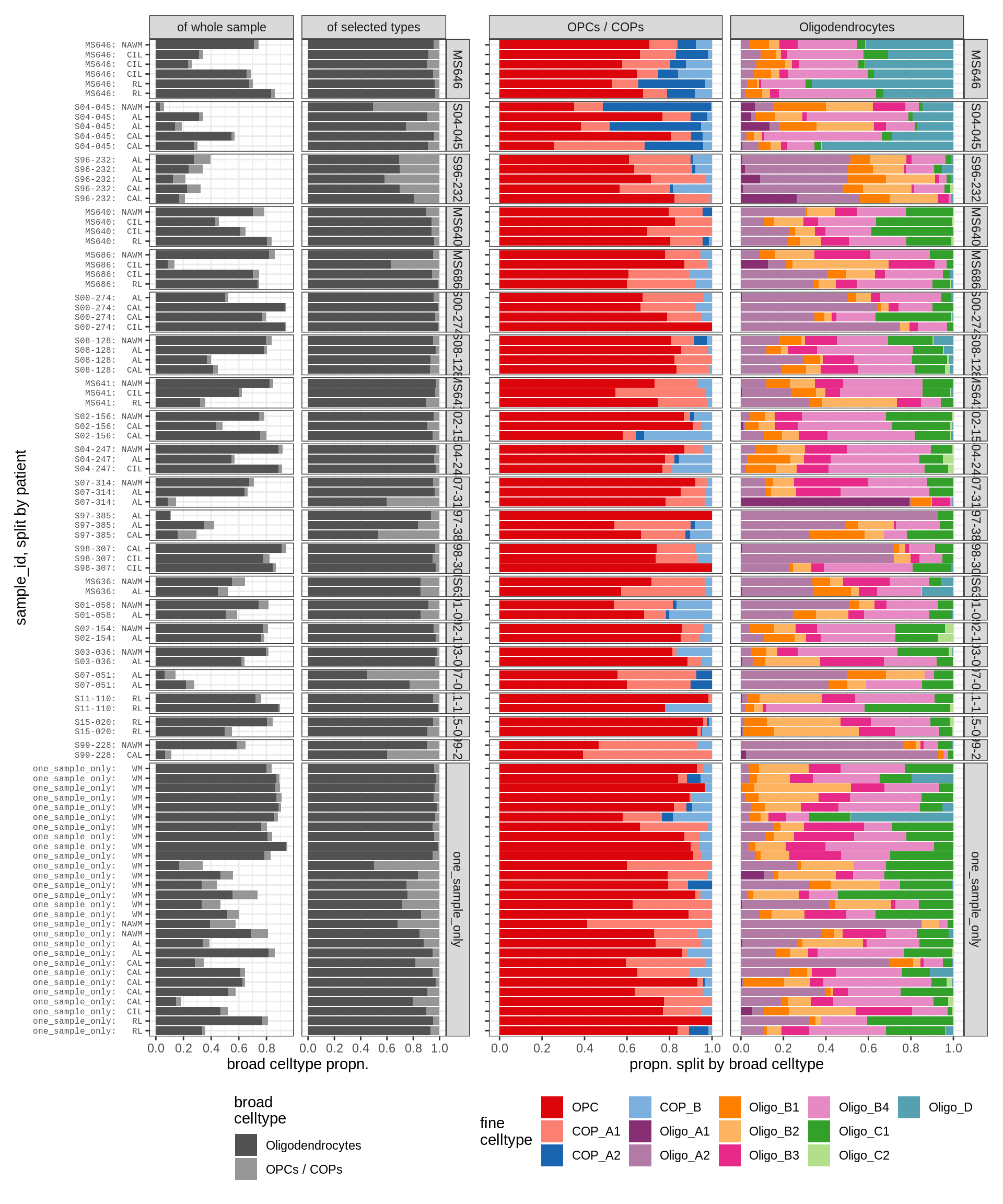
oligo_opc , GM
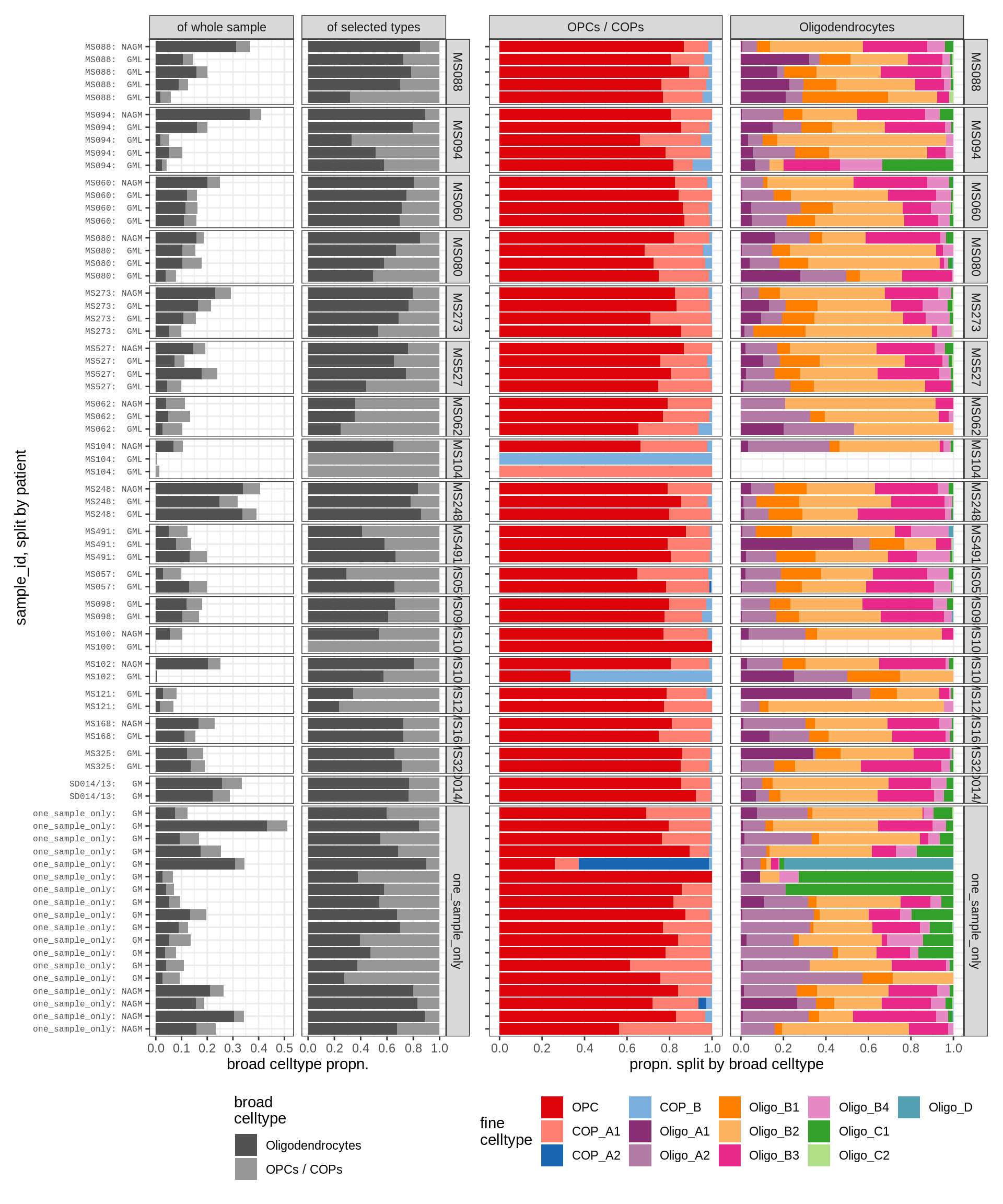
neurons , WM
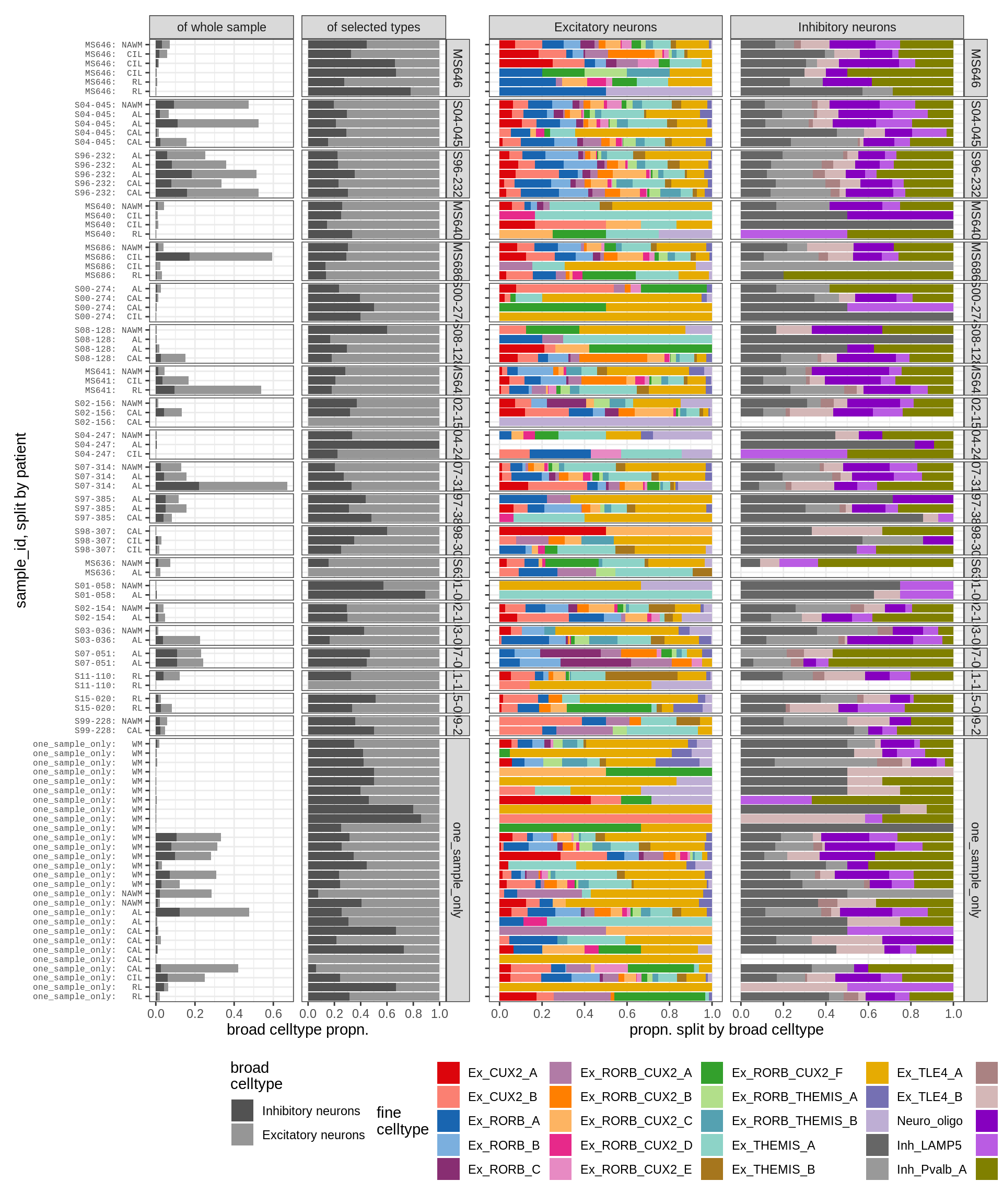
neurons , GM
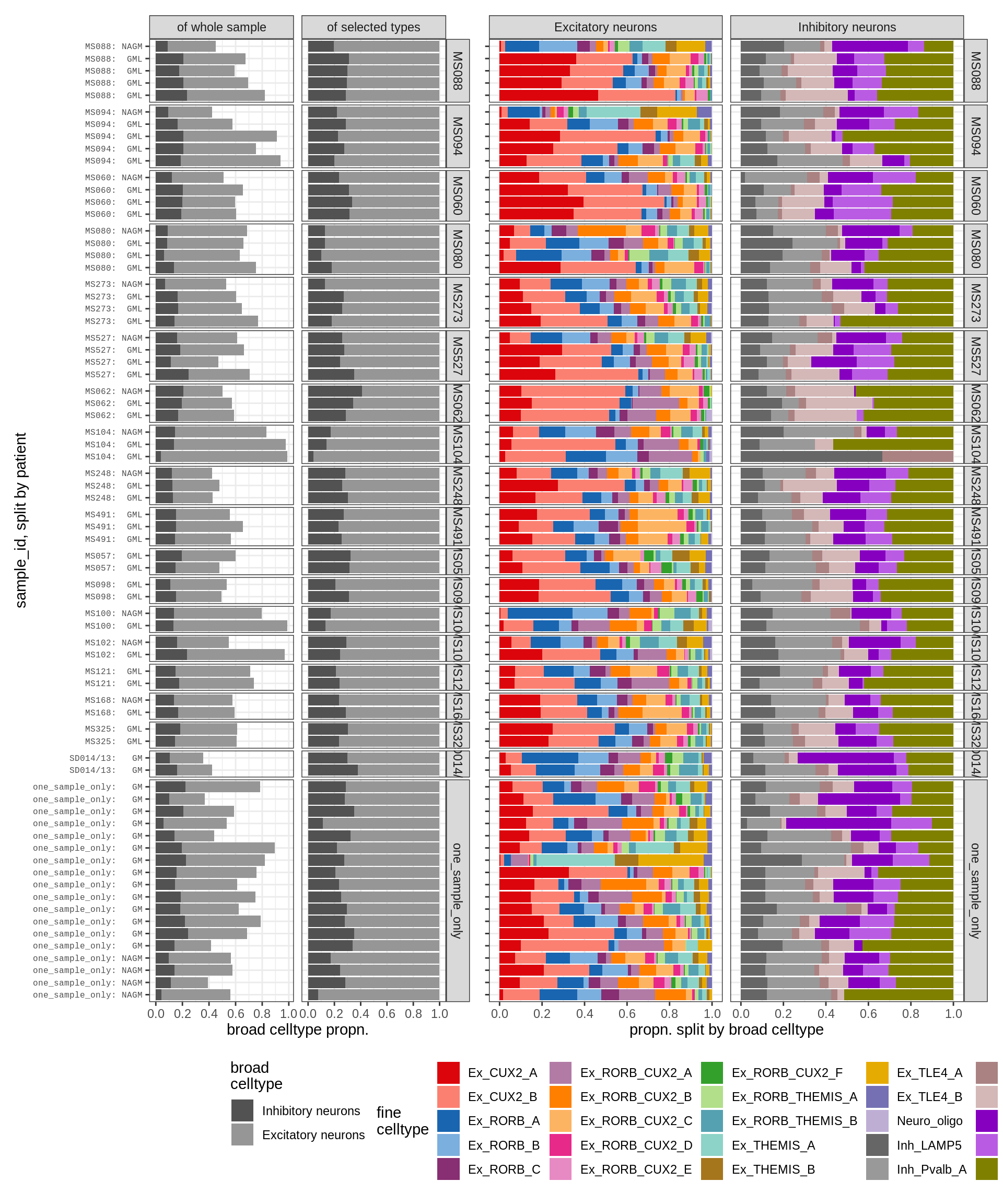
micro_immune , WM
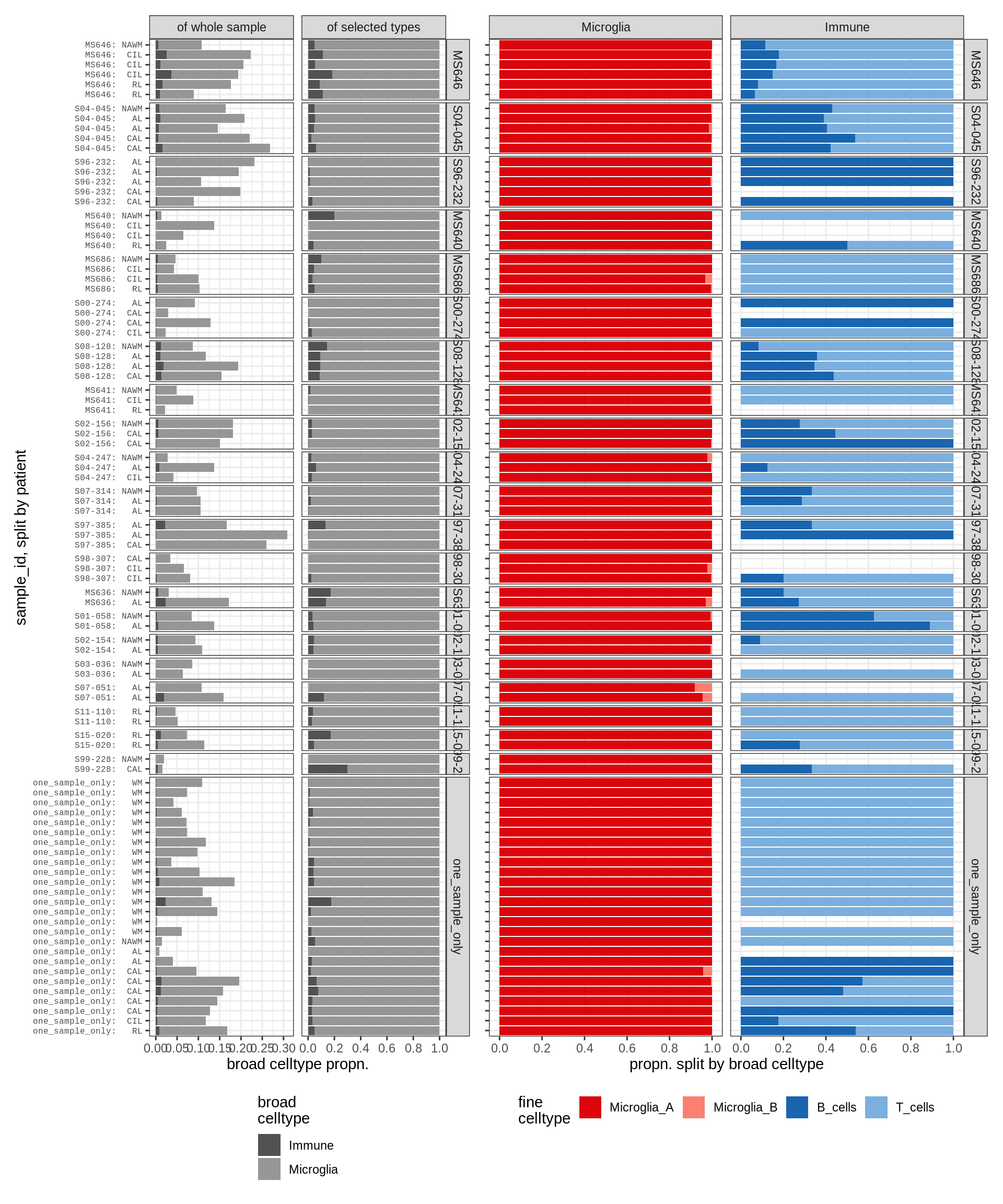
micro_immune , GM
astro_opc , WM
astro_opc , GM
endo_peri , WM
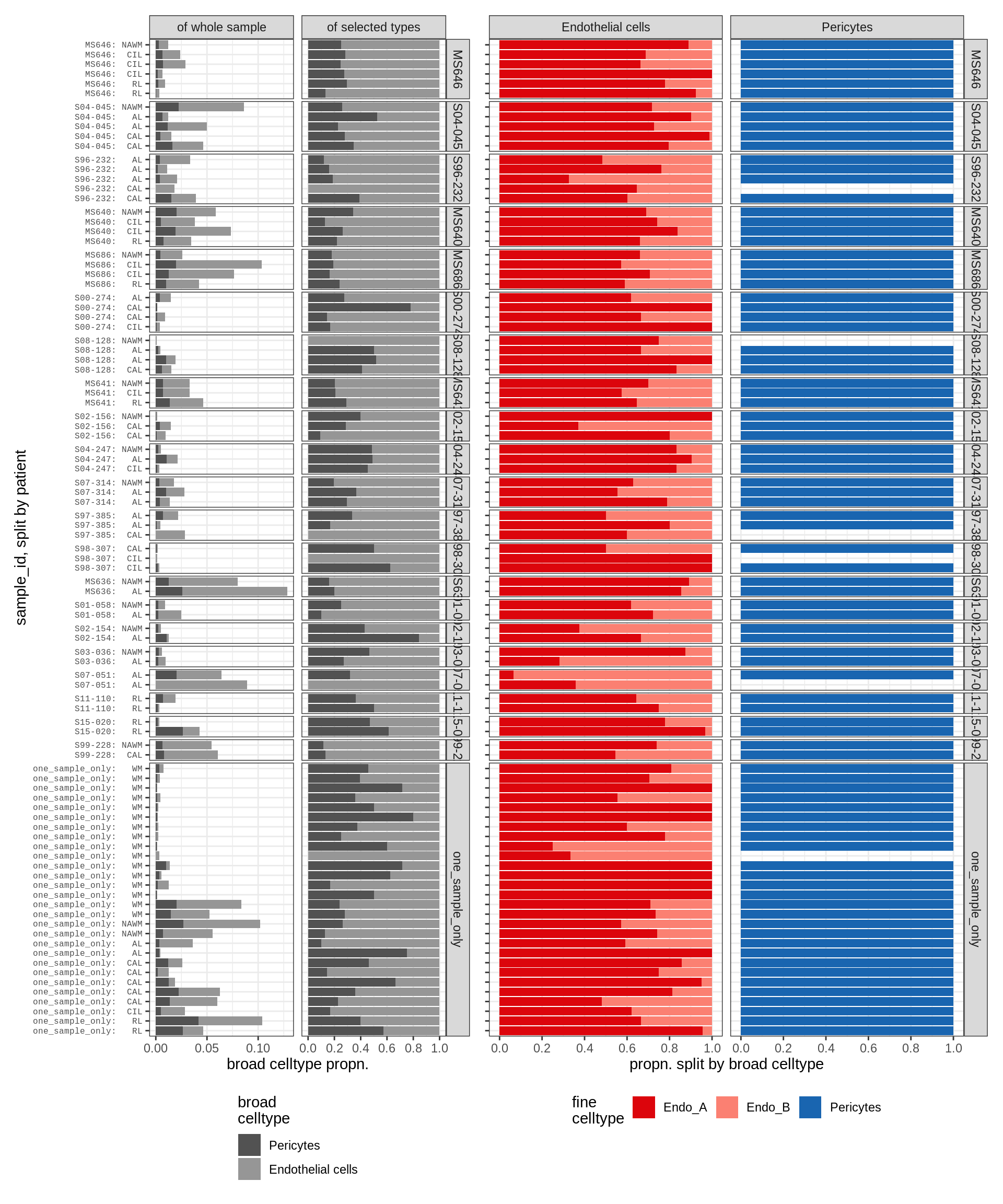
endo_peri , GM
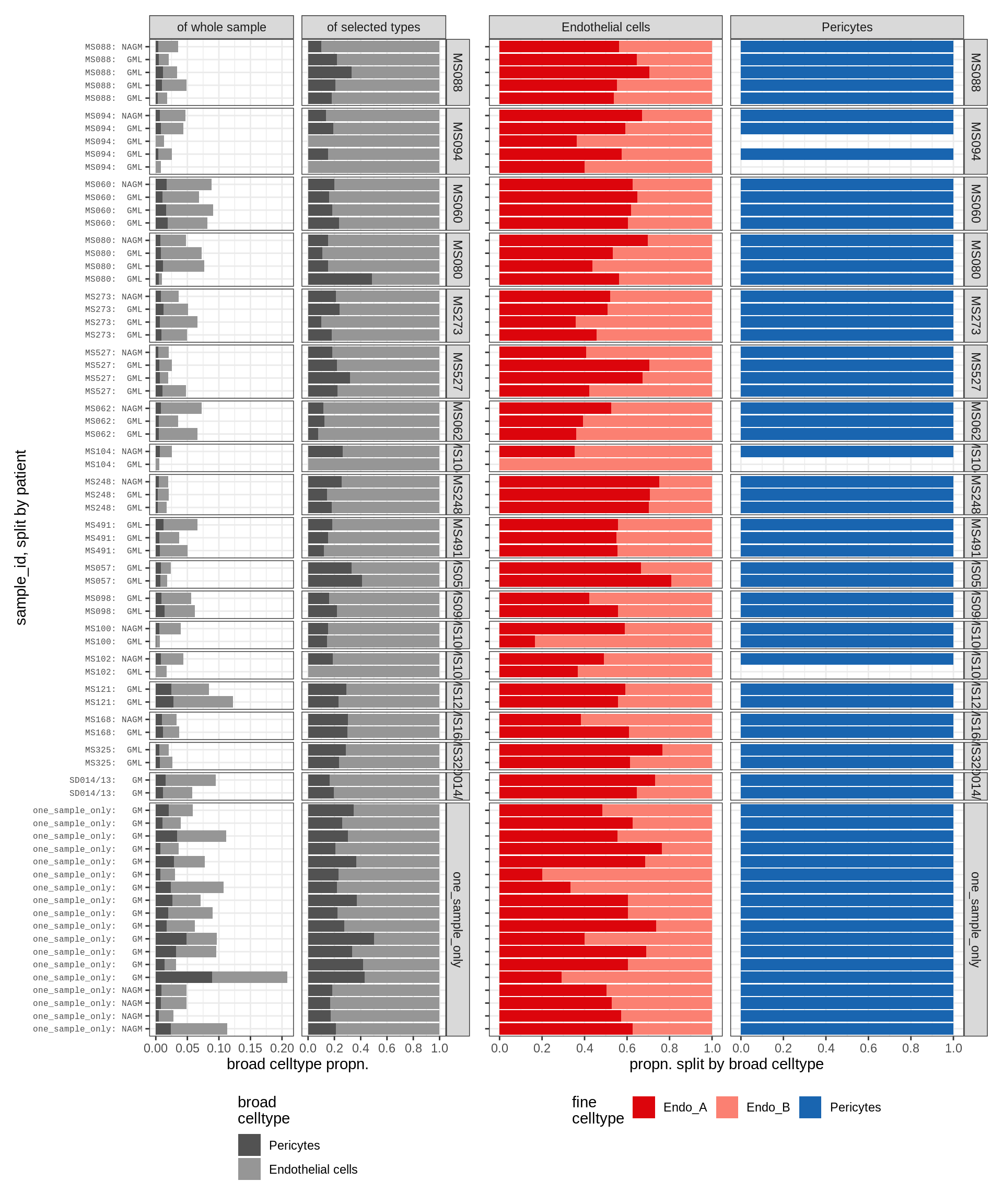
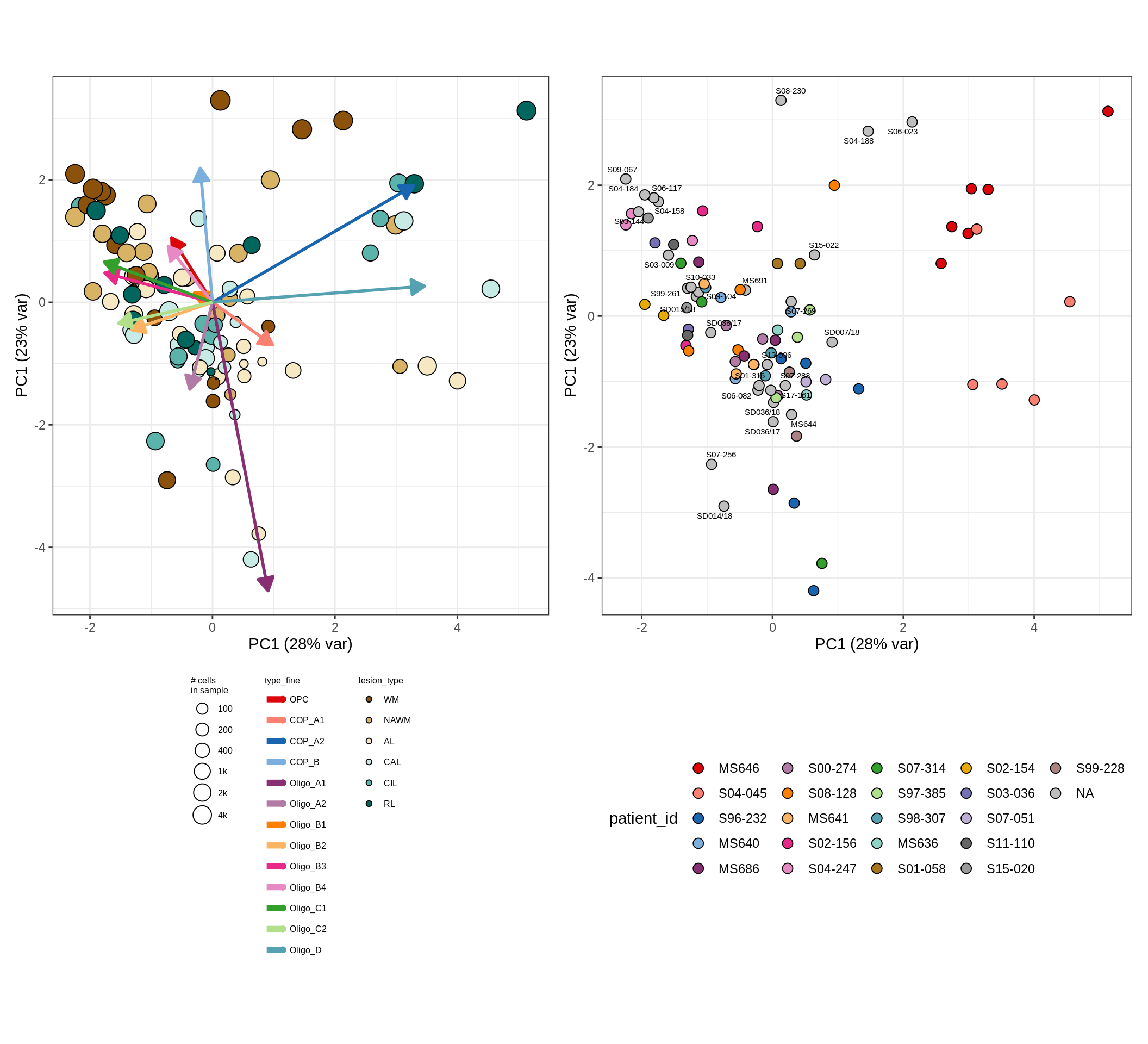
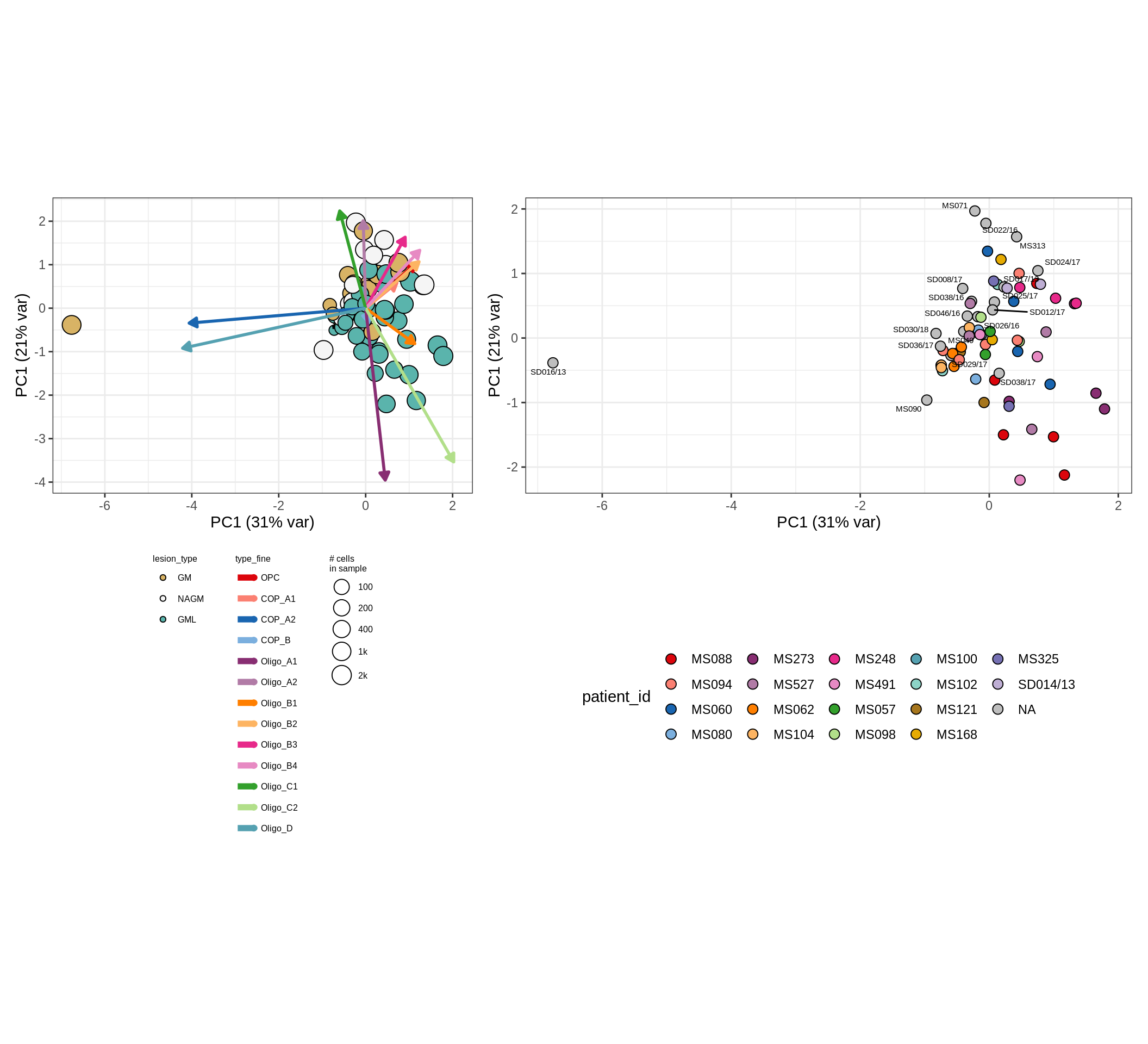
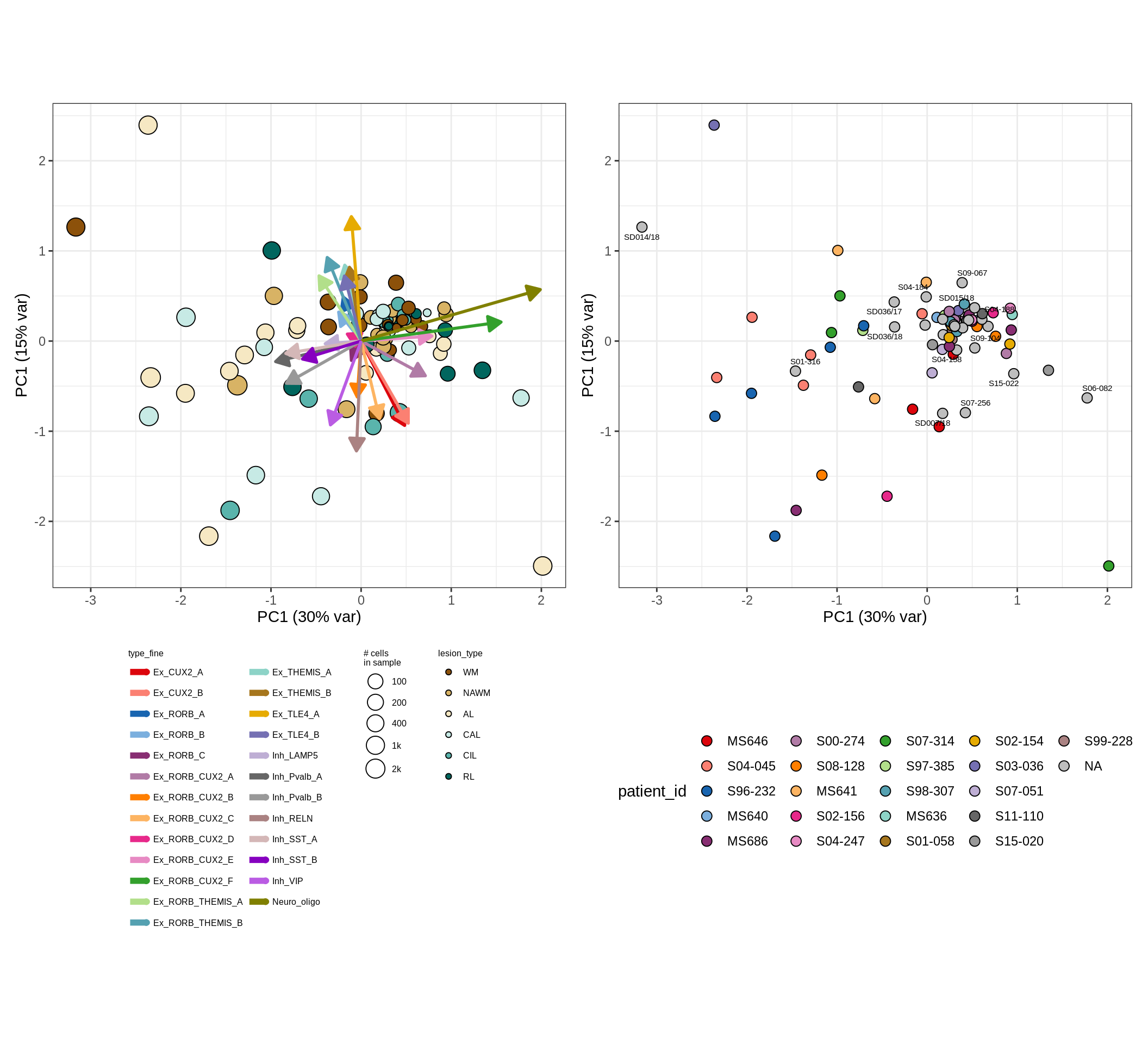
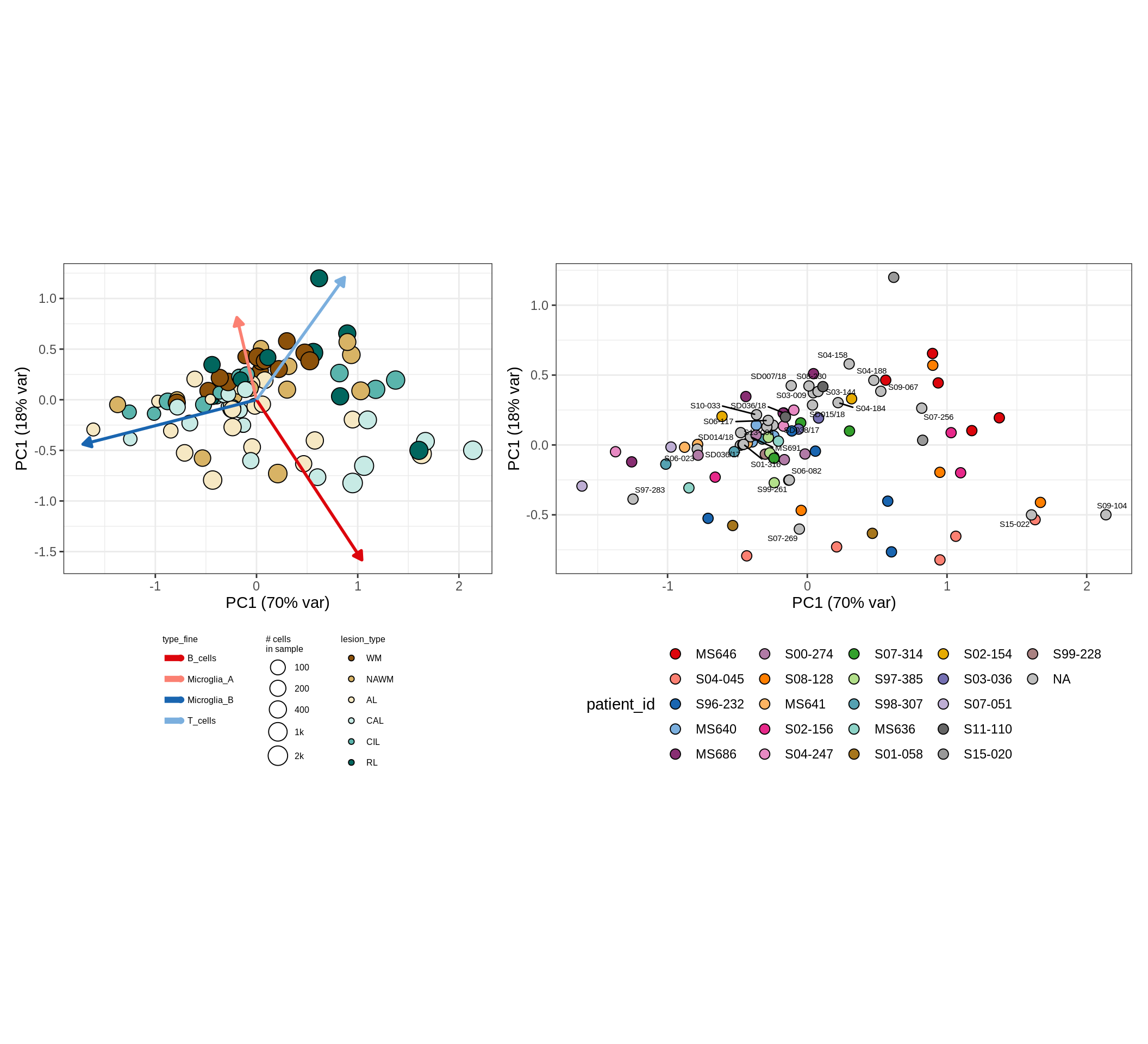
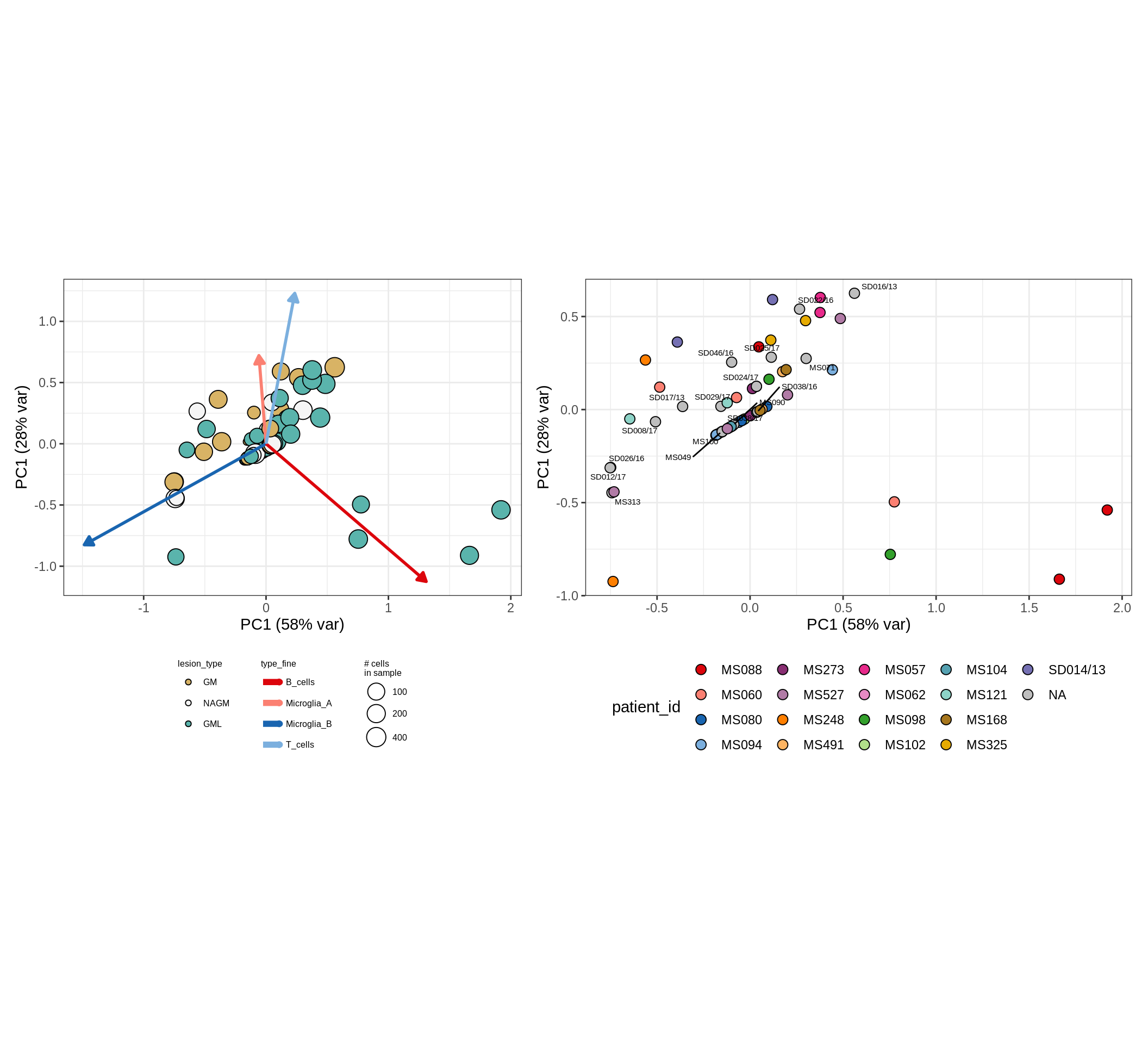
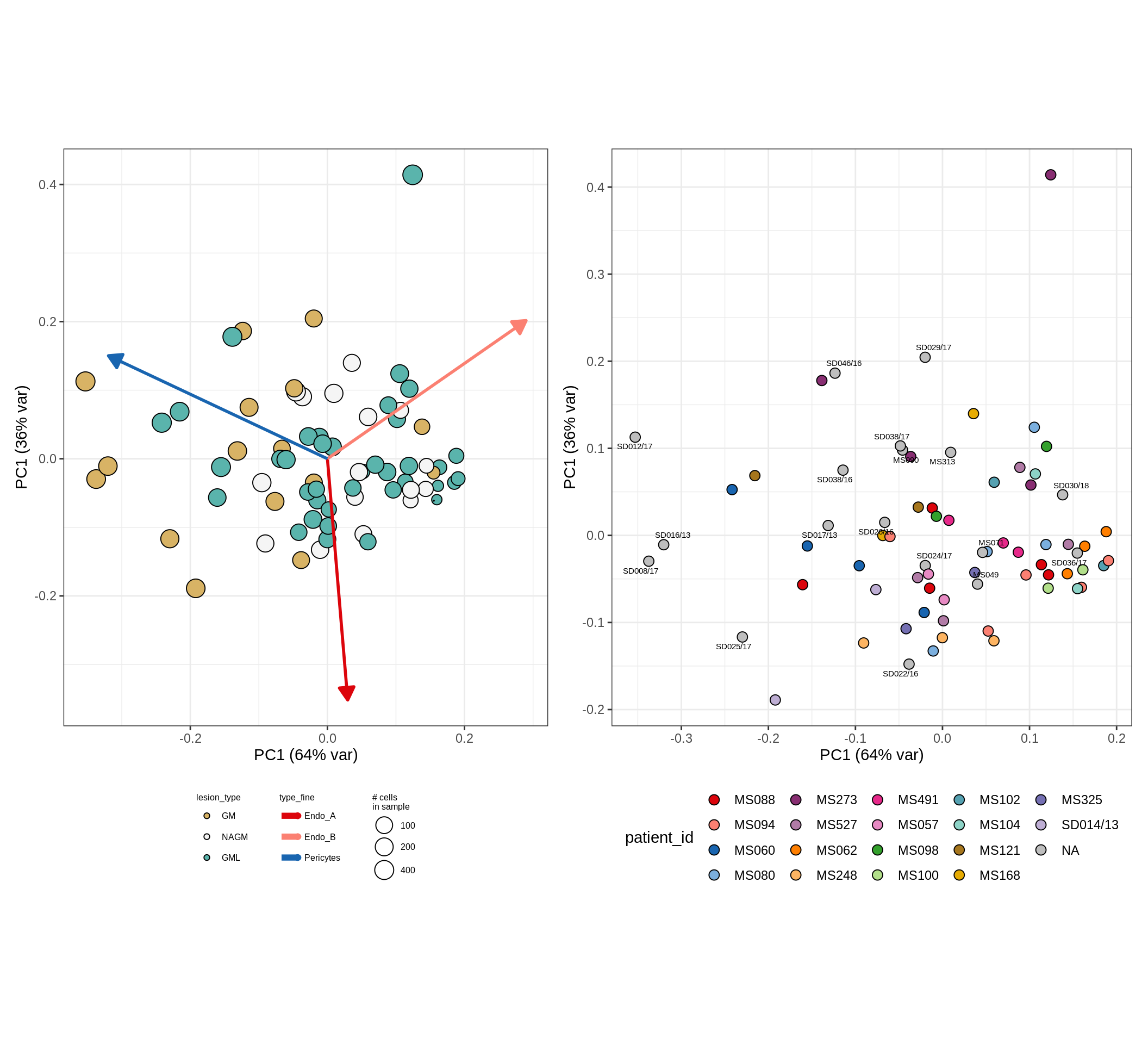
CLR plots of celltype proportions
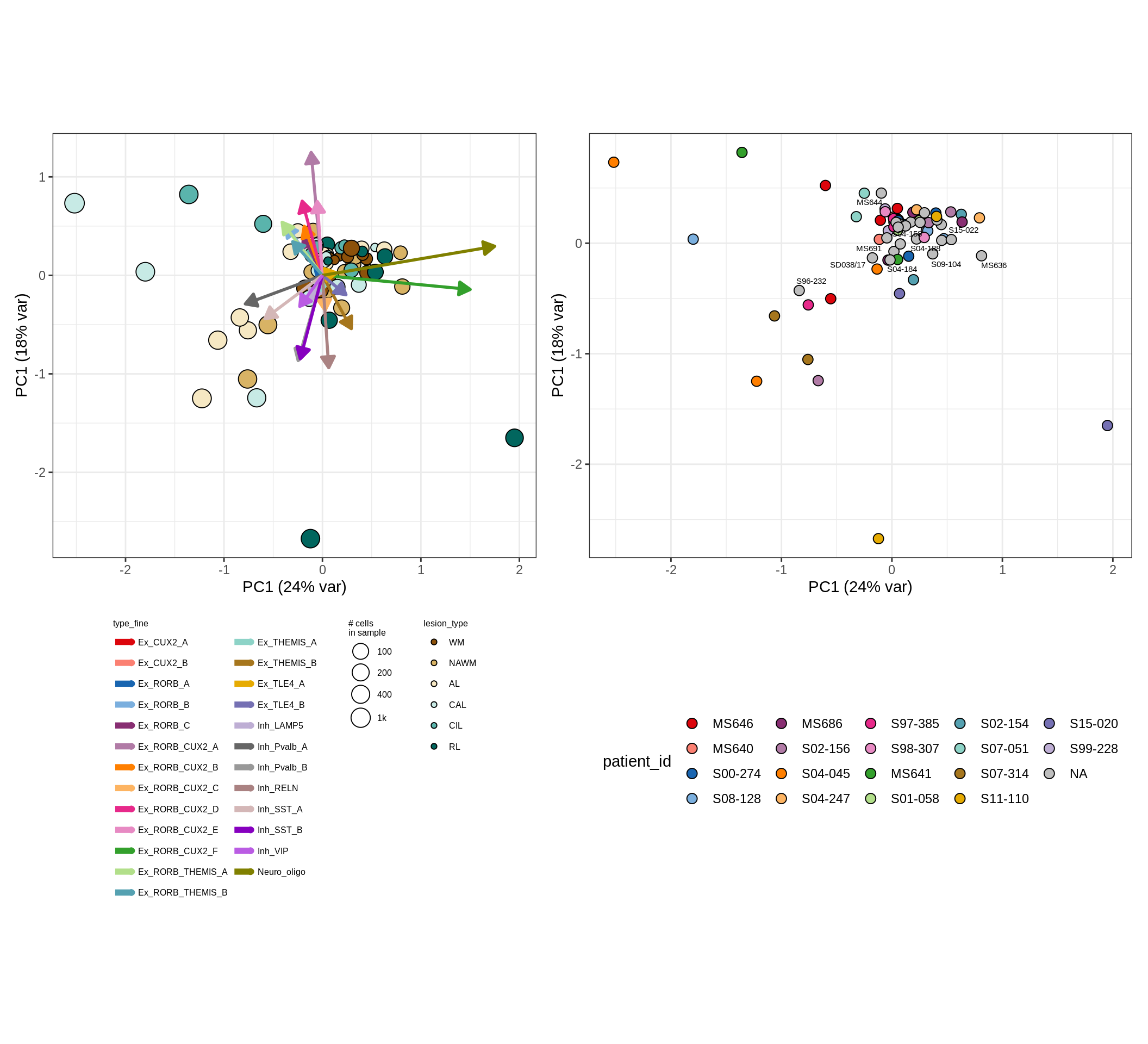
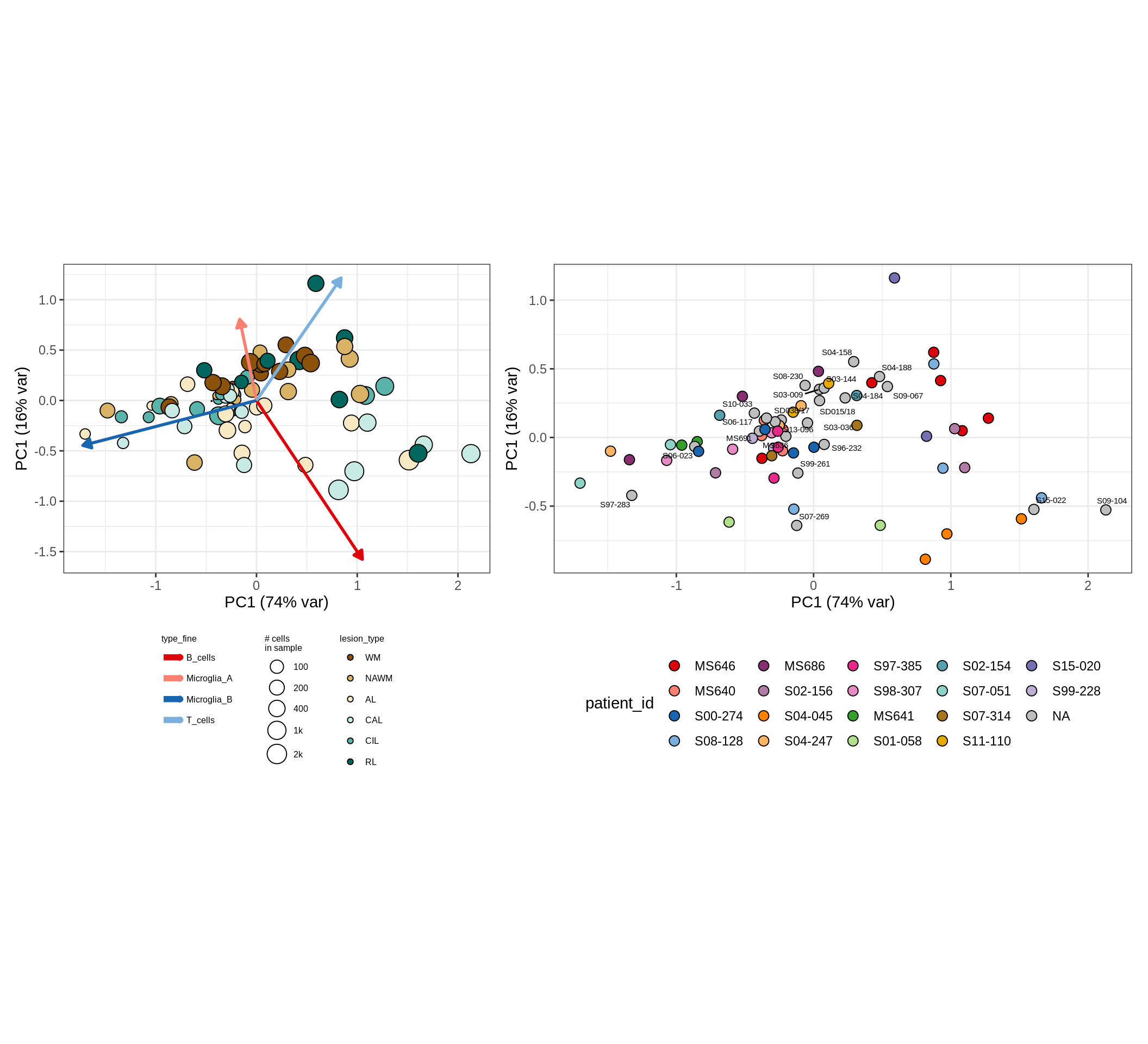
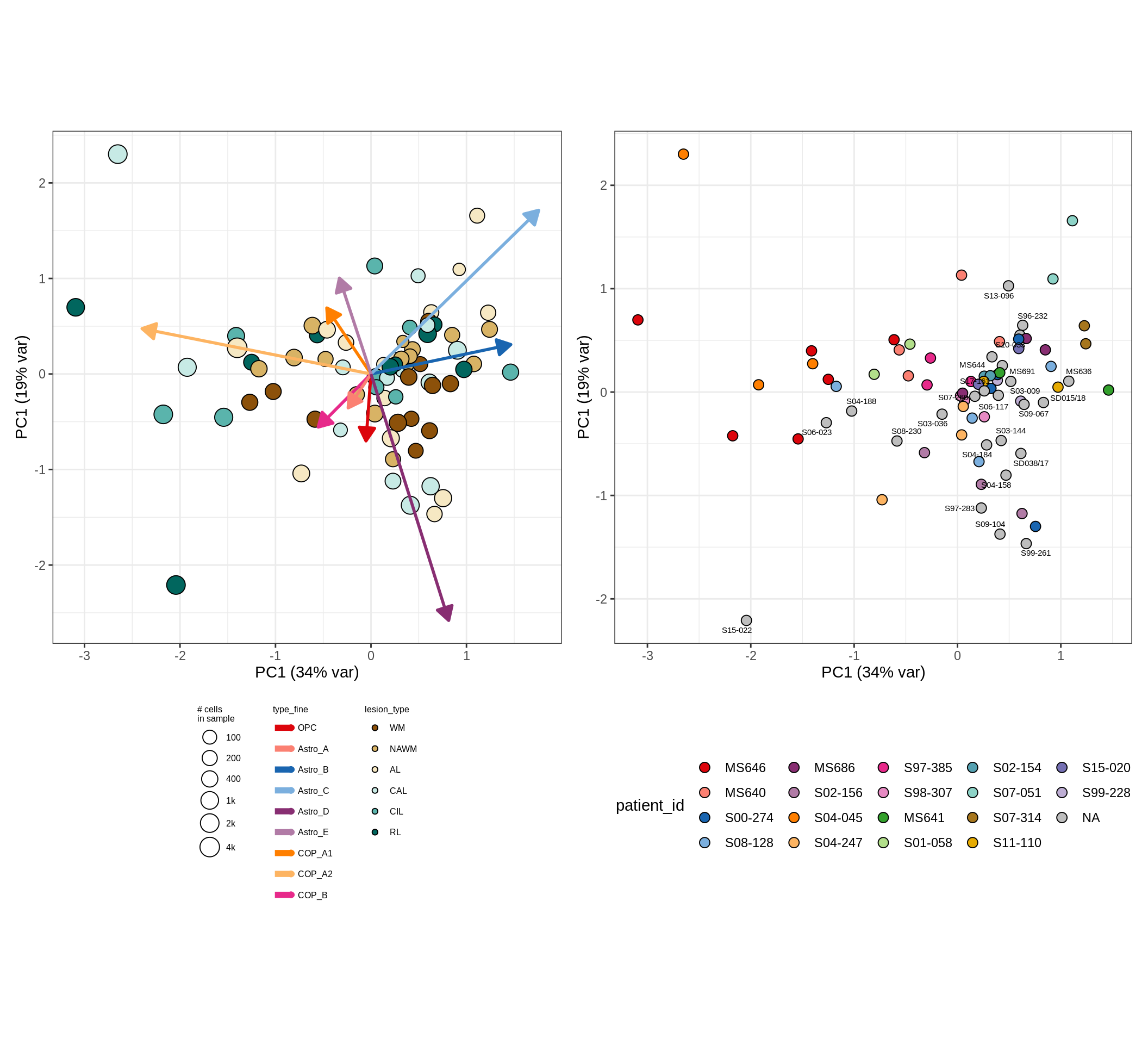
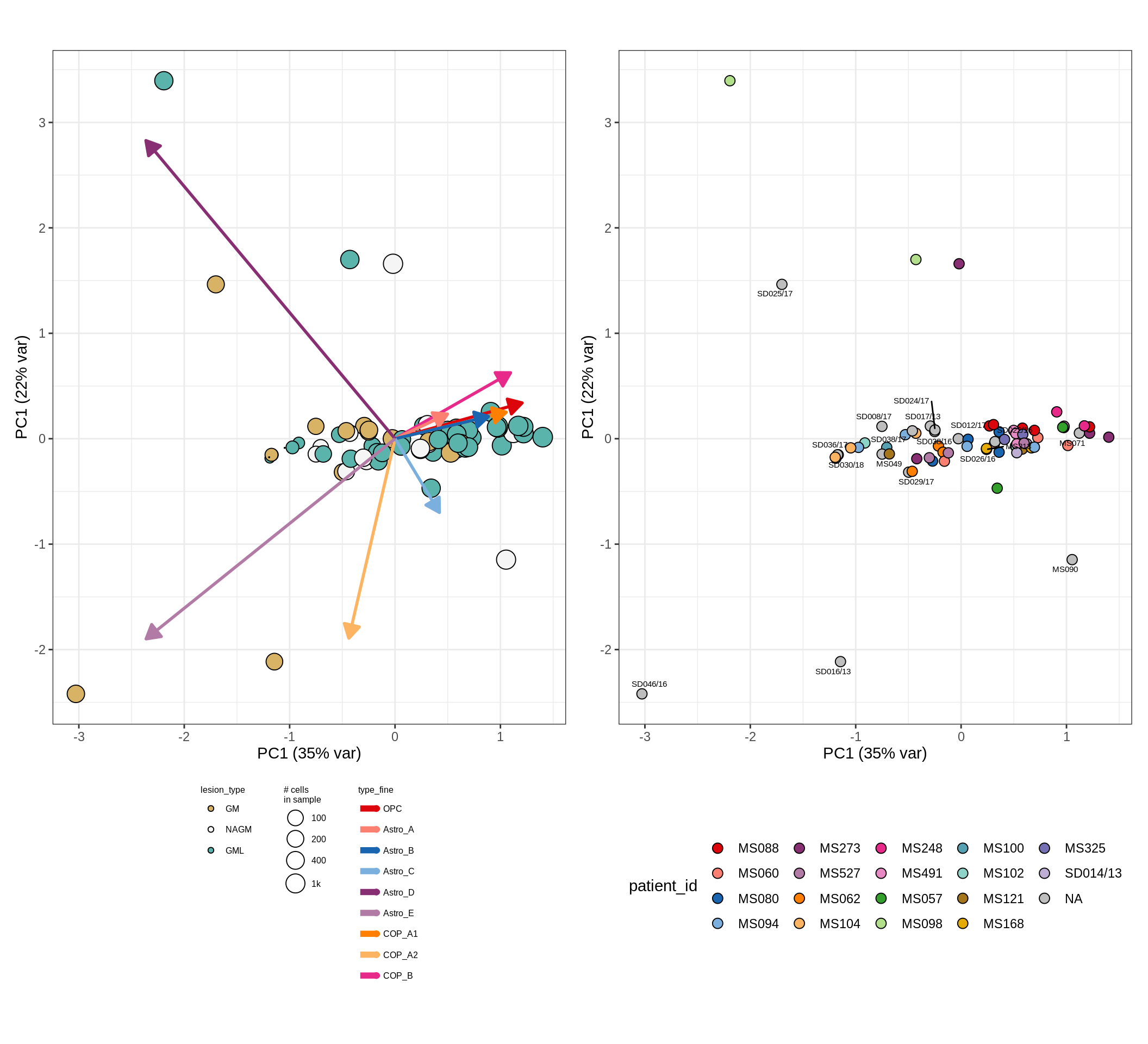
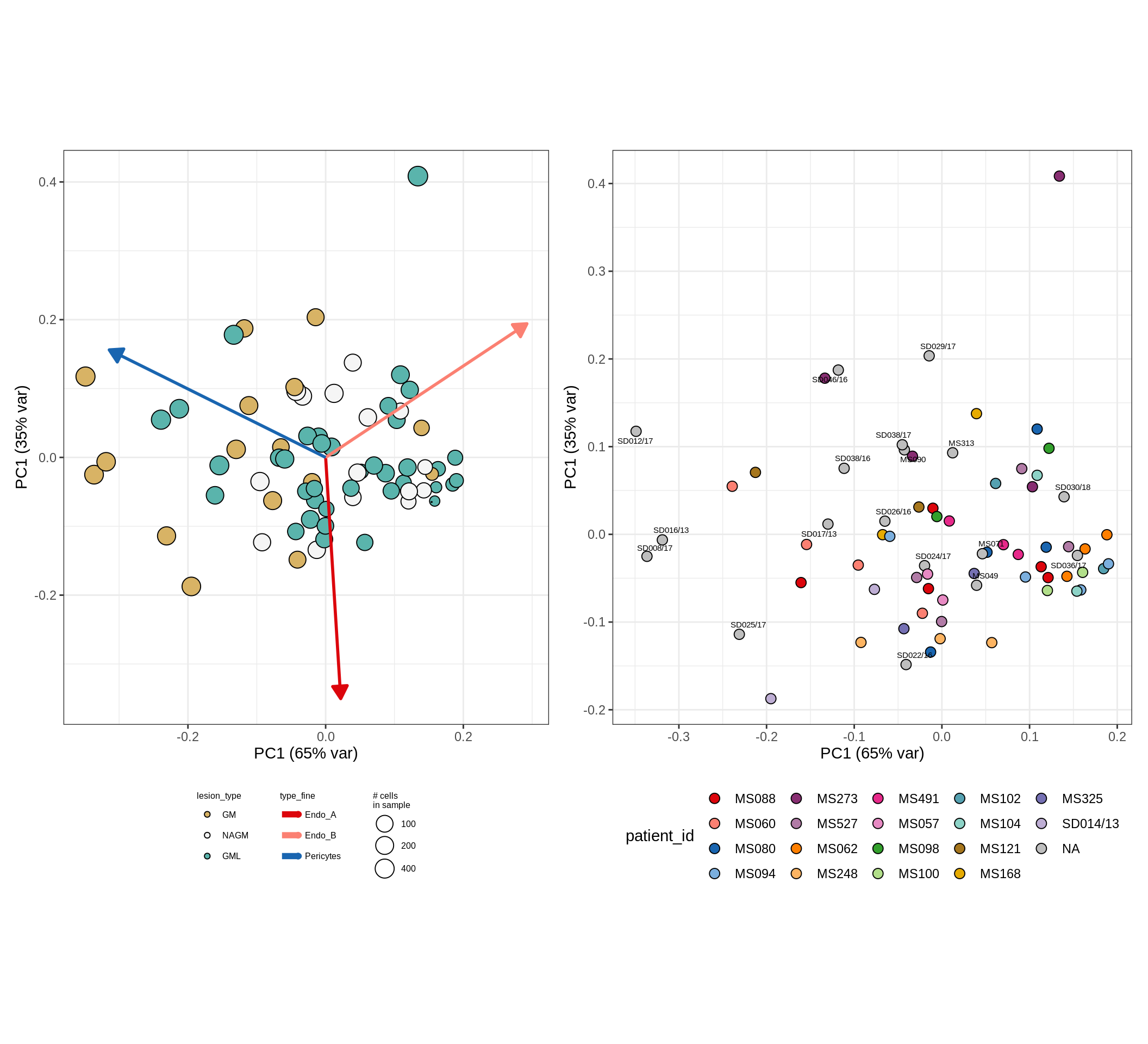
These plots show the broad variability between sample compositions, viewed at the log scale. A couple of notes:
- The aspect ratio of the plot reflects how much variance each principal component accounts for.
- The arrows show the directions in the plot corresponding to strongest positive change in celltype proportion.
- Patients with more than one sample are labelled by colour; patients with only one are labelled by text on the plot.
for (t in names(types_list)) {
for (m in c('WM', 'GM')) {
cat('### ', t, ', ', m, '\n')
types = types_list[[t]]
input_dt = conos_dt[(matter == m) & (type_broad %in% types)]
suppressWarnings(print(plot_sample_clrs(input_dt)))
cat('\n\n')
}
}oligo_opc , WM
oligo_opc , GM
neurons , WM
neurons , GM
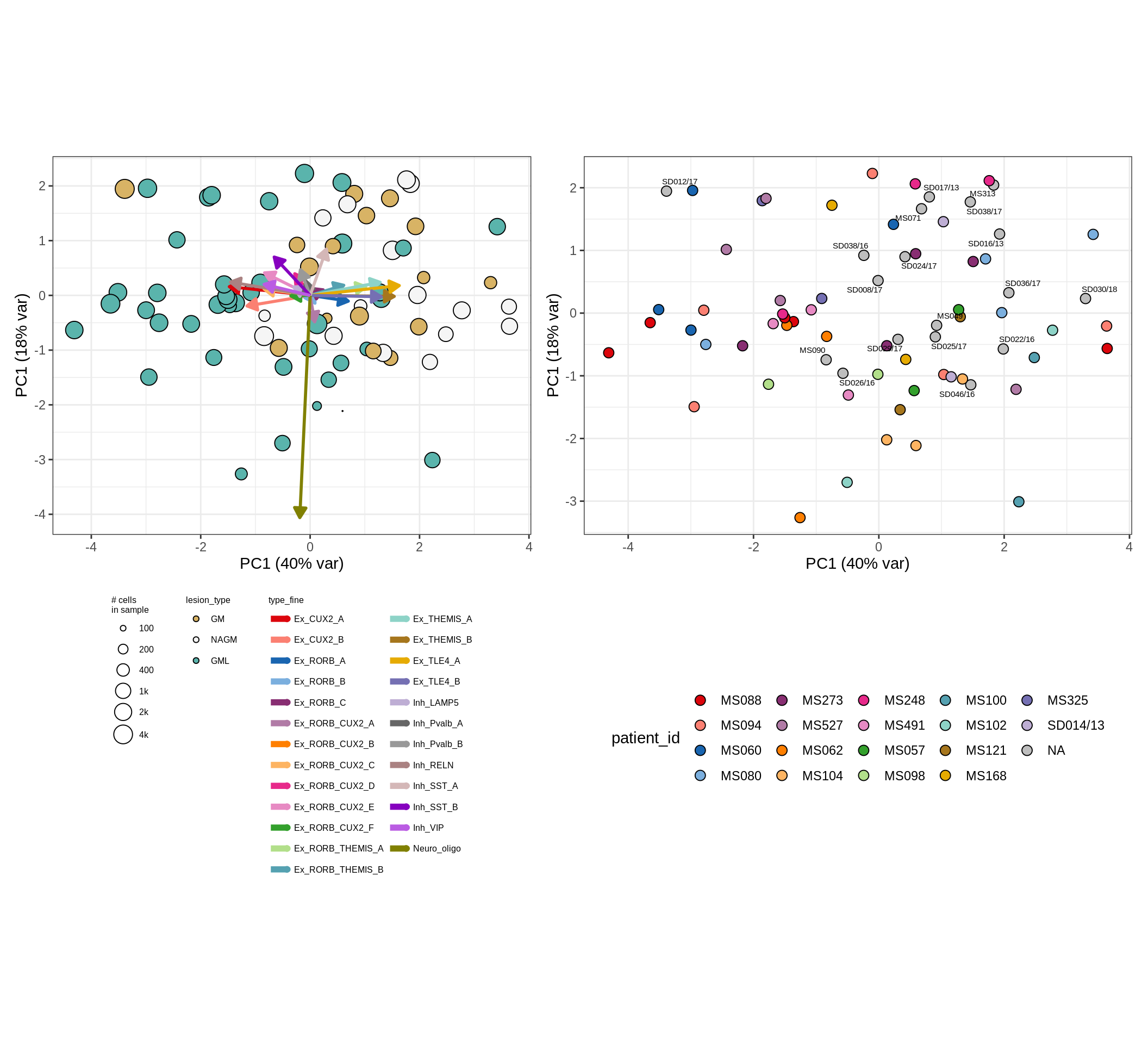
micro_immune , WM
micro_immune , GM
astro_opc , WM
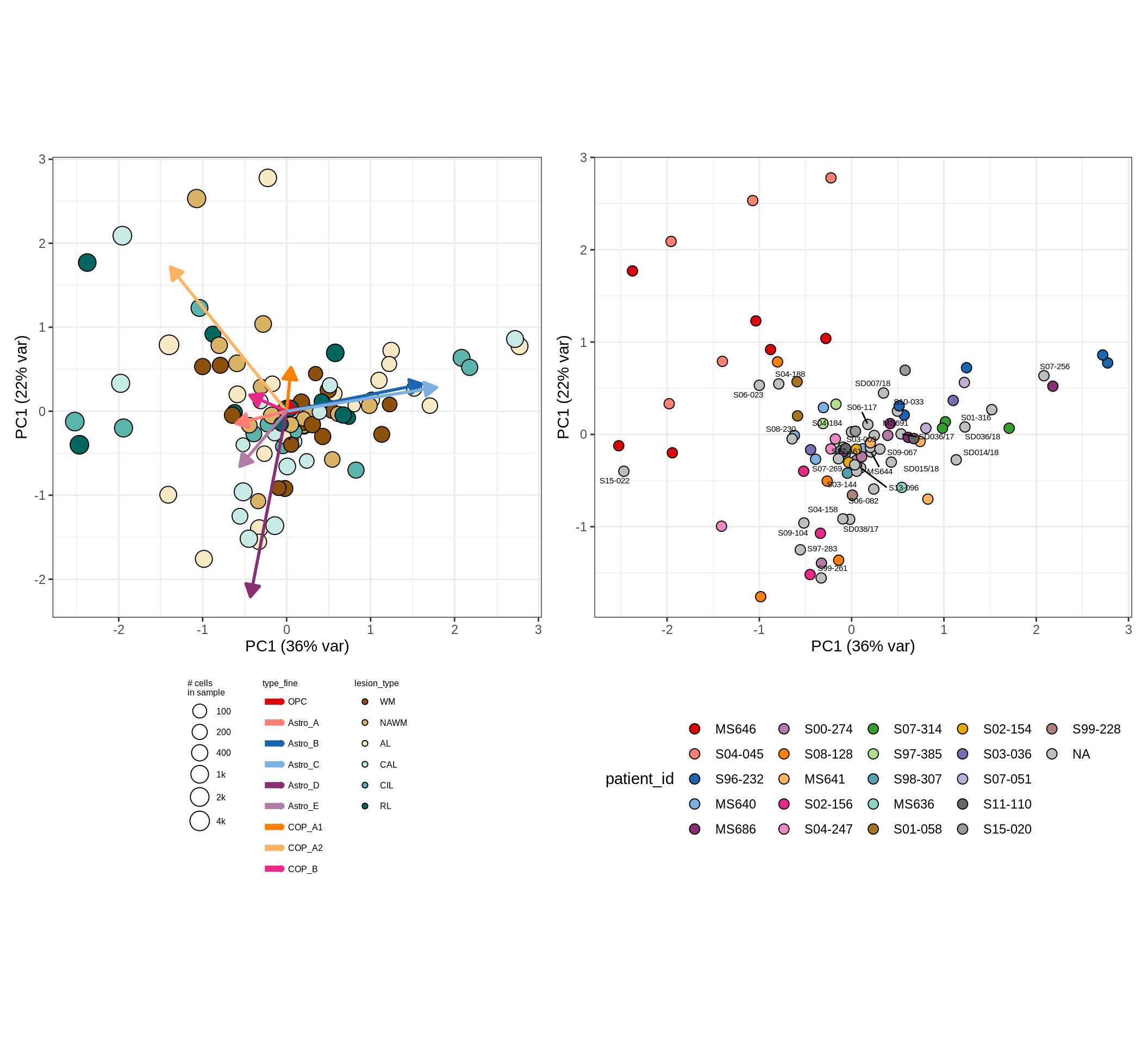
astro_opc , GM
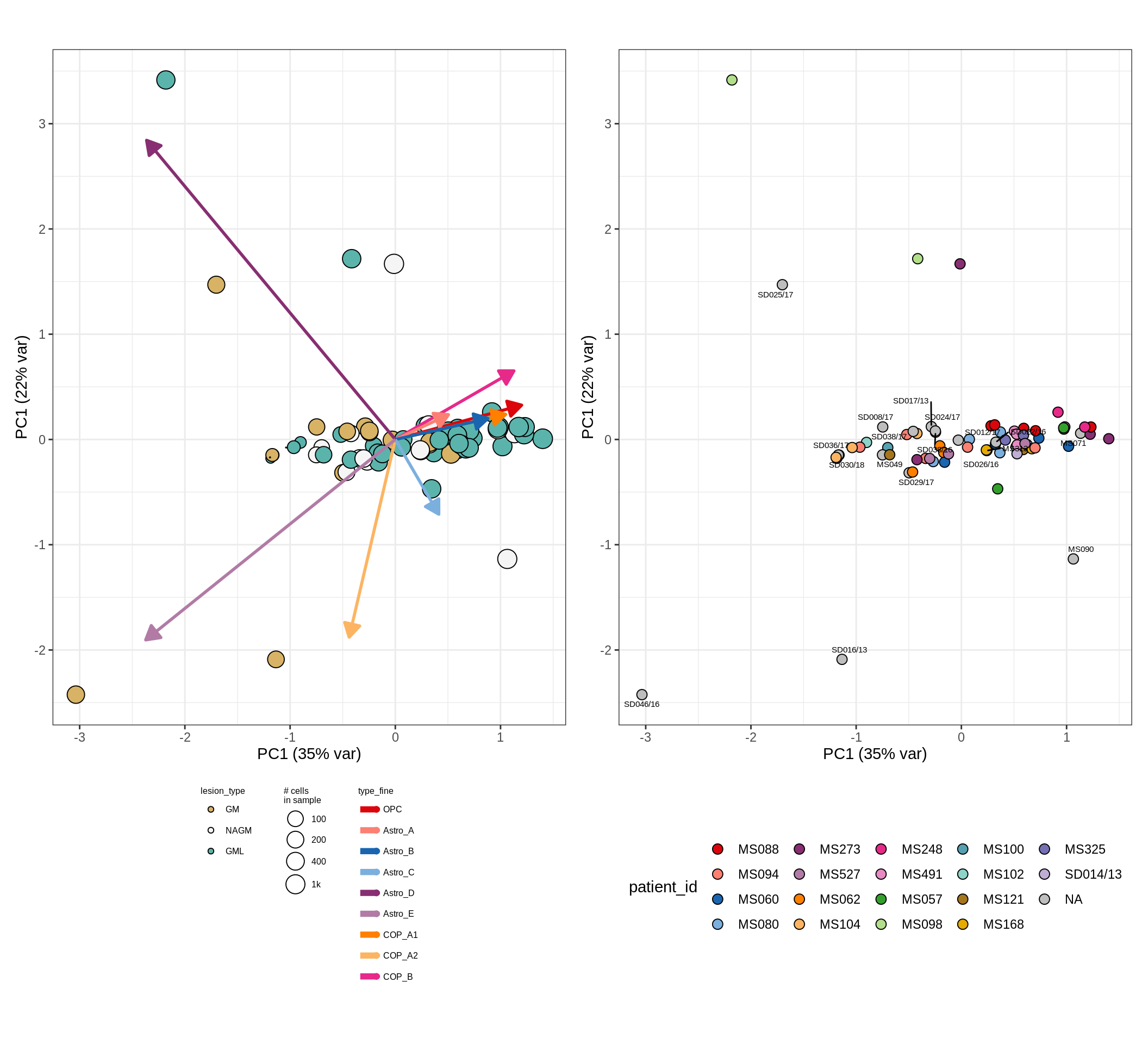
endo_peri , WM
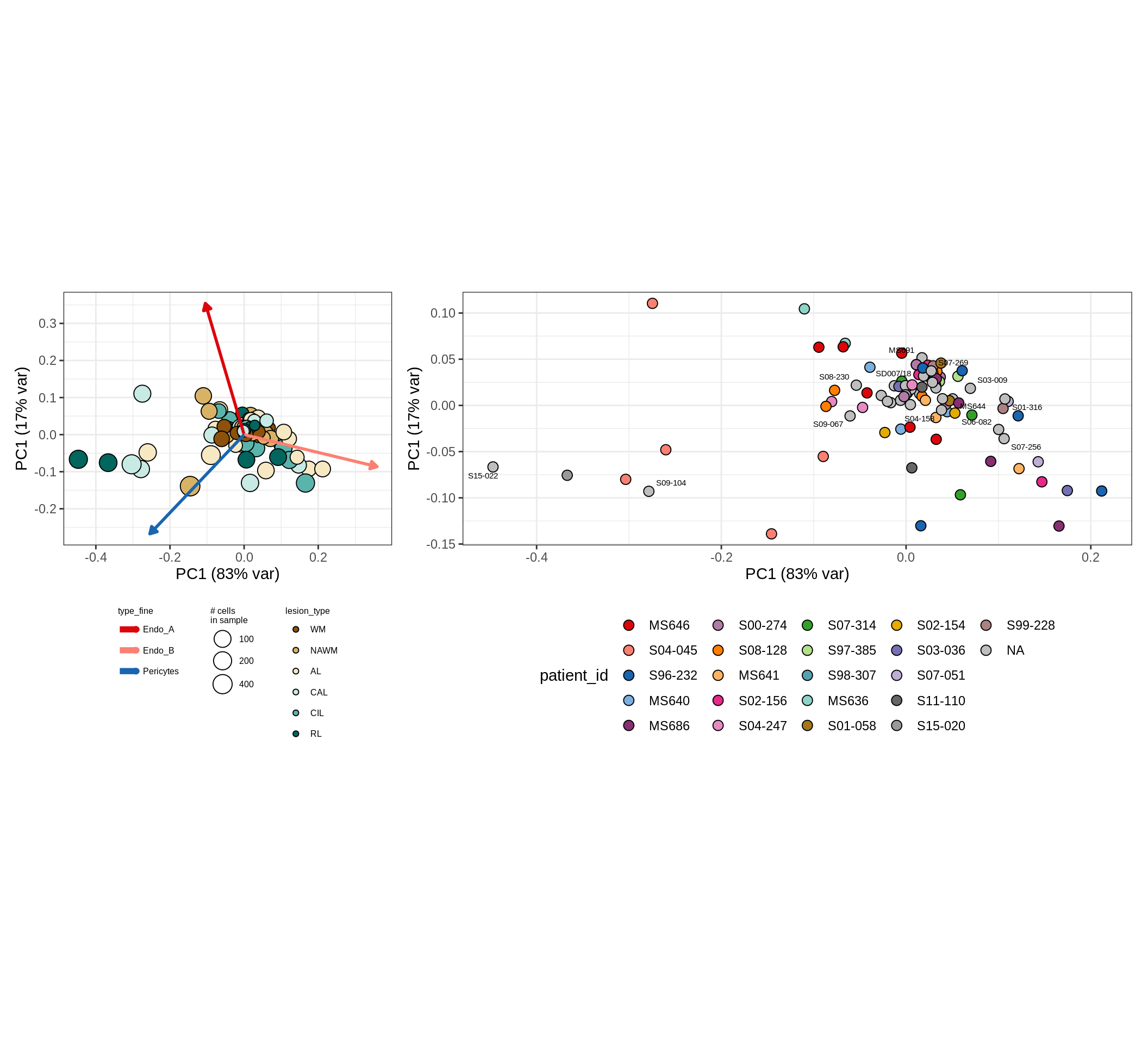
endo_peri , GM
CLR plots of celltype proportions, neuron proportion outliers excluded
for (t in names(types_list)) {
for (m in c('WM', 'GM')) {
cat('### ', t, ', ', m, '\n')
types = types_list[[t]]
input_dt = conos_dt[(matter == m) & (neuro_ok == TRUE) &
type_broad %in% types]
suppressWarnings(print(plot_sample_clrs(input_dt)))
cat('\n\n')
}
}oligo_opc , WM
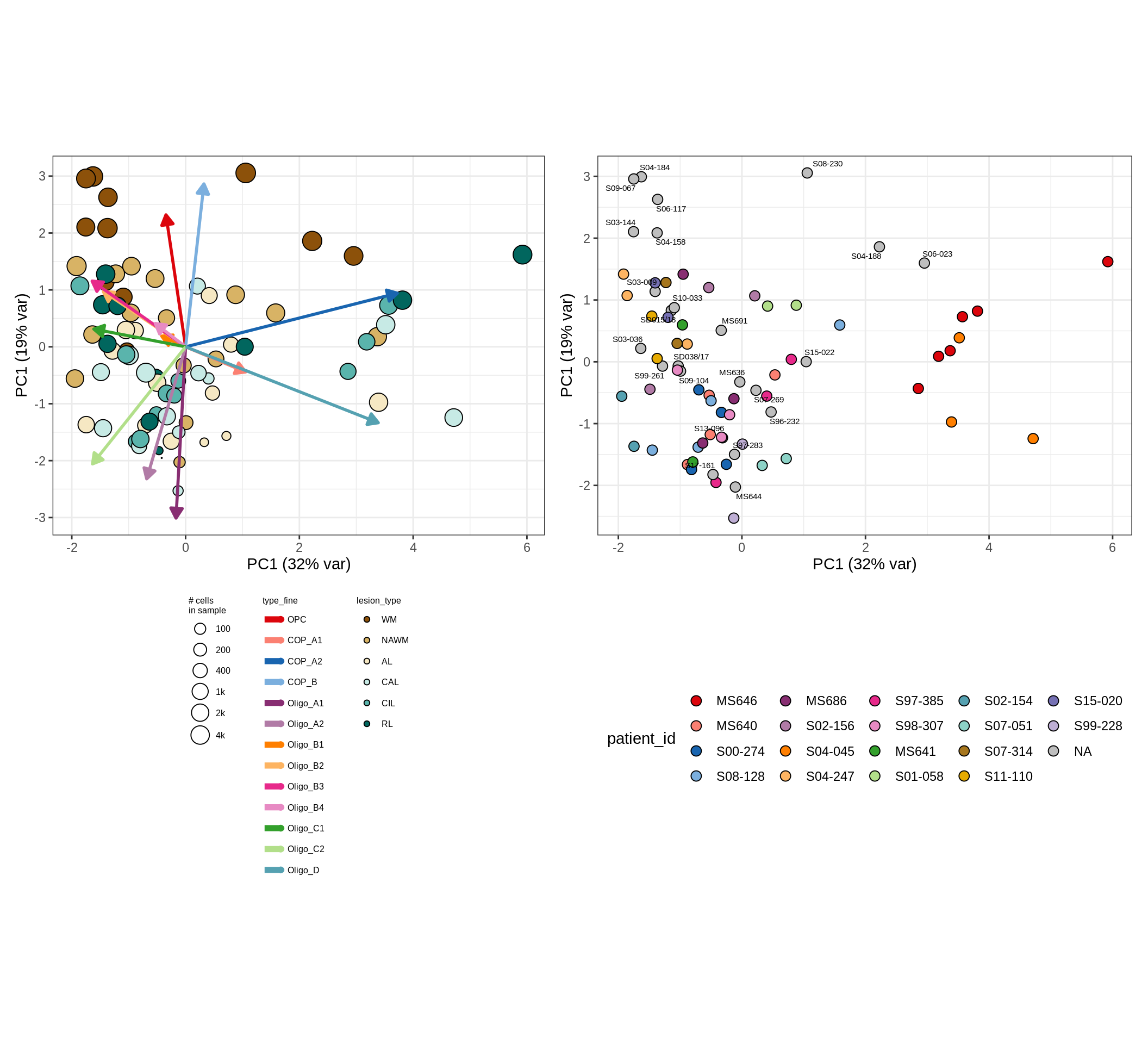
oligo_opc , GM
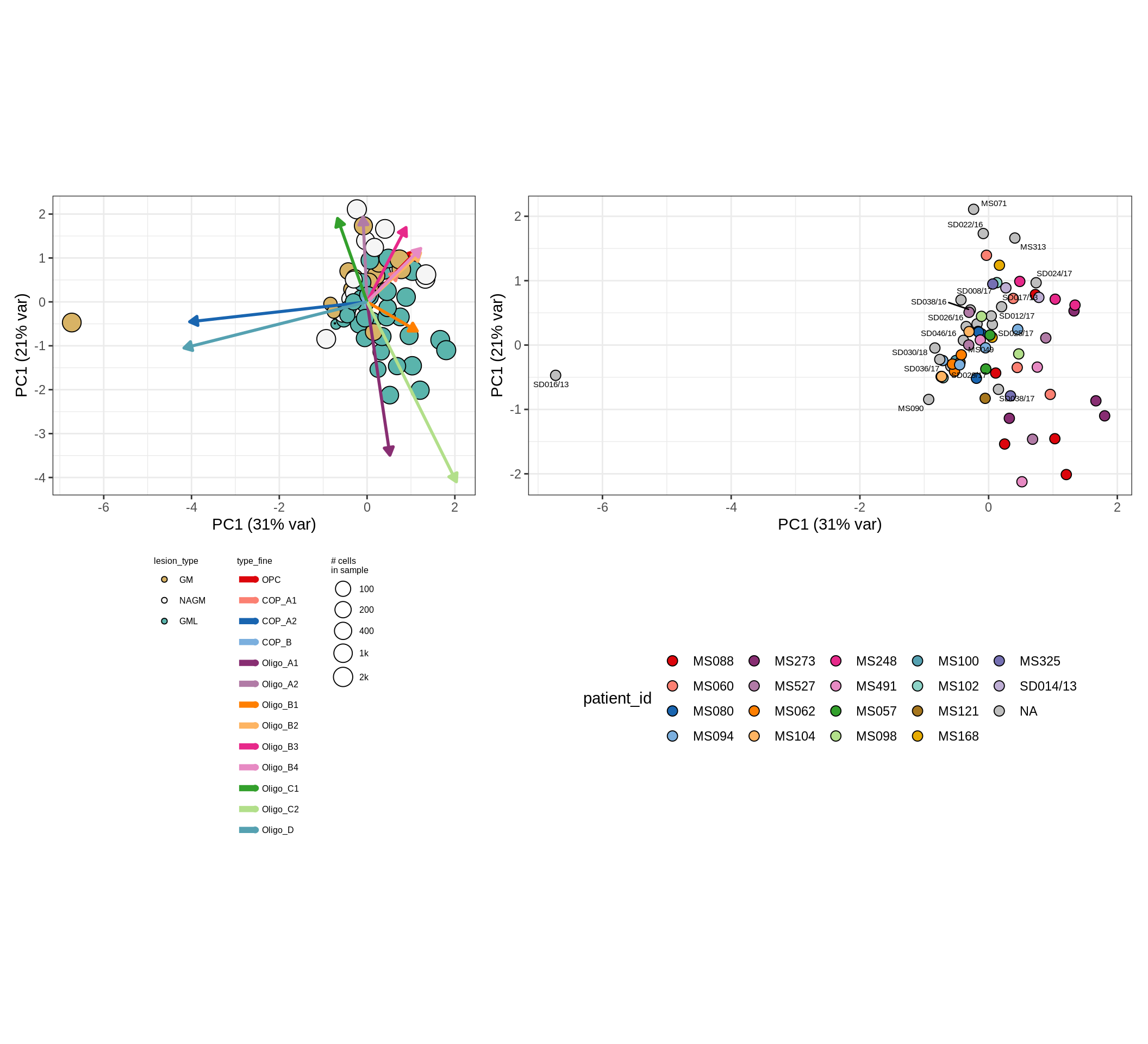
neurons , WM
neurons , GM
micro_immune , WM
micro_immune , GM
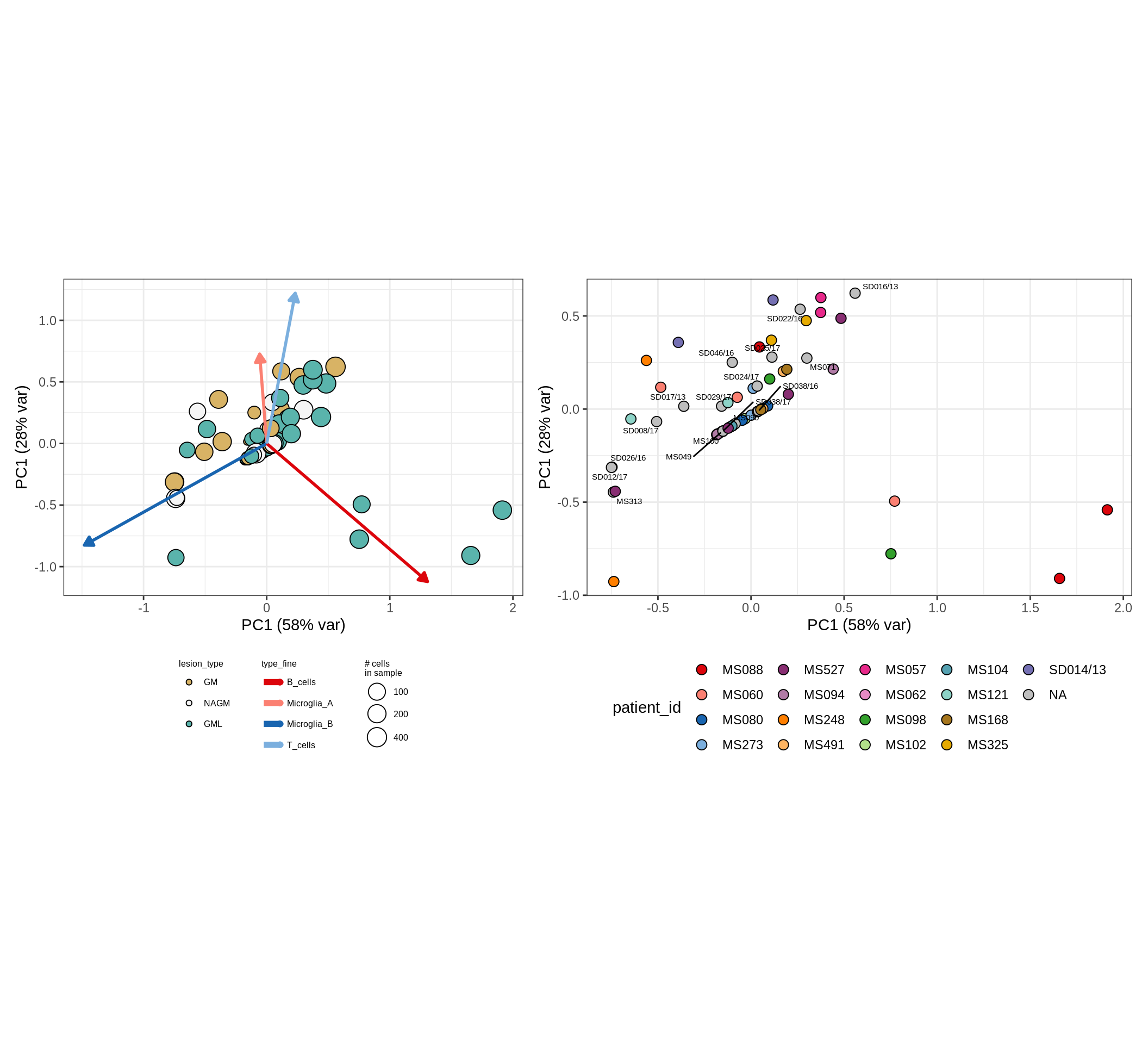
astro_opc , WM
astro_opc , GM
endo_peri , WM
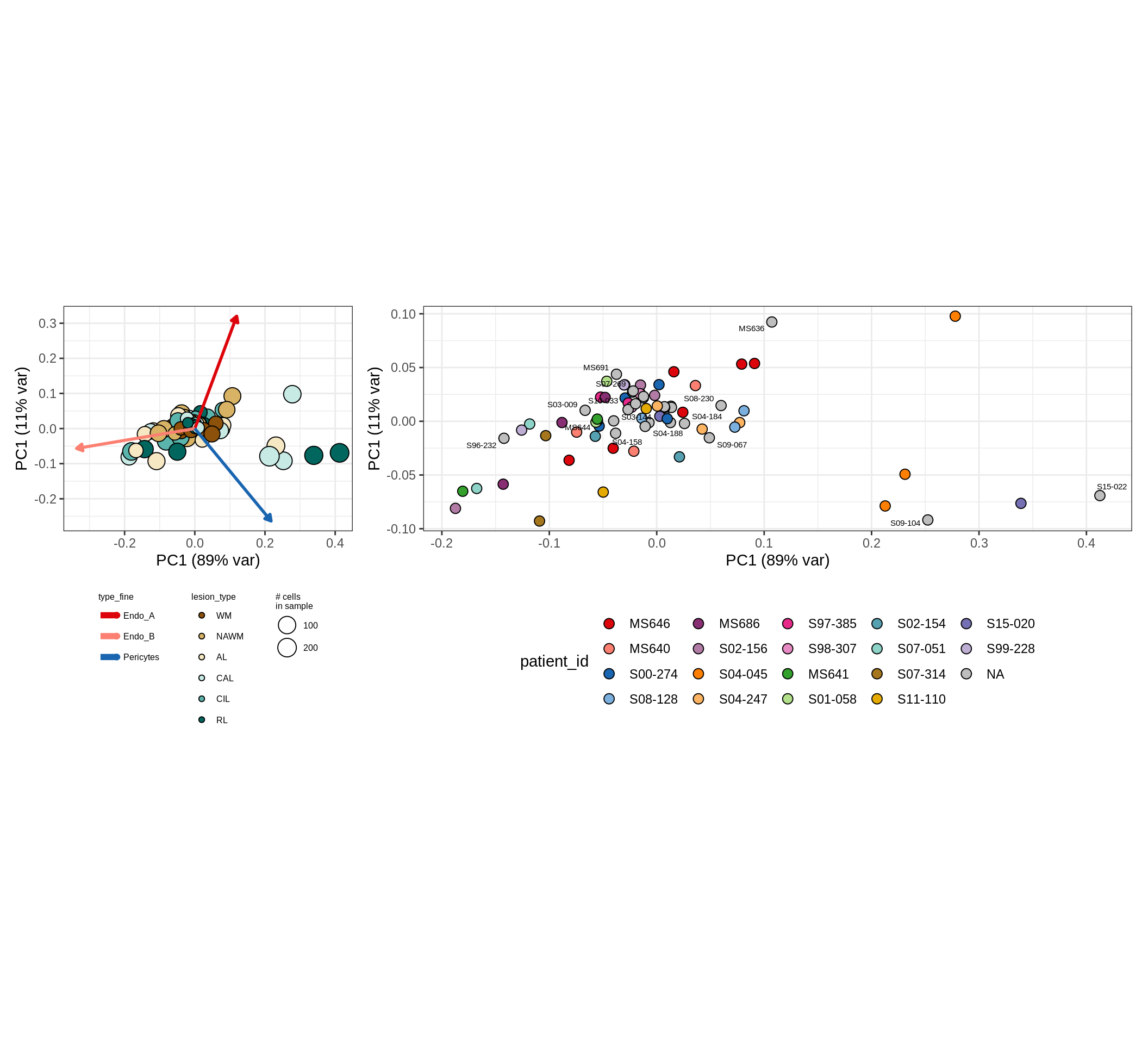
endo_peri , GM
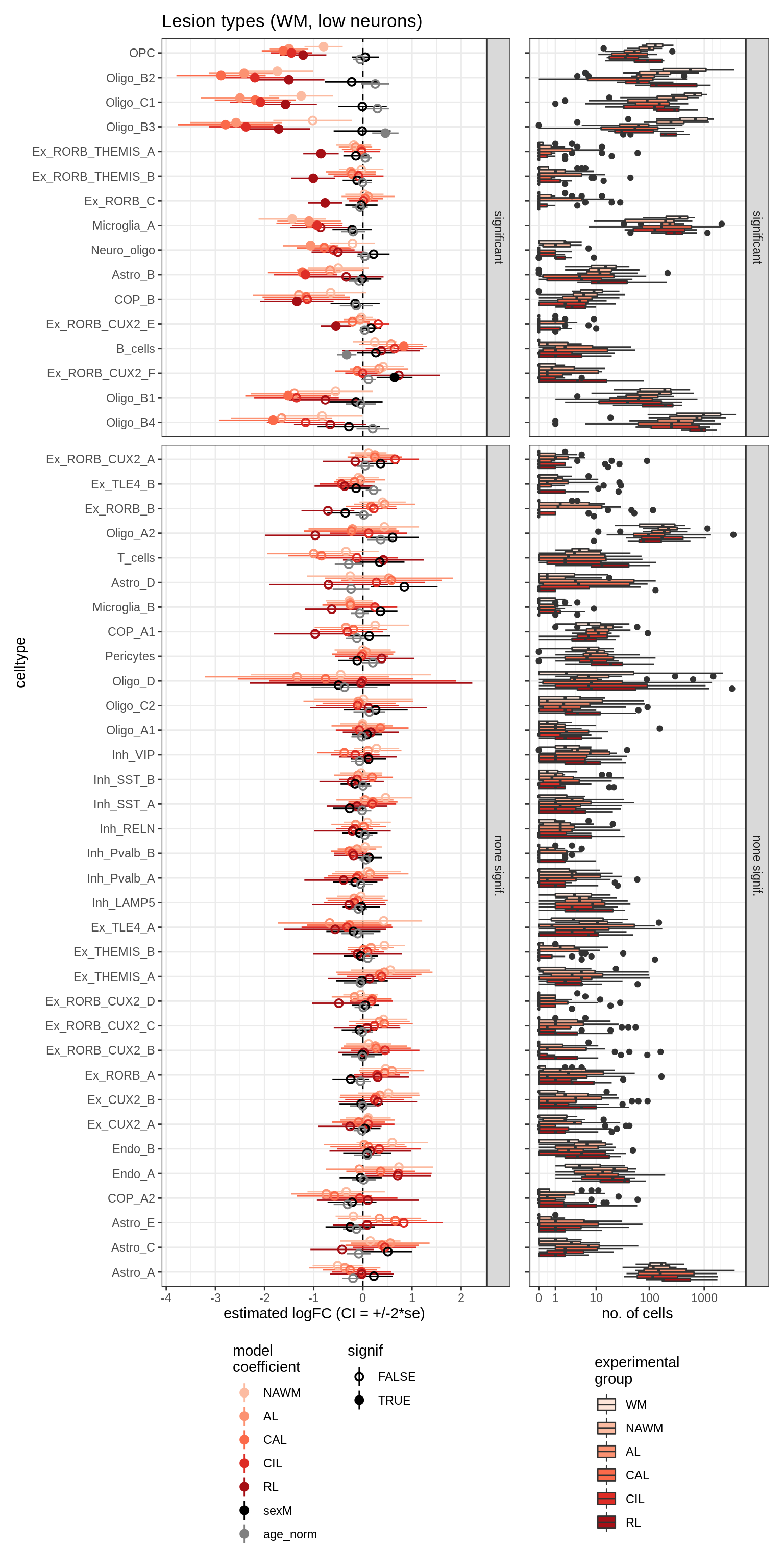
ANCOM CIs (signif only)
for (nn in model_names) {
cat('### ', nn, '\n')
print(plot_ancombc_ci(ancom_list[[nn]], counts_wide, names_list[[nn]],
whatplot = whatplot_list[[nn]], reported_only = TRUE))
cat('\n\n')
}lesions_WM_mad
lesions_WM_big_mad
lesions_GM_mad
lesions_GM_big_mad
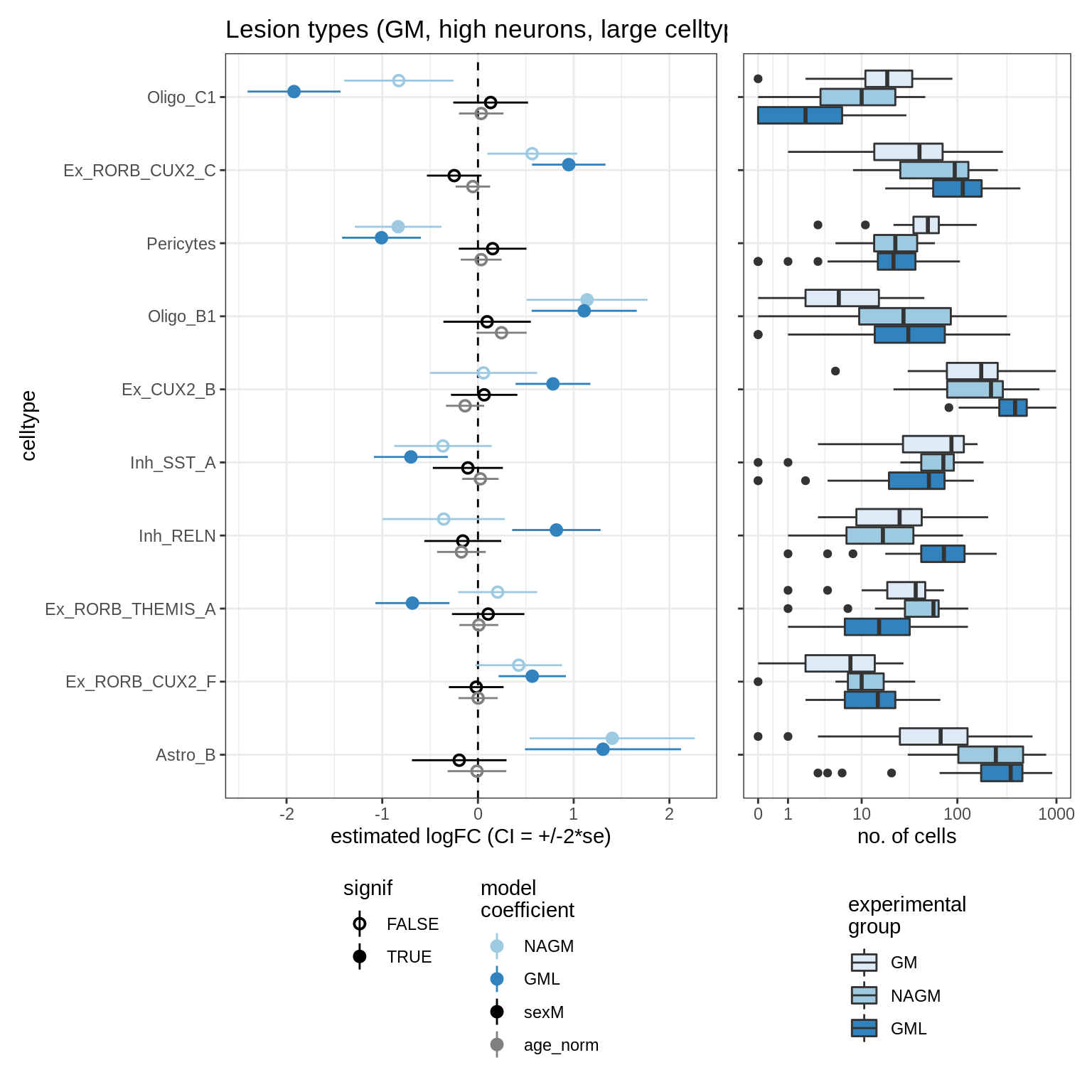
lesions_WM_no_neuro
lesions_GM_neuro
lesions_WM_all
lesions_GM_all
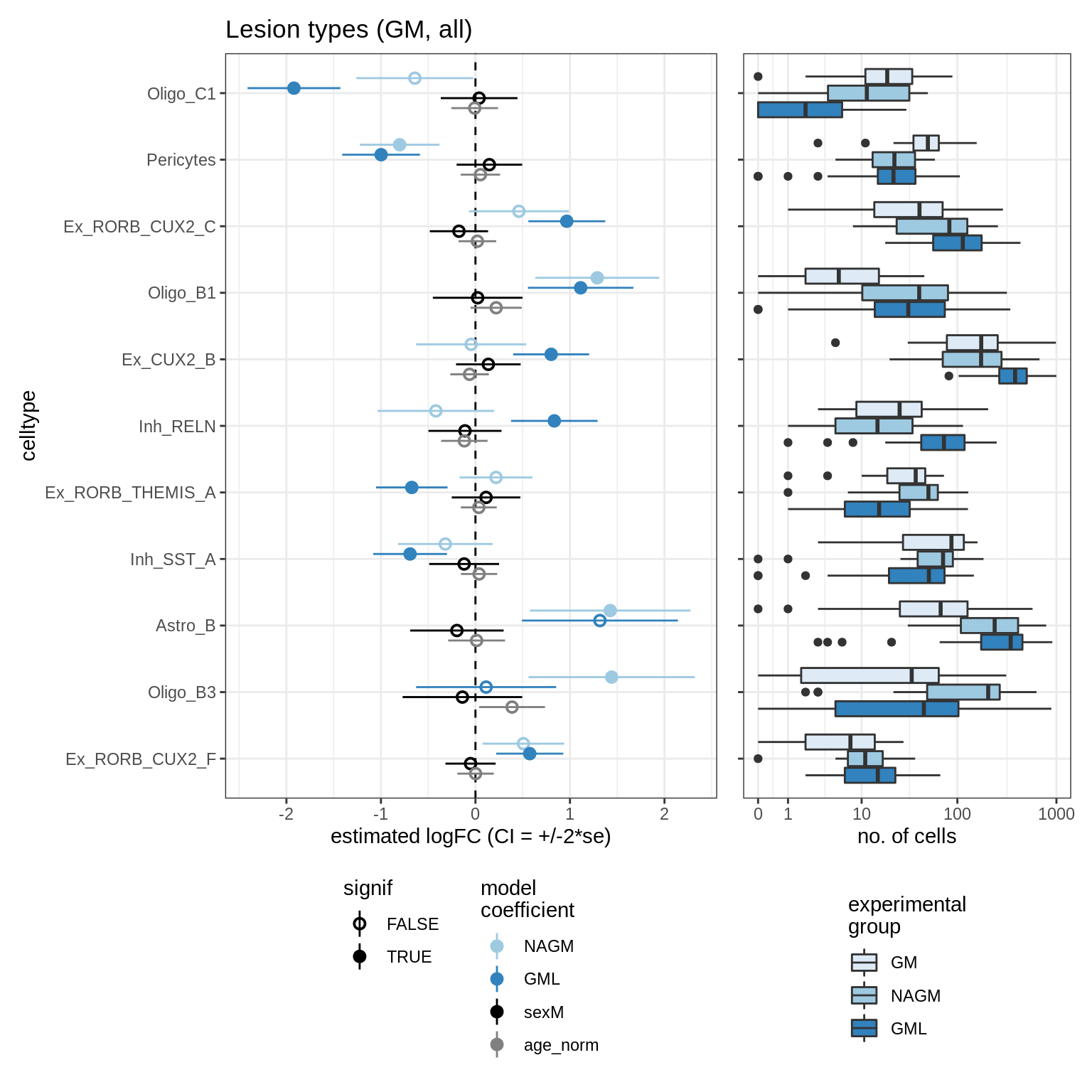
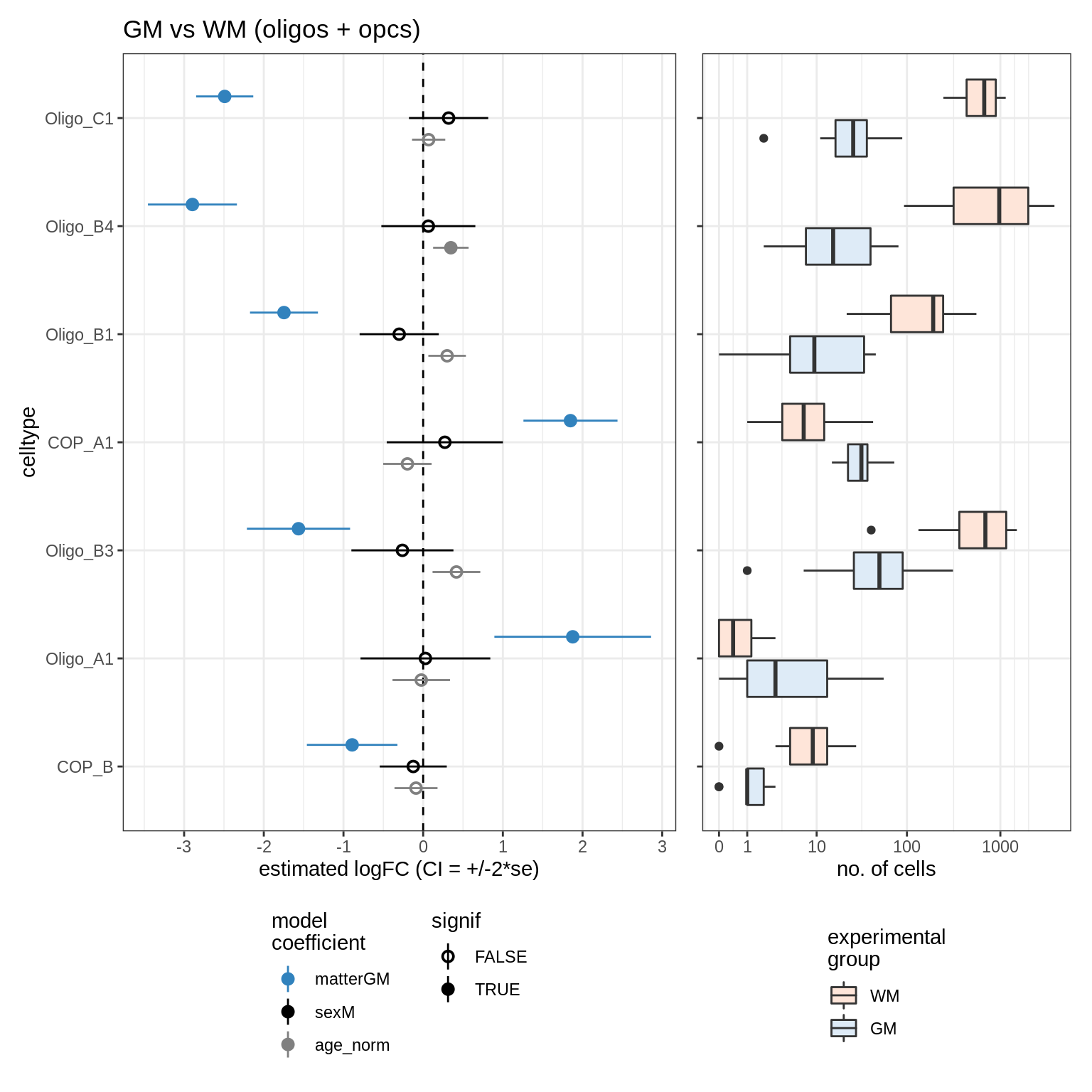
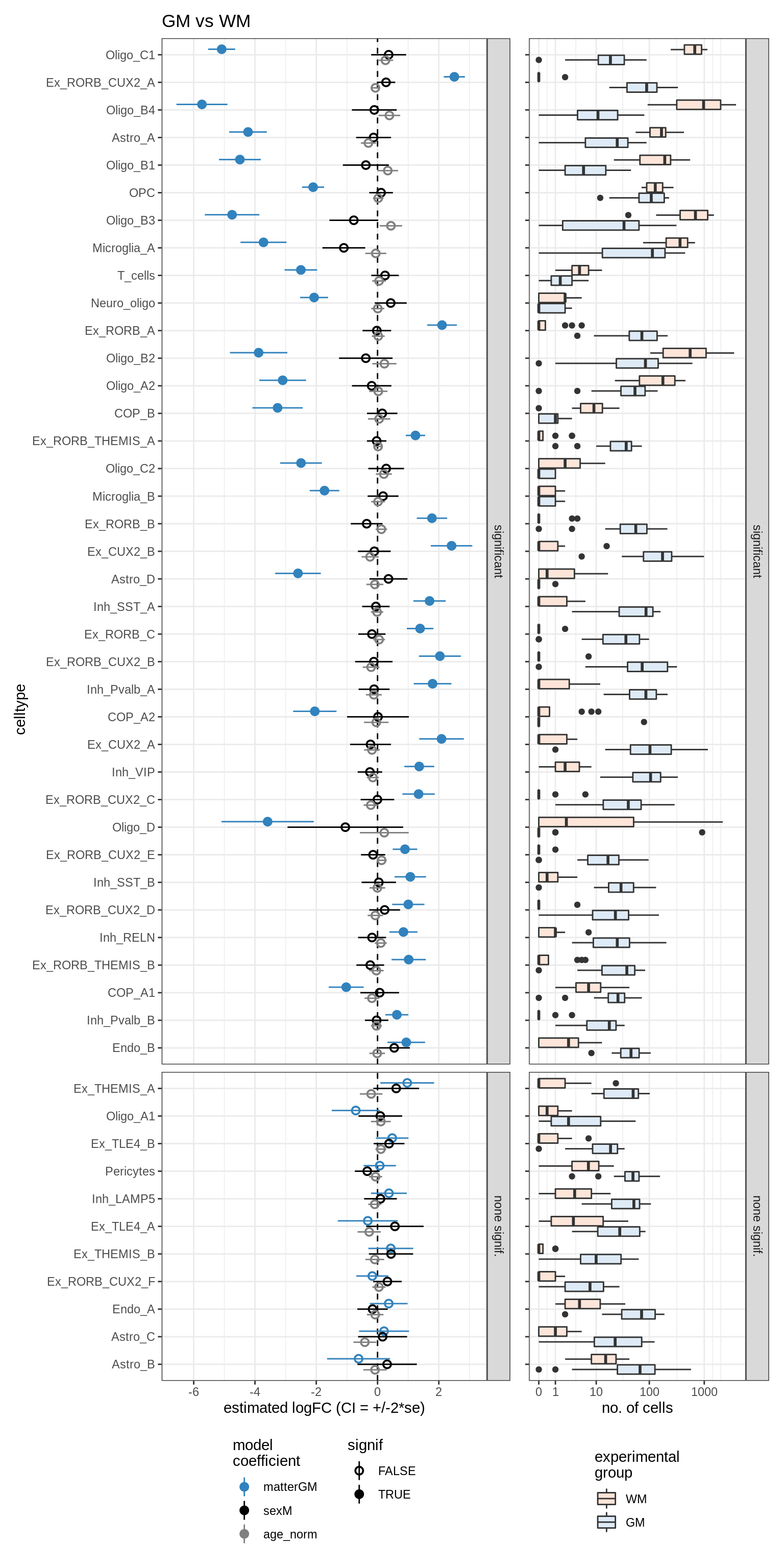
GM_vs_WM
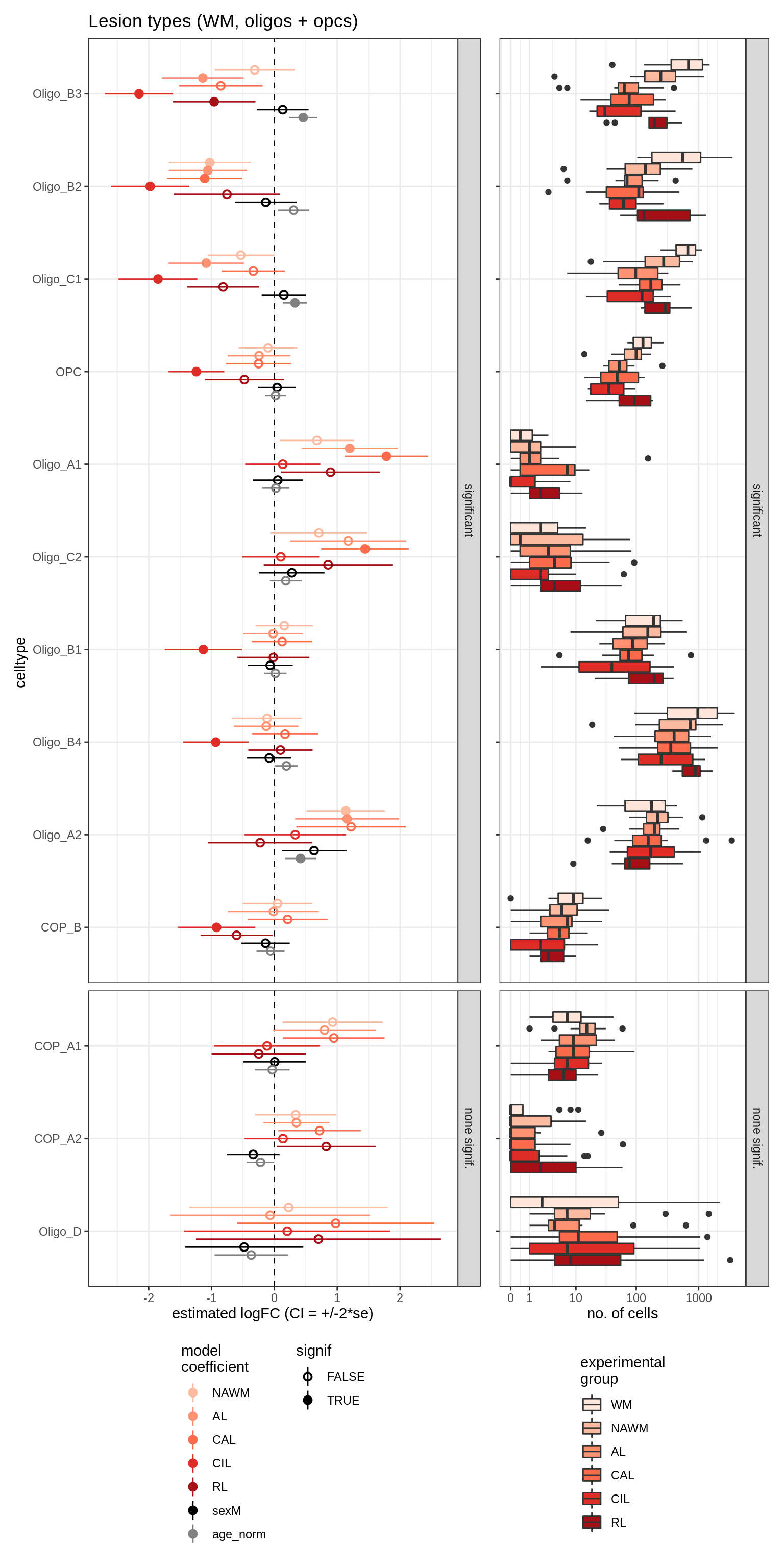
lesions_WM_oligos
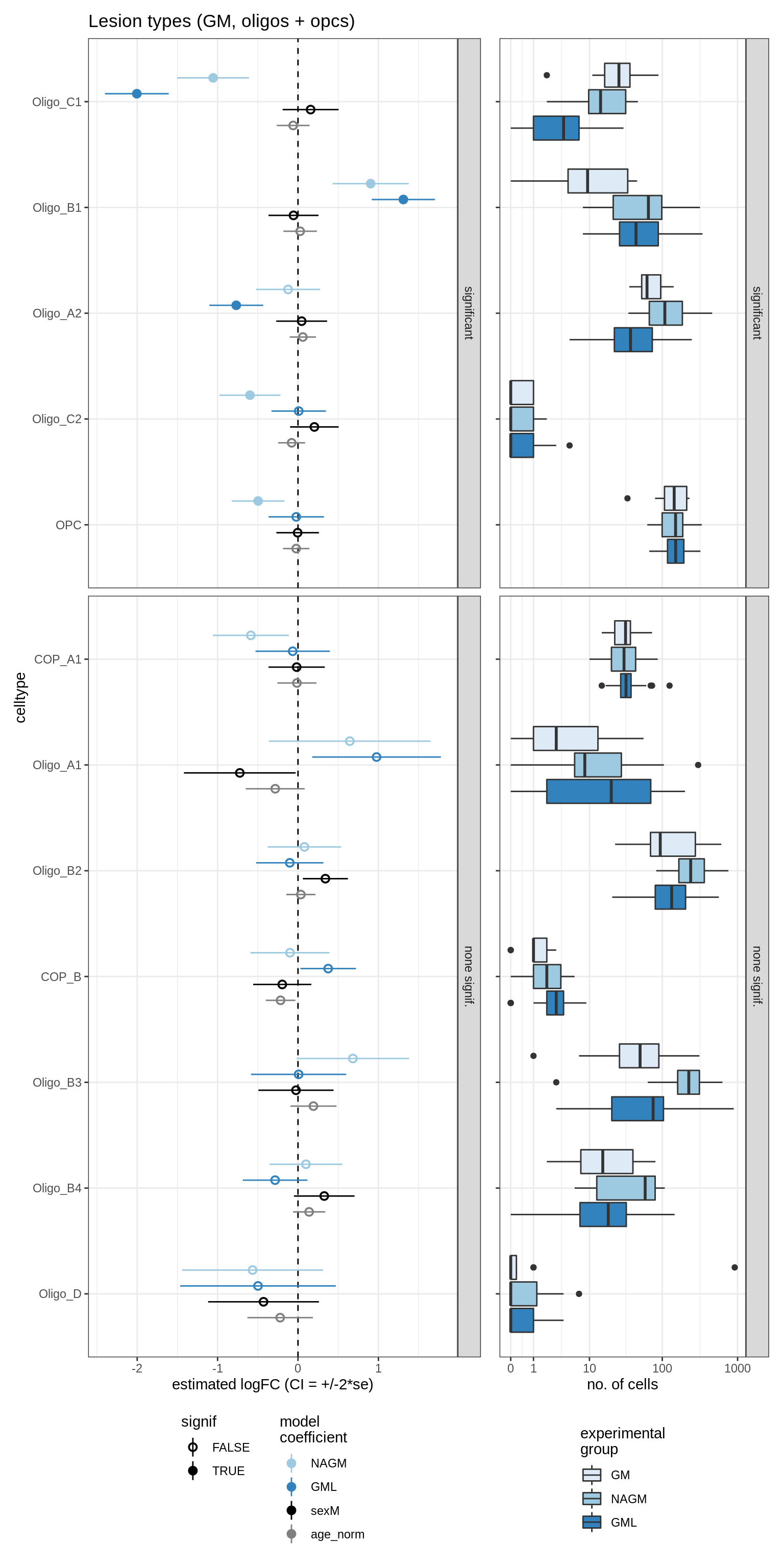
lesions_GM_oligos
GM_vs_WM_oligos
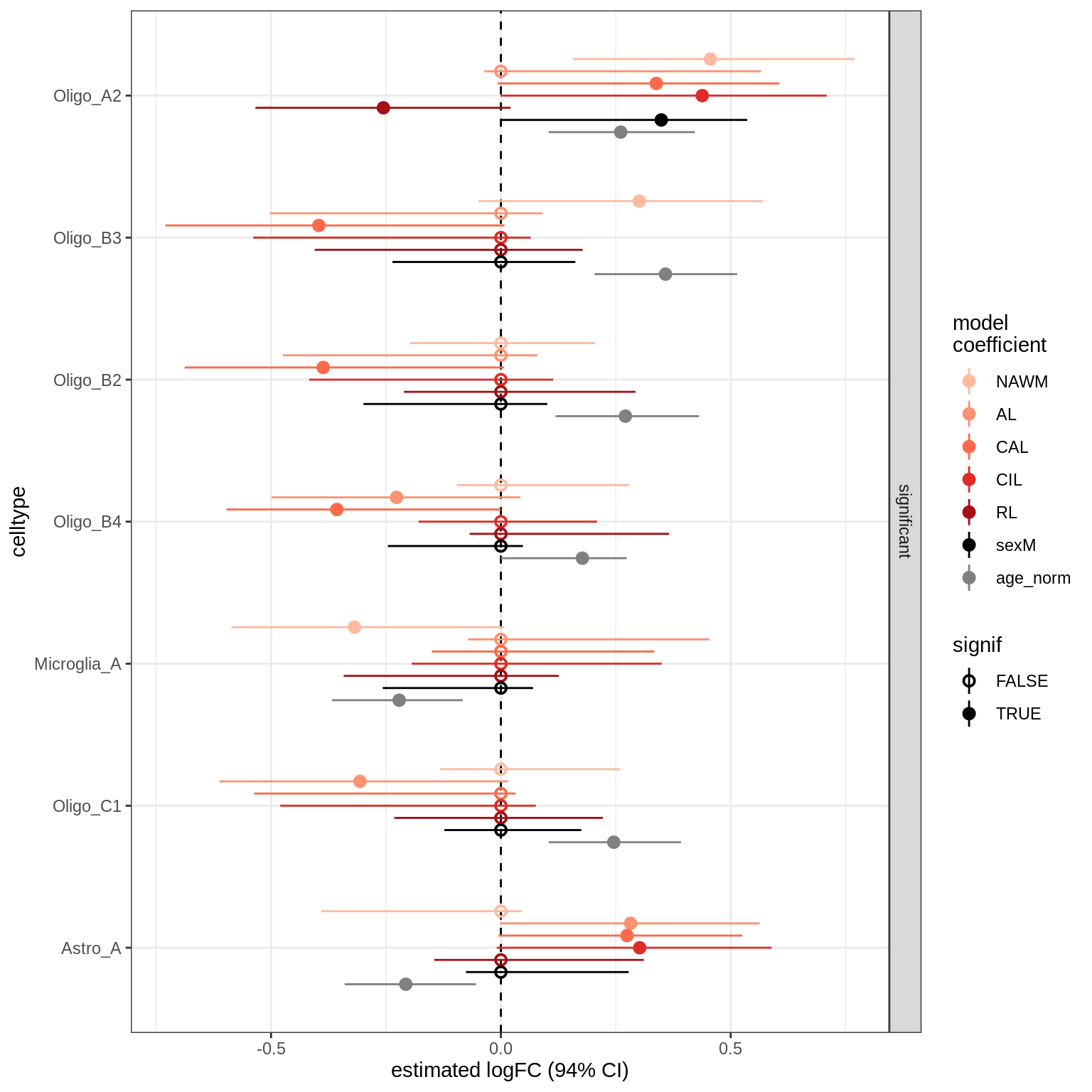
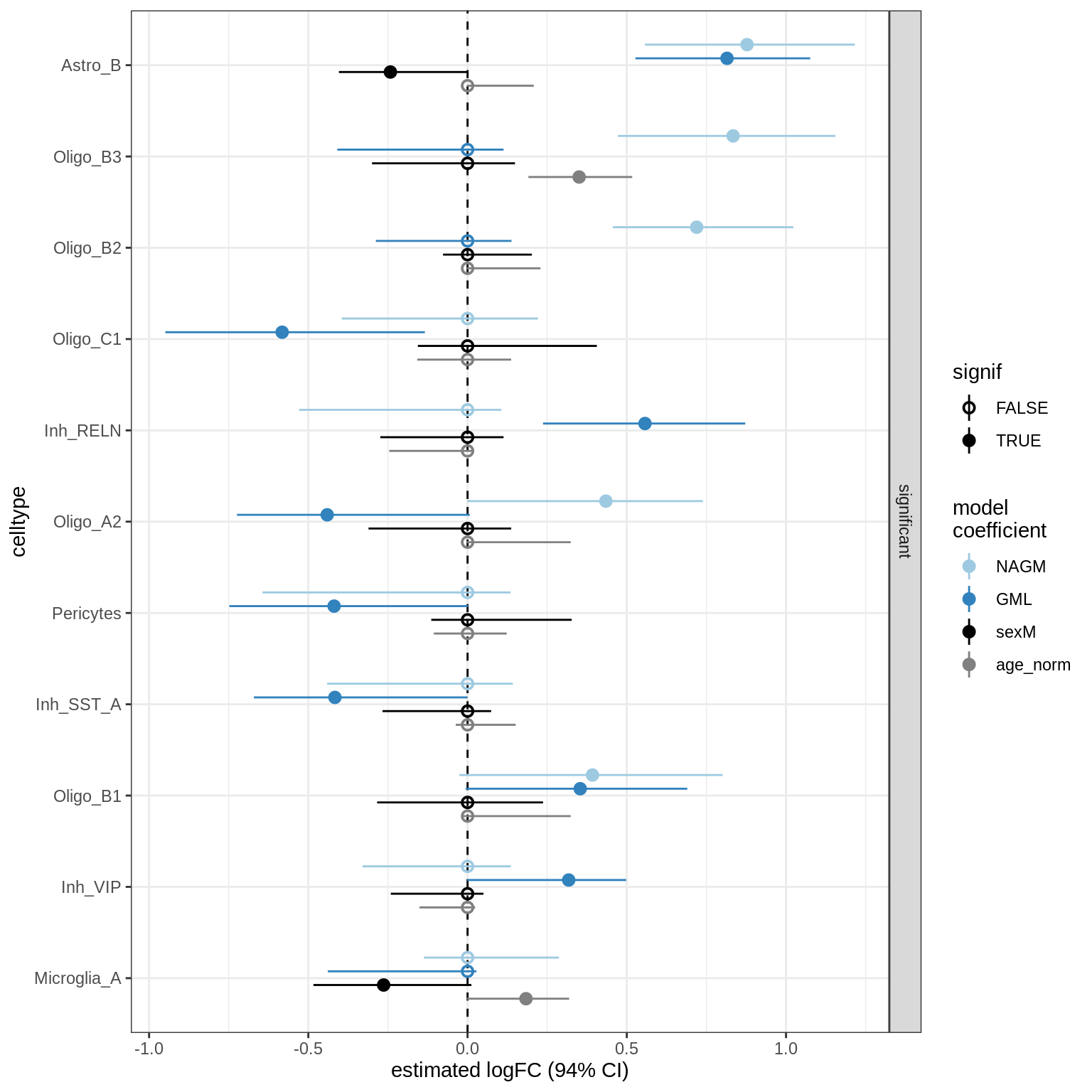
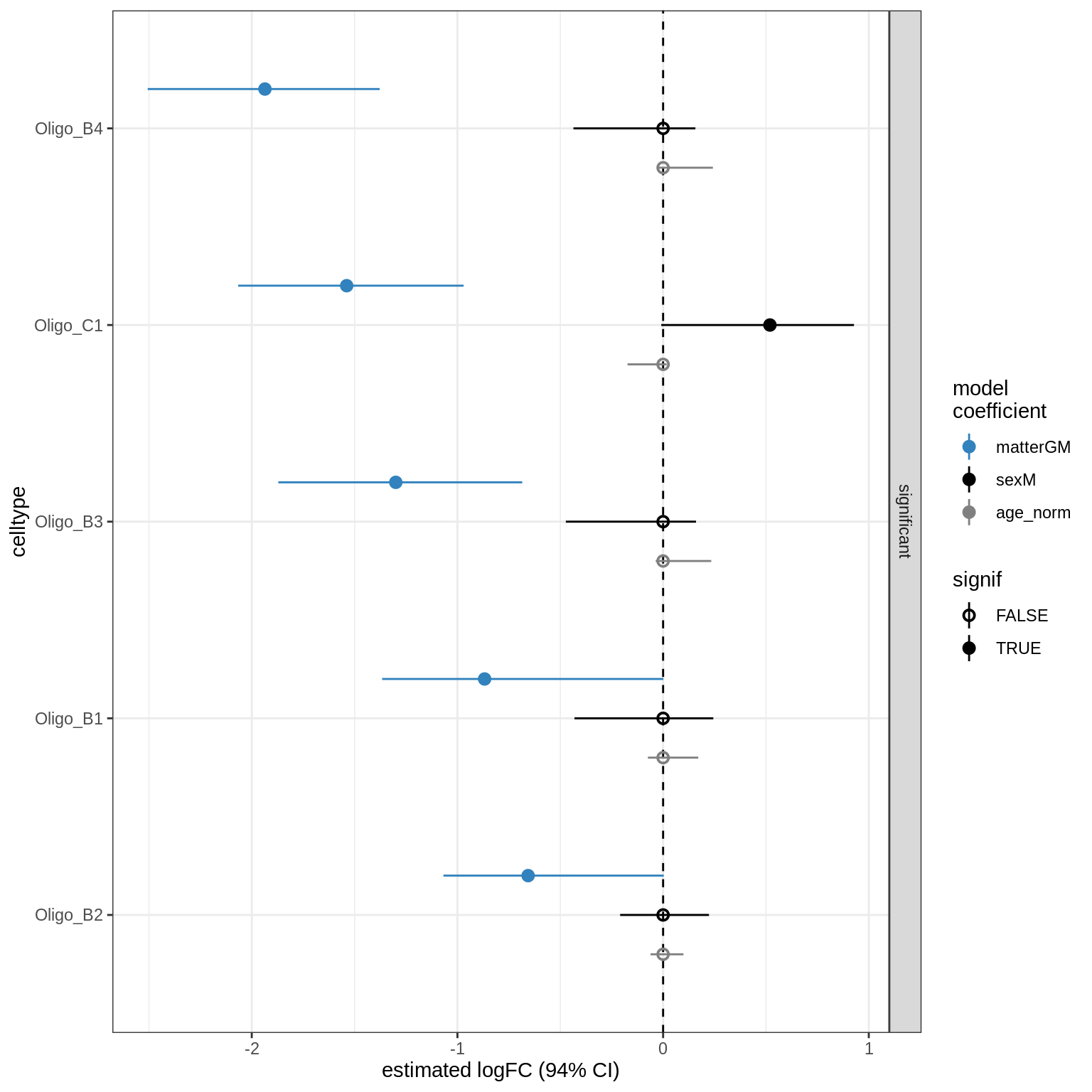
scCODA CIs (signif only)
for (nn in model_names) {
# extract this model
cat('### ', nn, '\n')
print(plot_sccoda_ci(coda_list[[nn]], reported_only = TRUE))
cat('\n\n')
}lesions_WM_mad
lesions_WM_big_mad
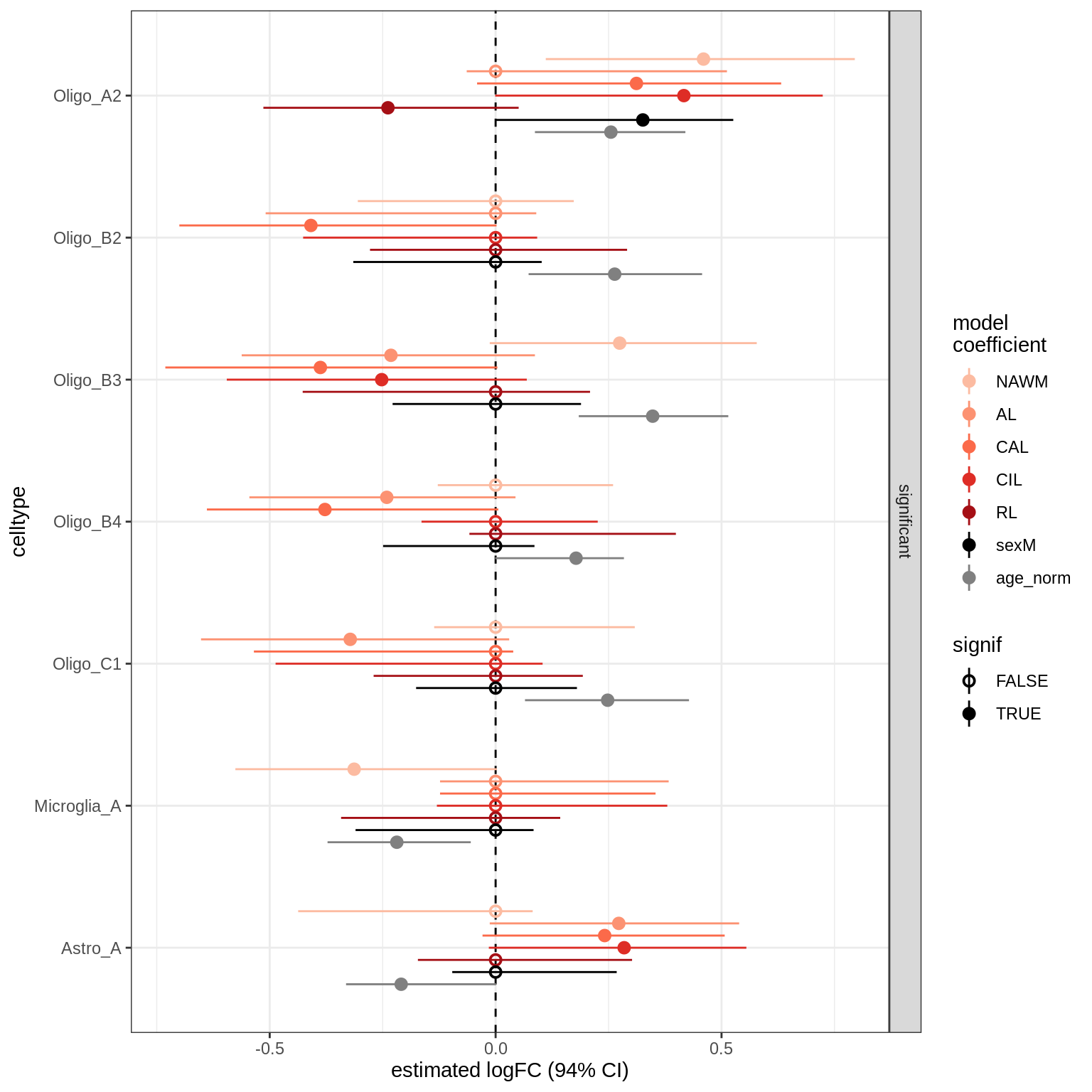
lesions_GM_mad
lesions_GM_big_mad
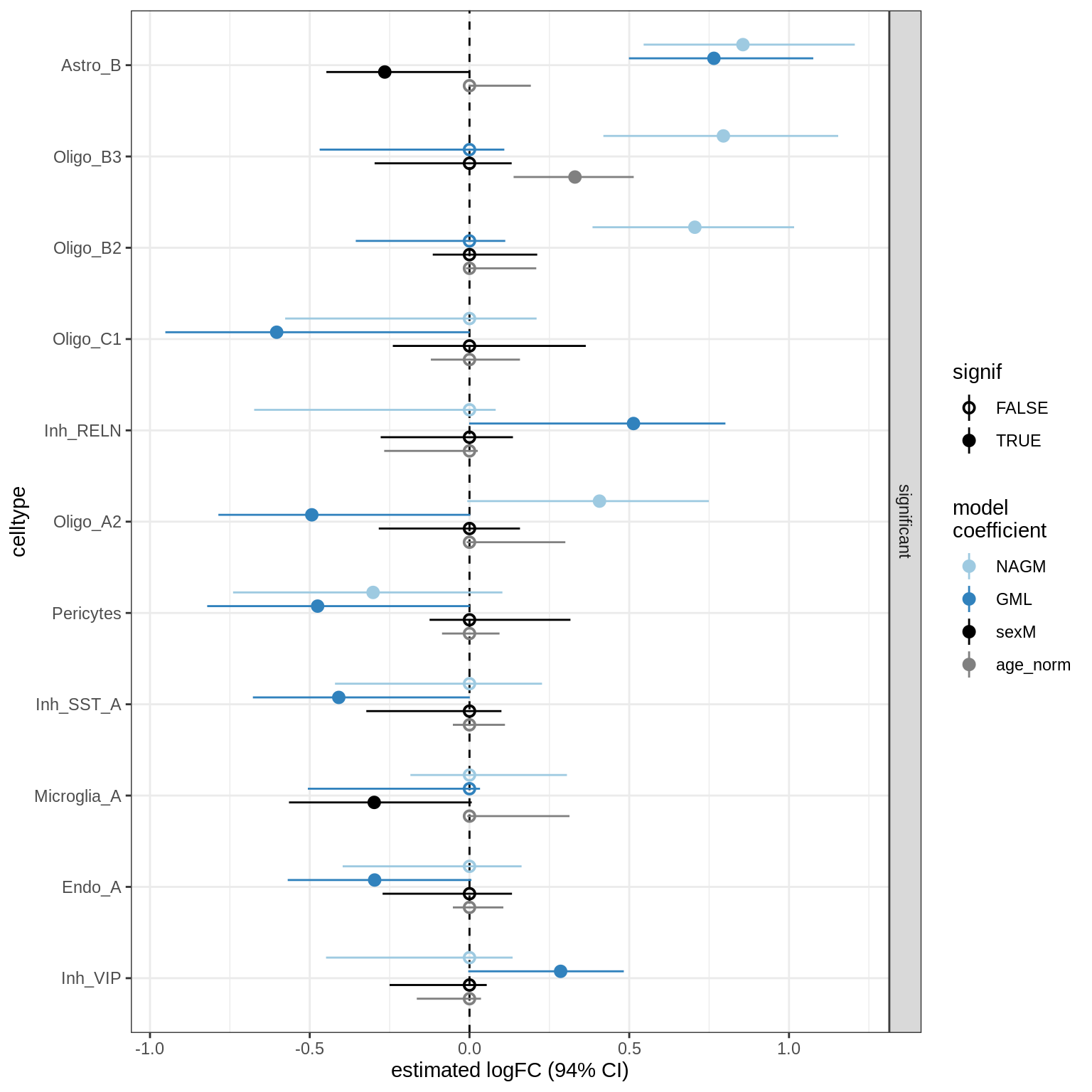
lesions_WM_no_neuro
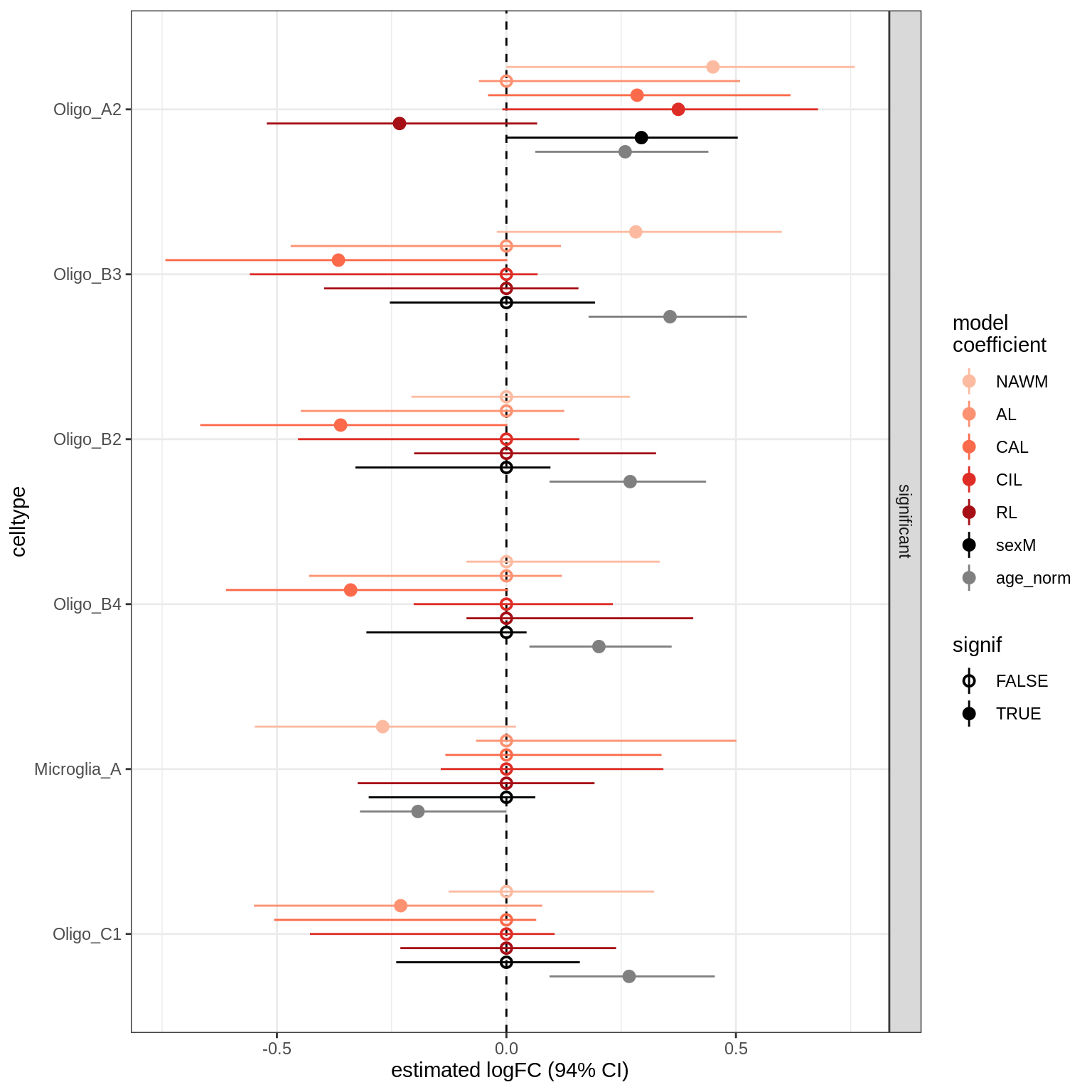
lesions_GM_neuro
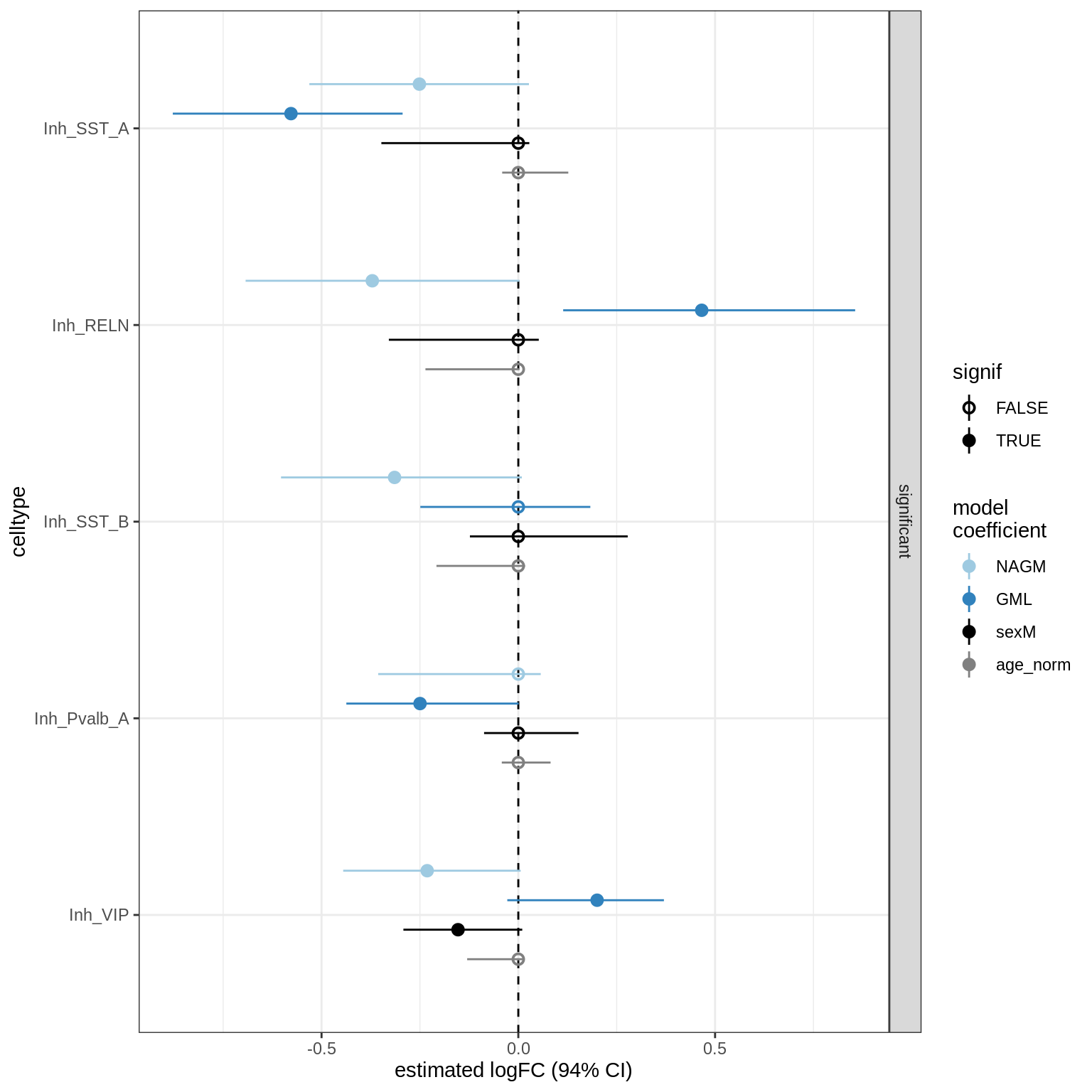
lesions_WM_all
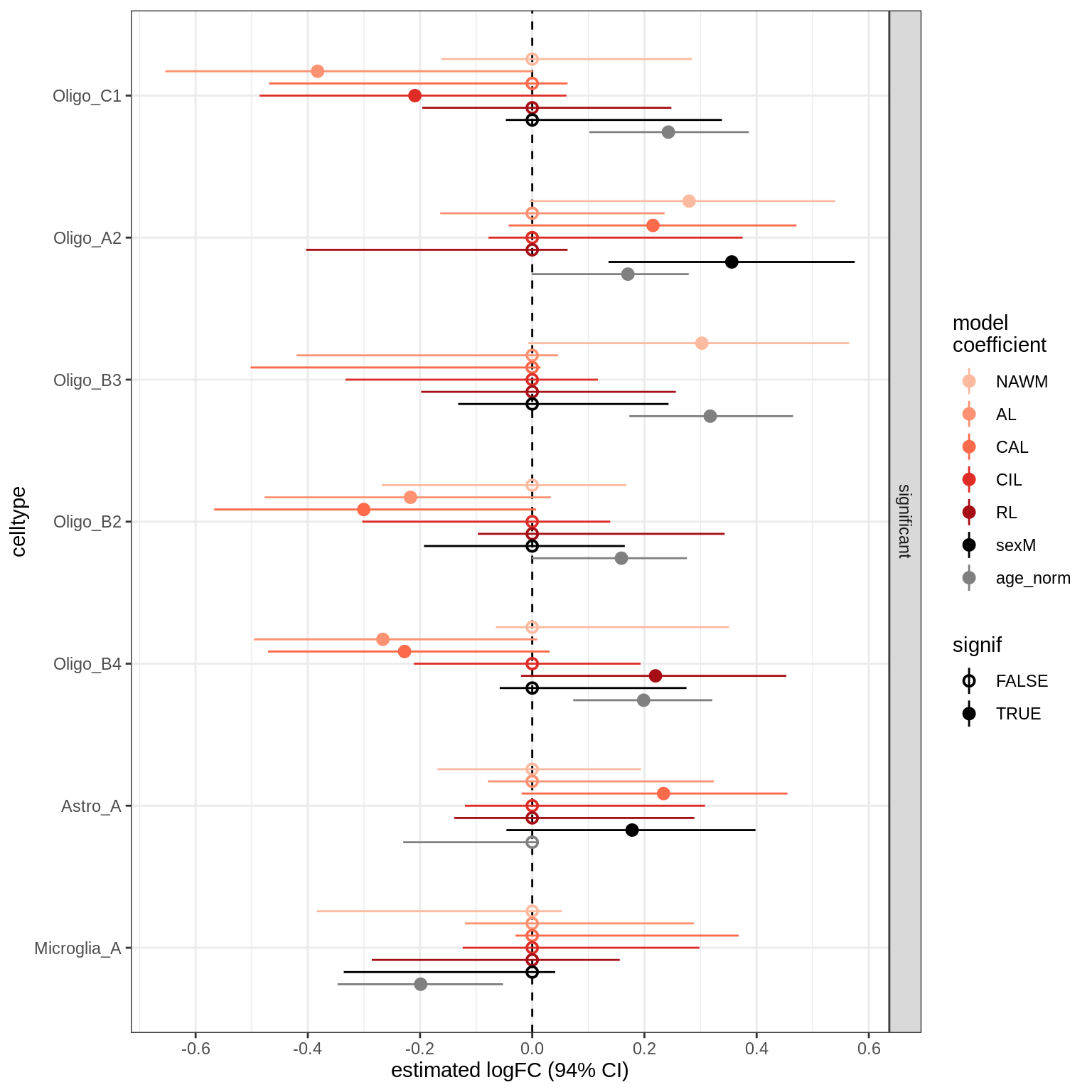
lesions_GM_all
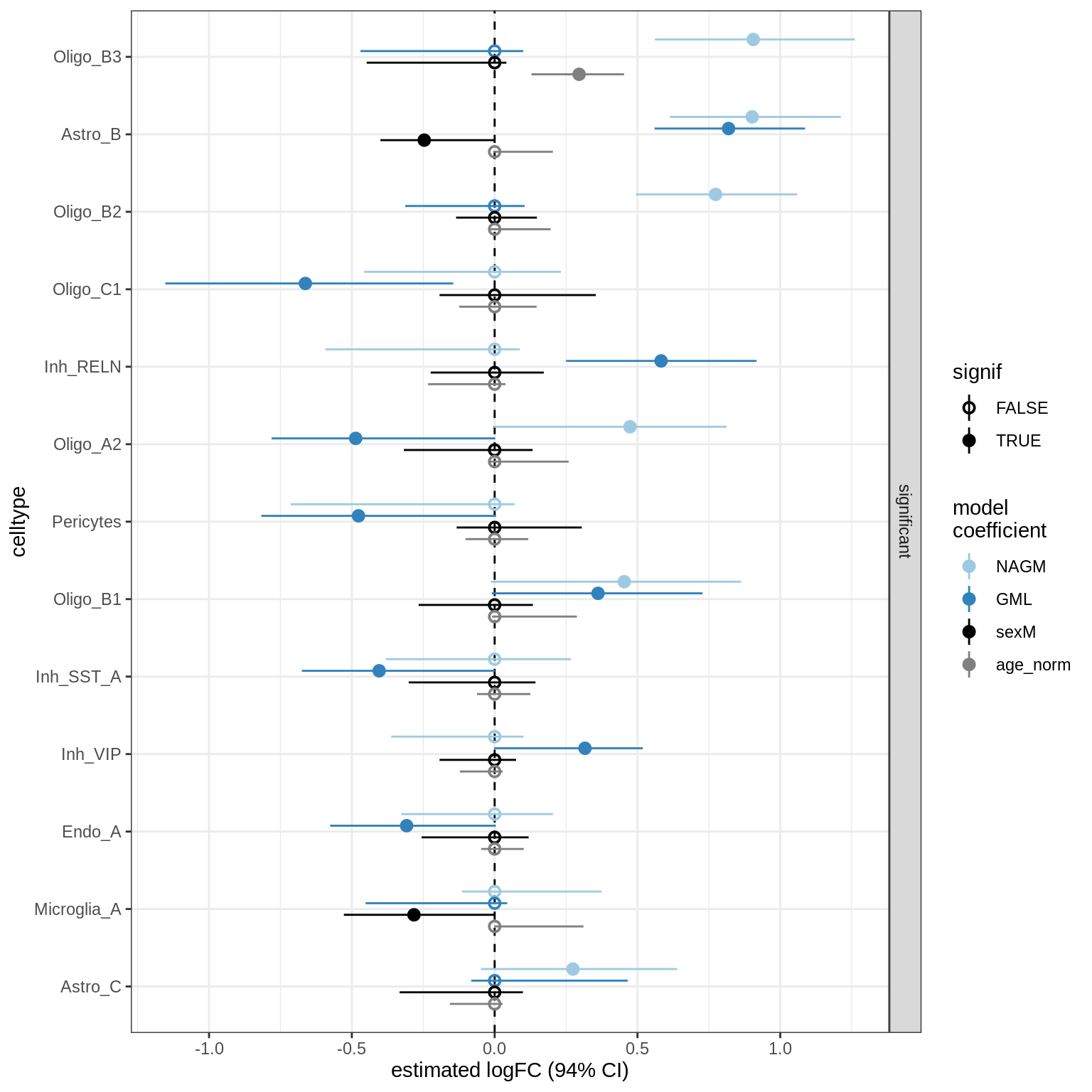
GM_vs_WM
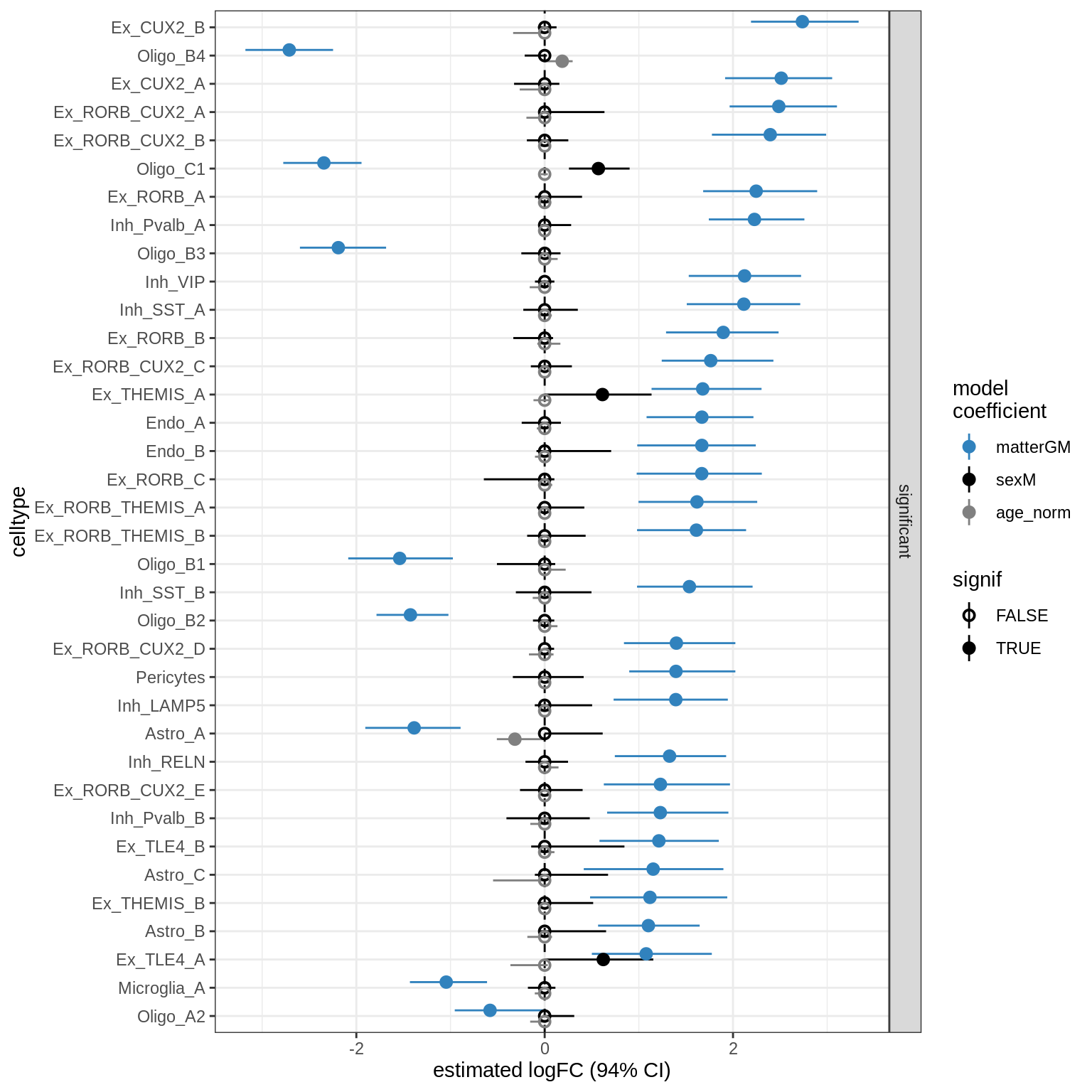
lesions_WM_oligos
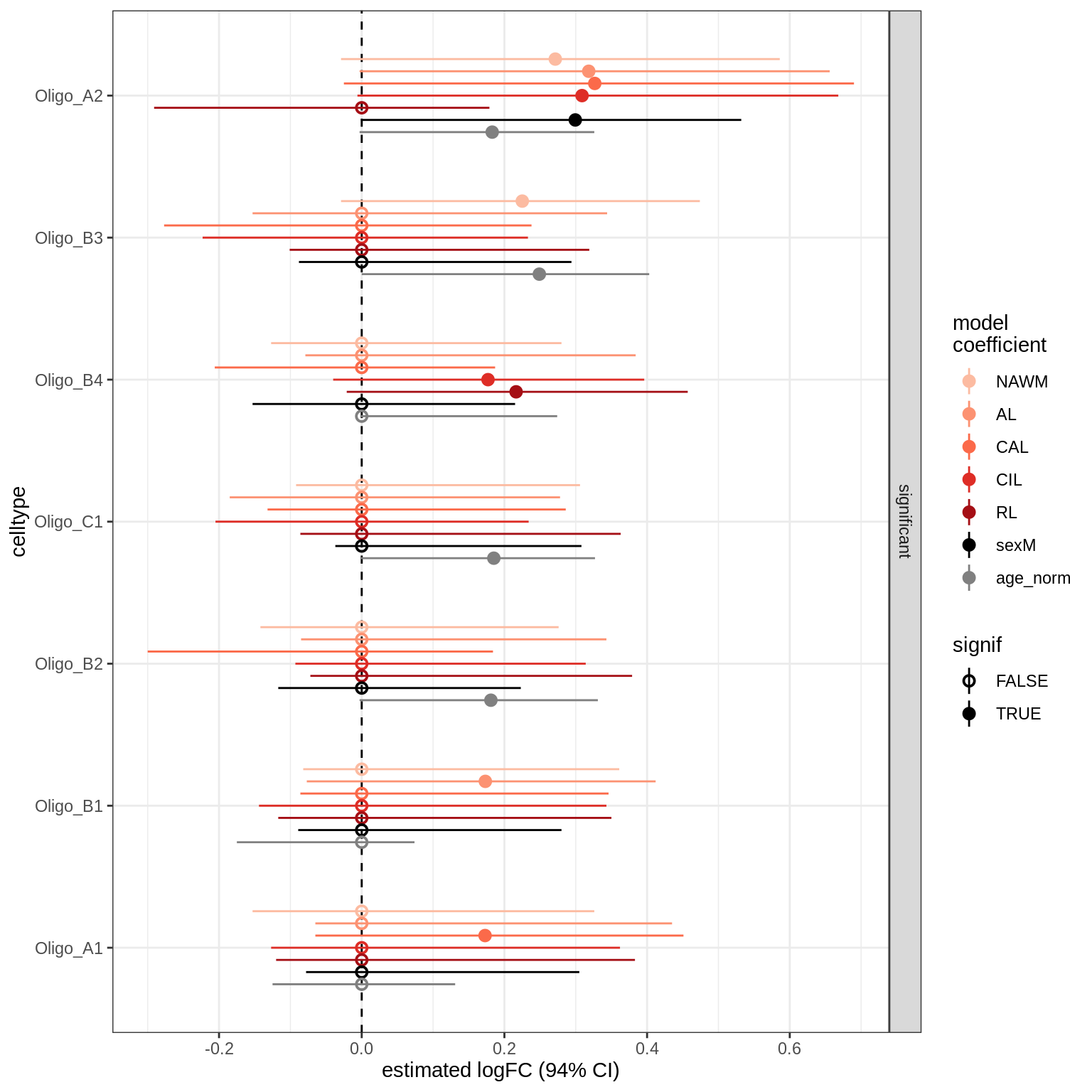
lesions_GM_oligos
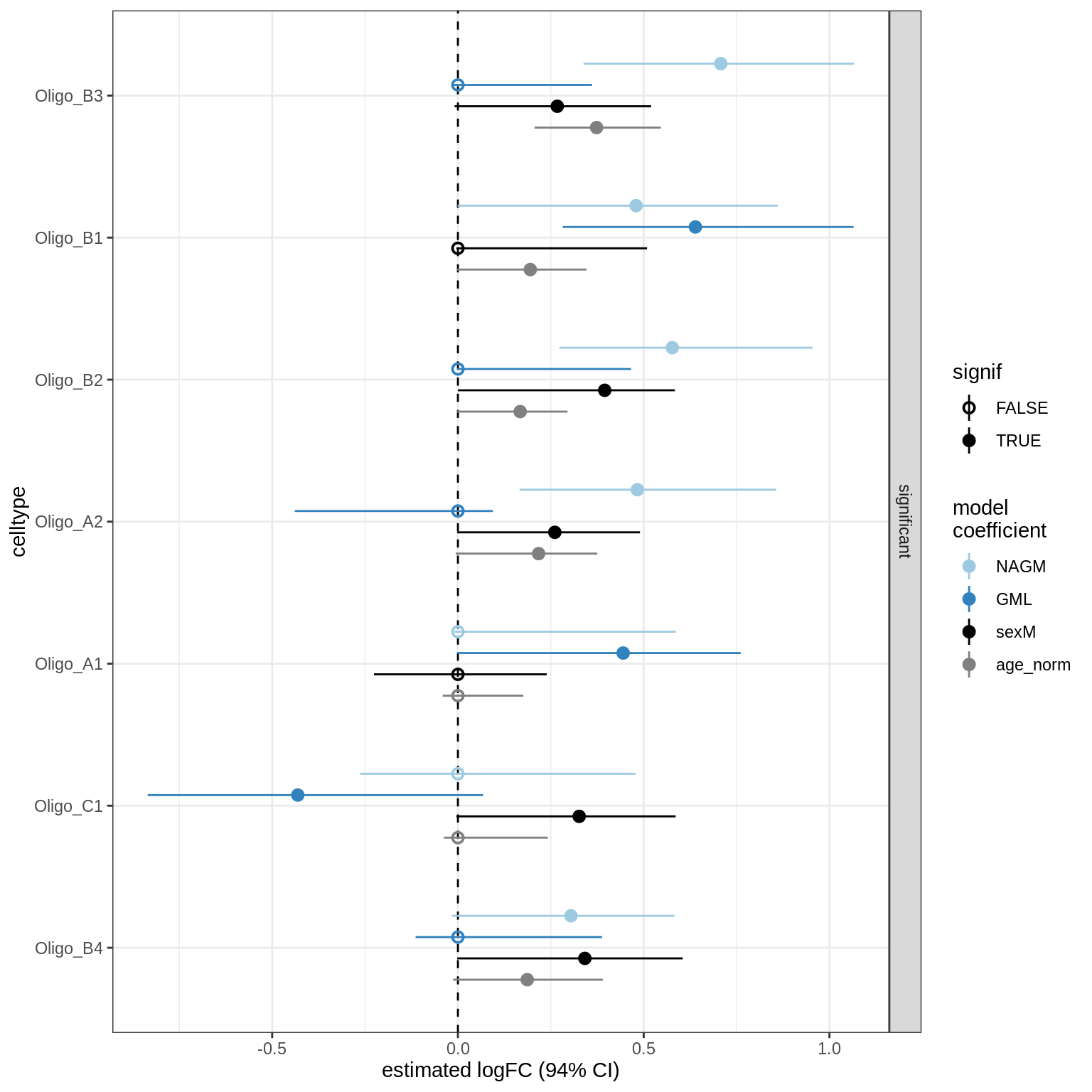
GM_vs_WM_oligos
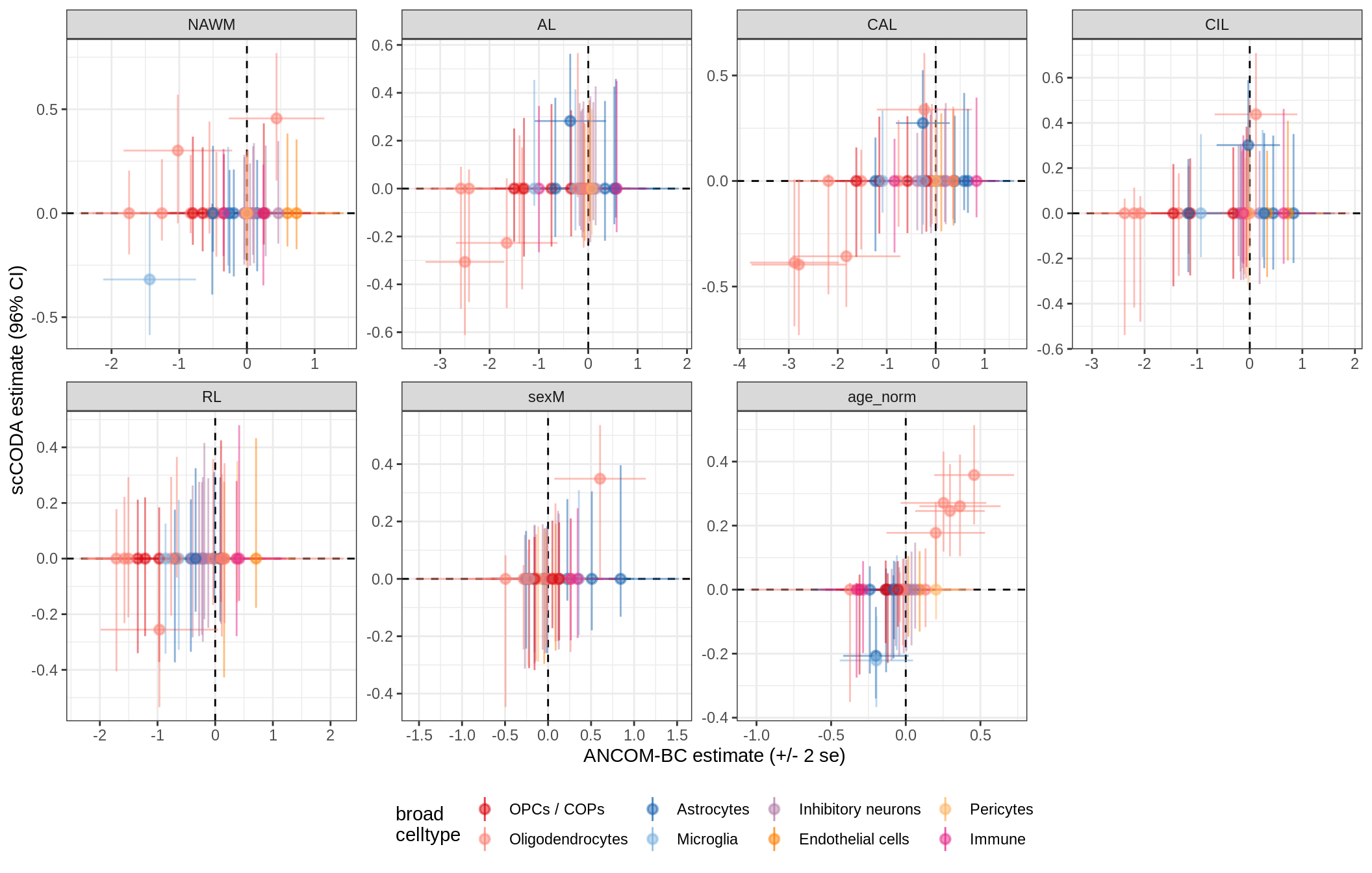
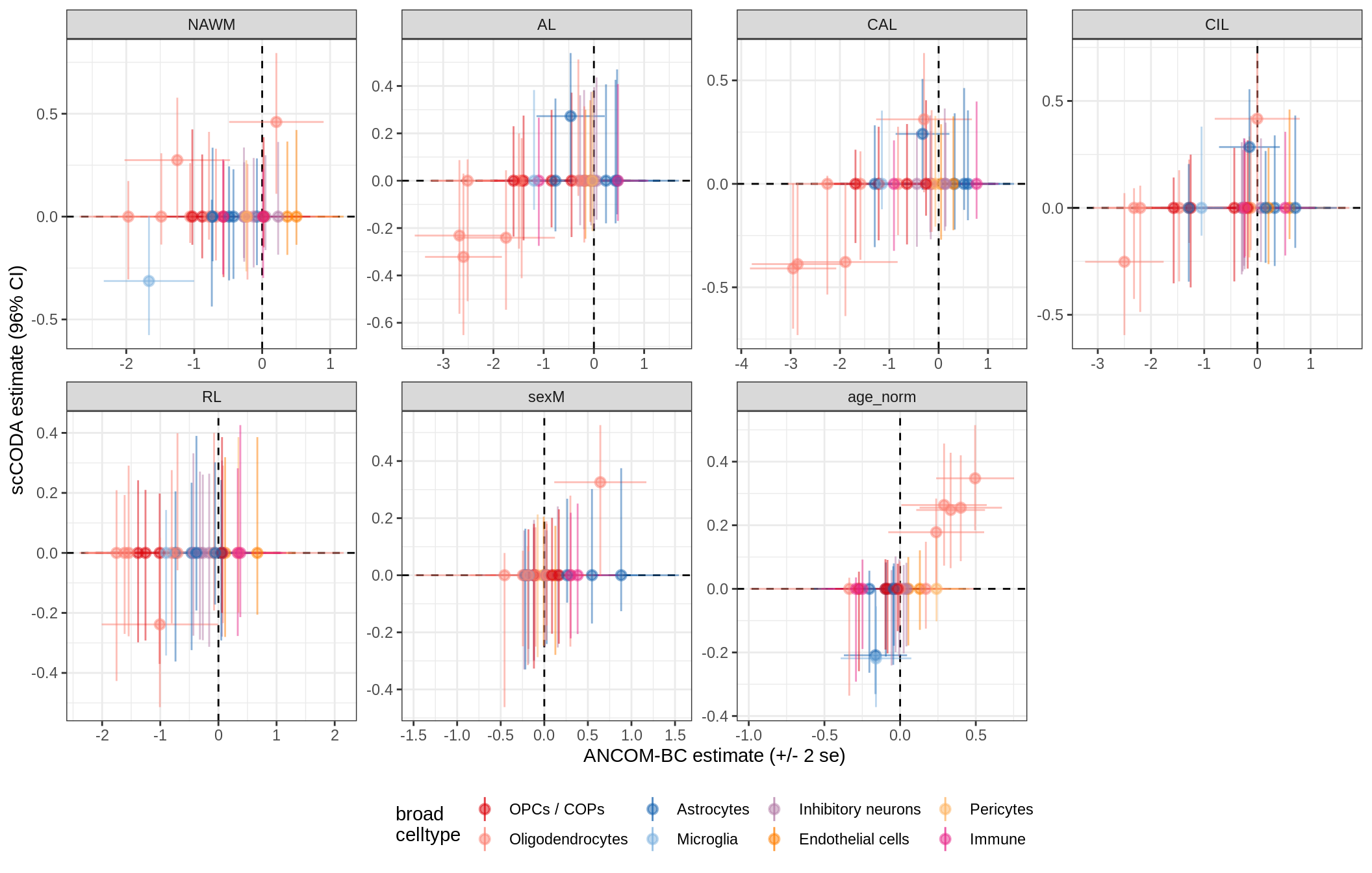
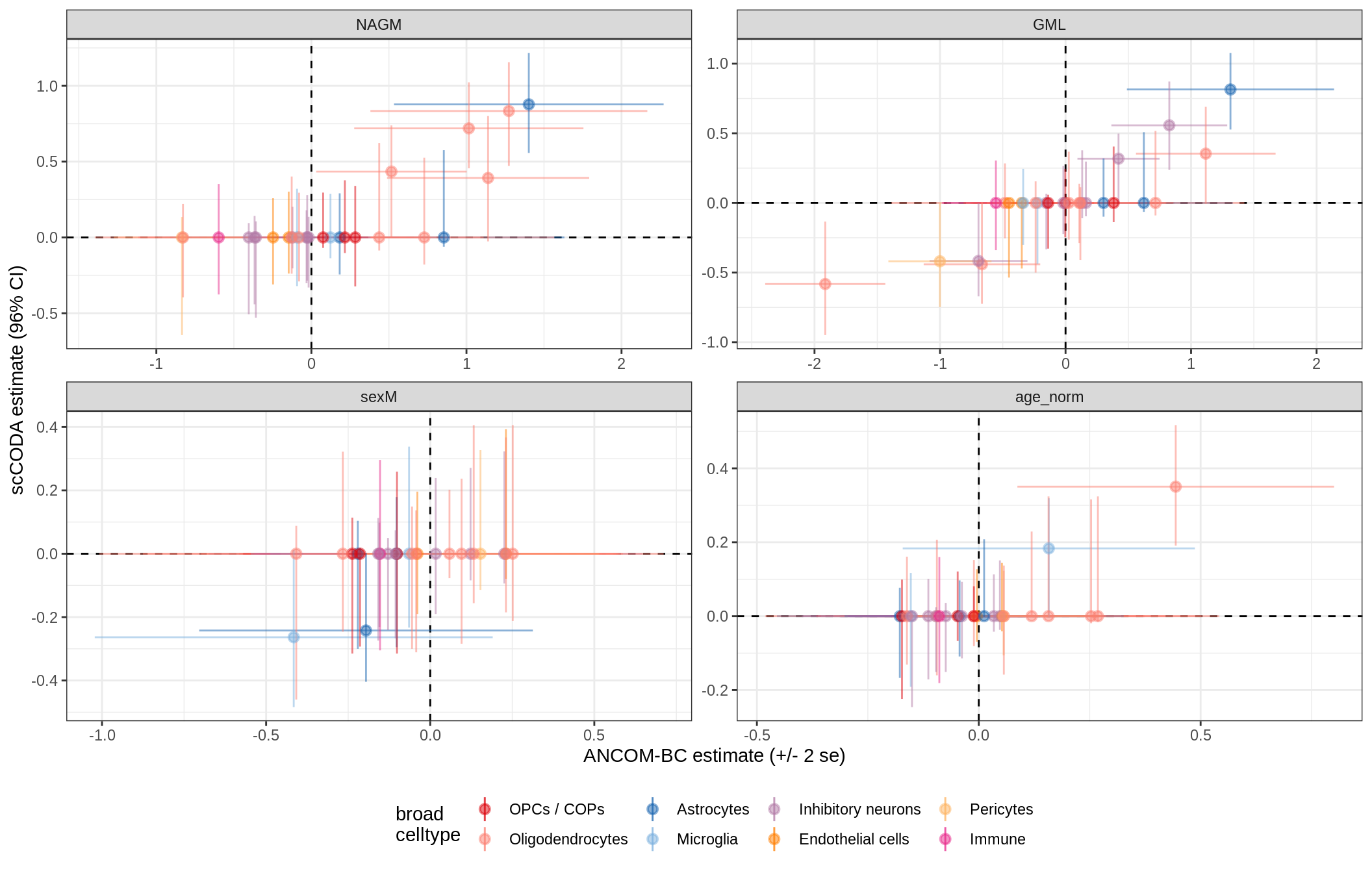
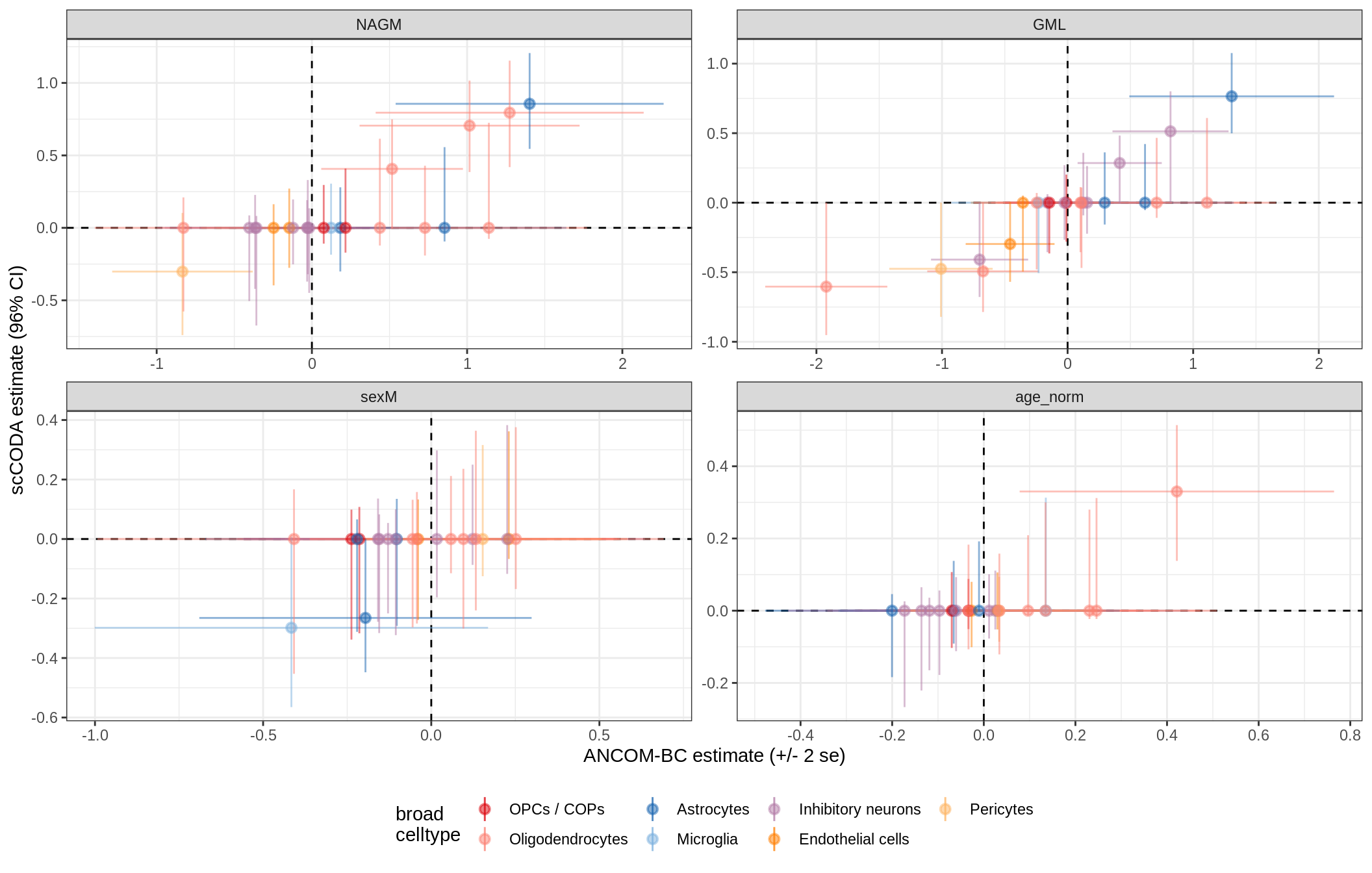
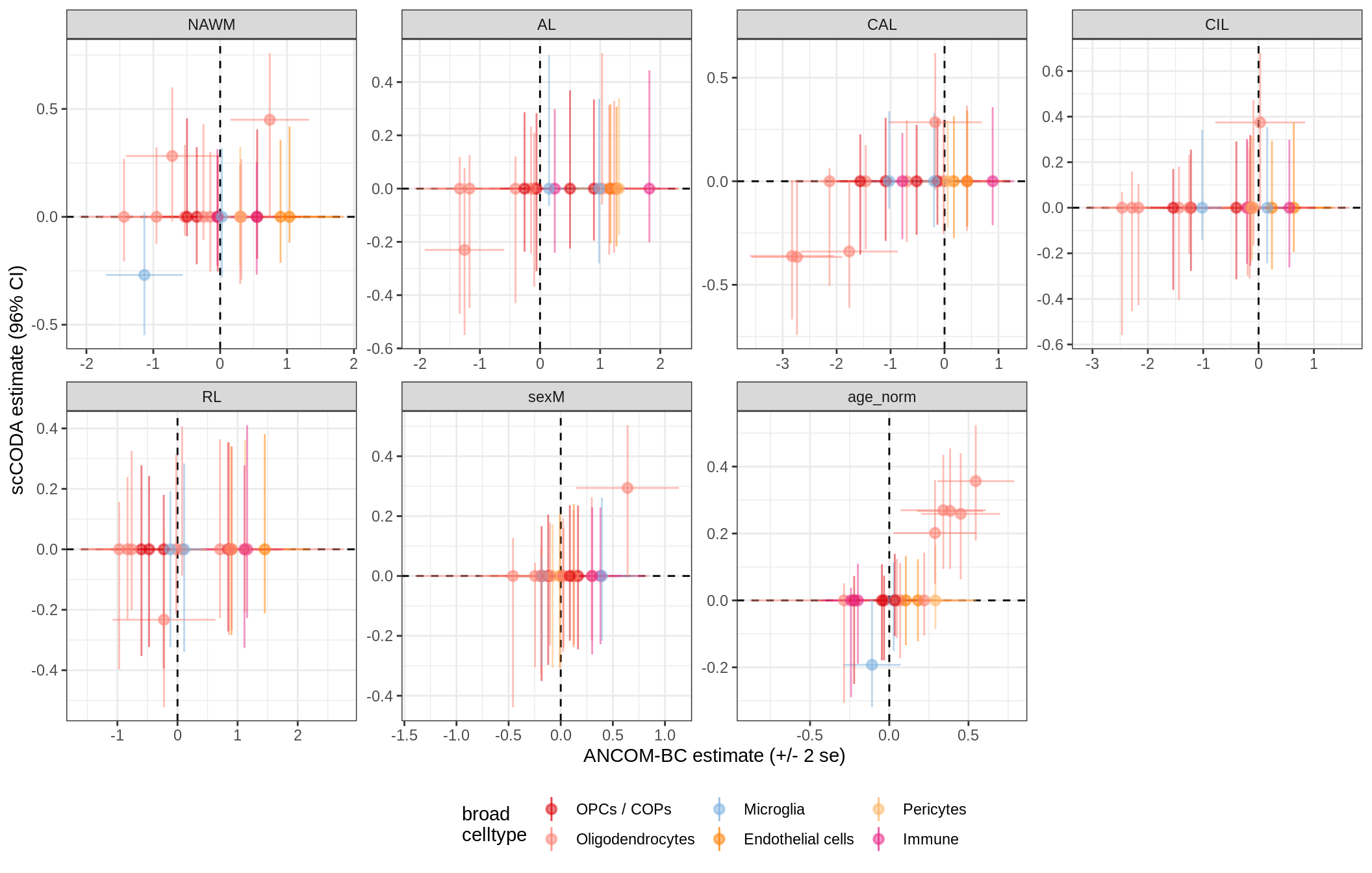
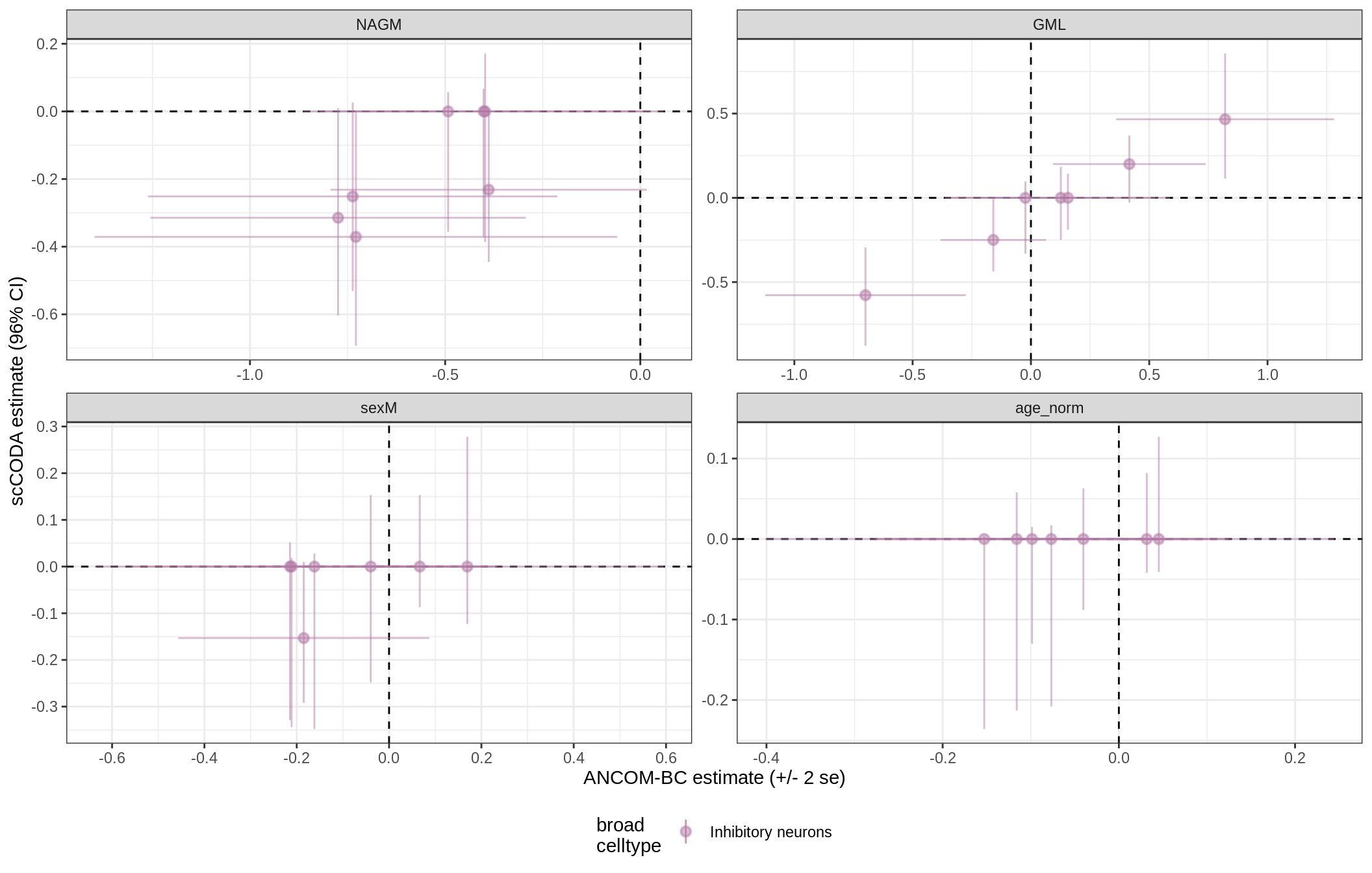
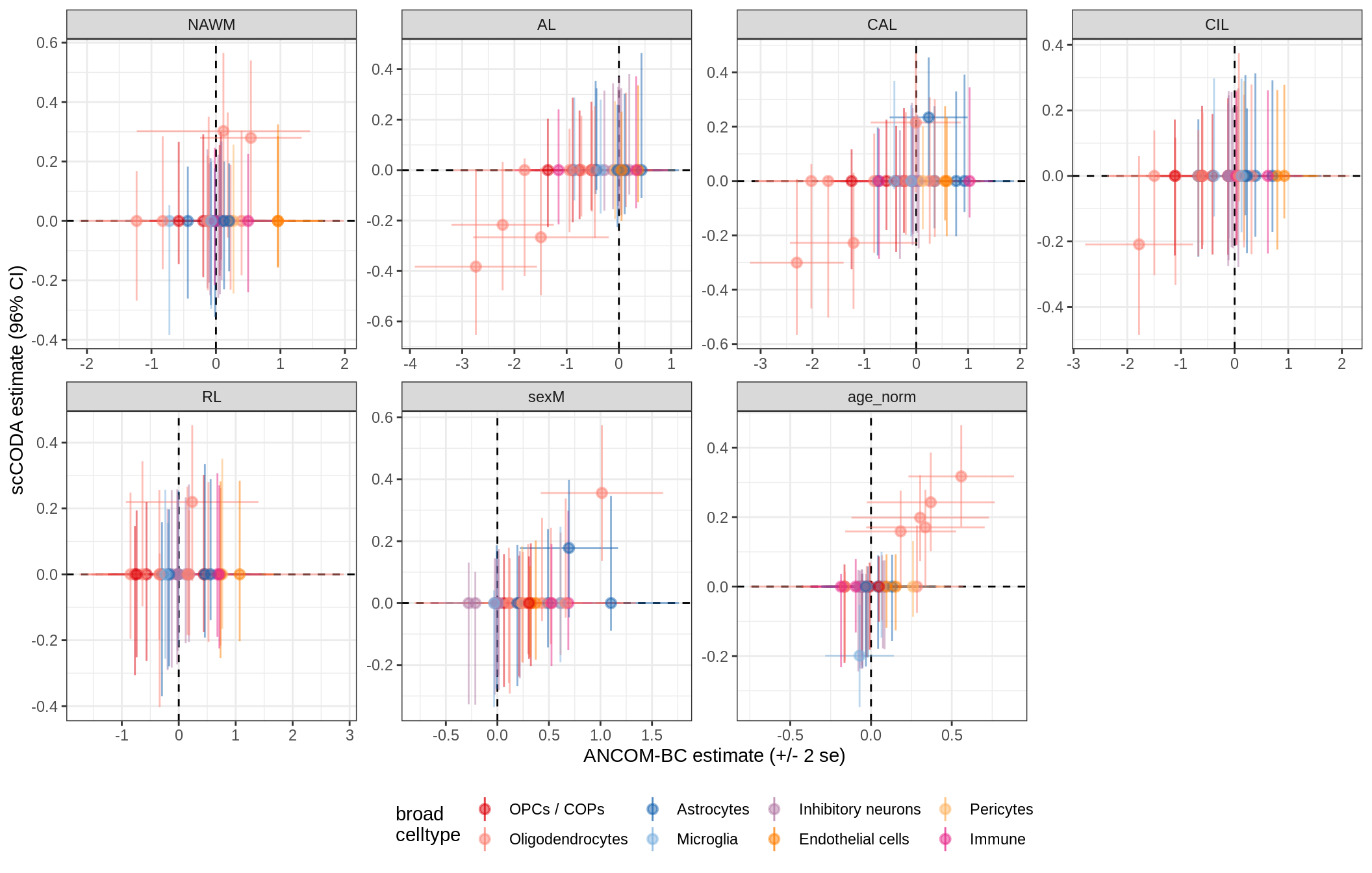
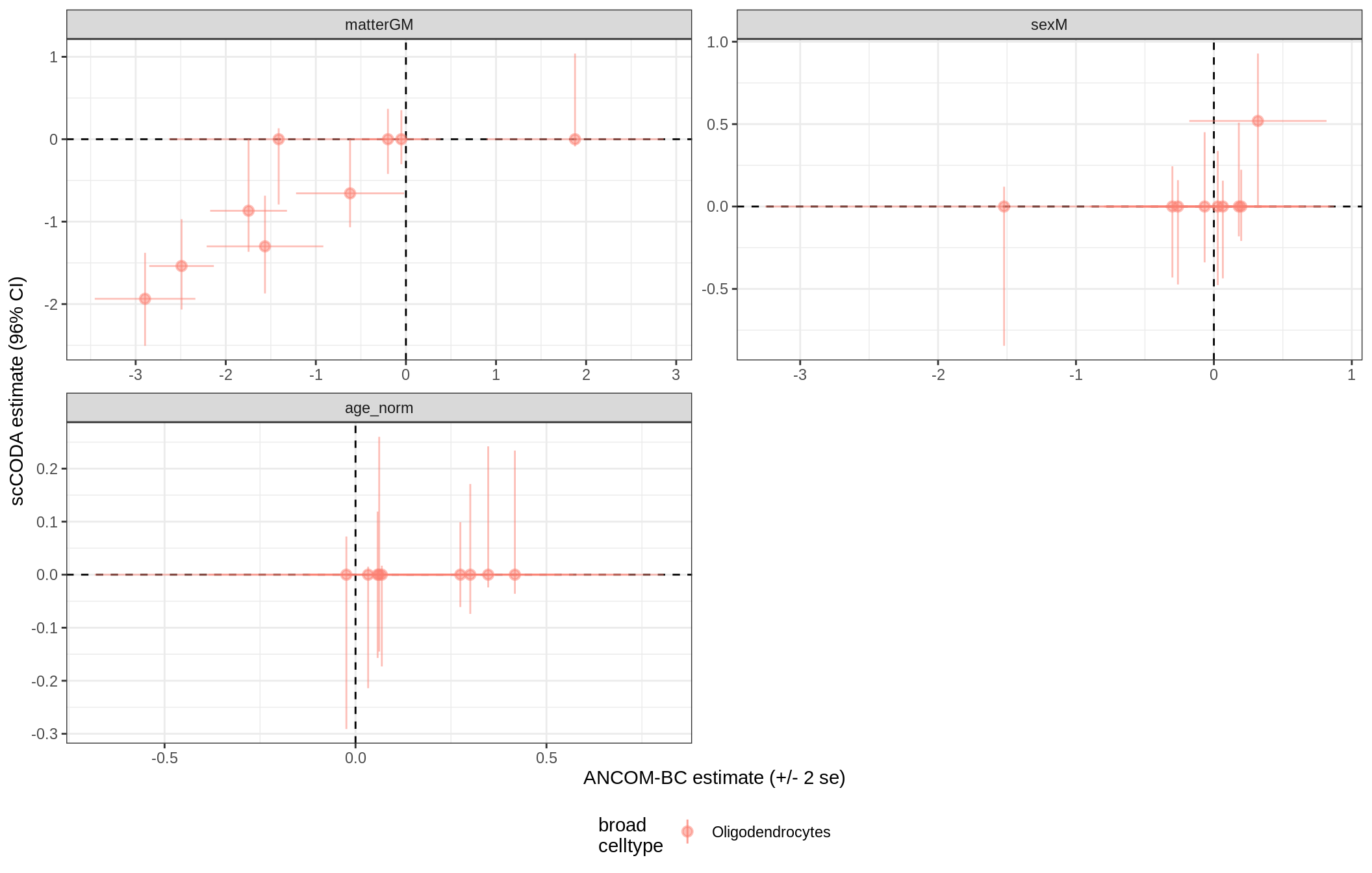
ANCOM-BC vs scCODA
for (nn in model_names) {
# extract this model
cat('### ', nn, '{.tabset}\n')
print(plot_ancom_vs_sccoda(ancom_list[[nn]], coda_list[[nn]], labels_dt))
cat('\n\n')
}lesions_WM_mad
lesions_WM_big_mad
lesions_GM_mad
lesions_GM_big_mad
lesions_WM_no_neuro
lesions_GM_neuro
lesions_WM_all
lesions_GM_all
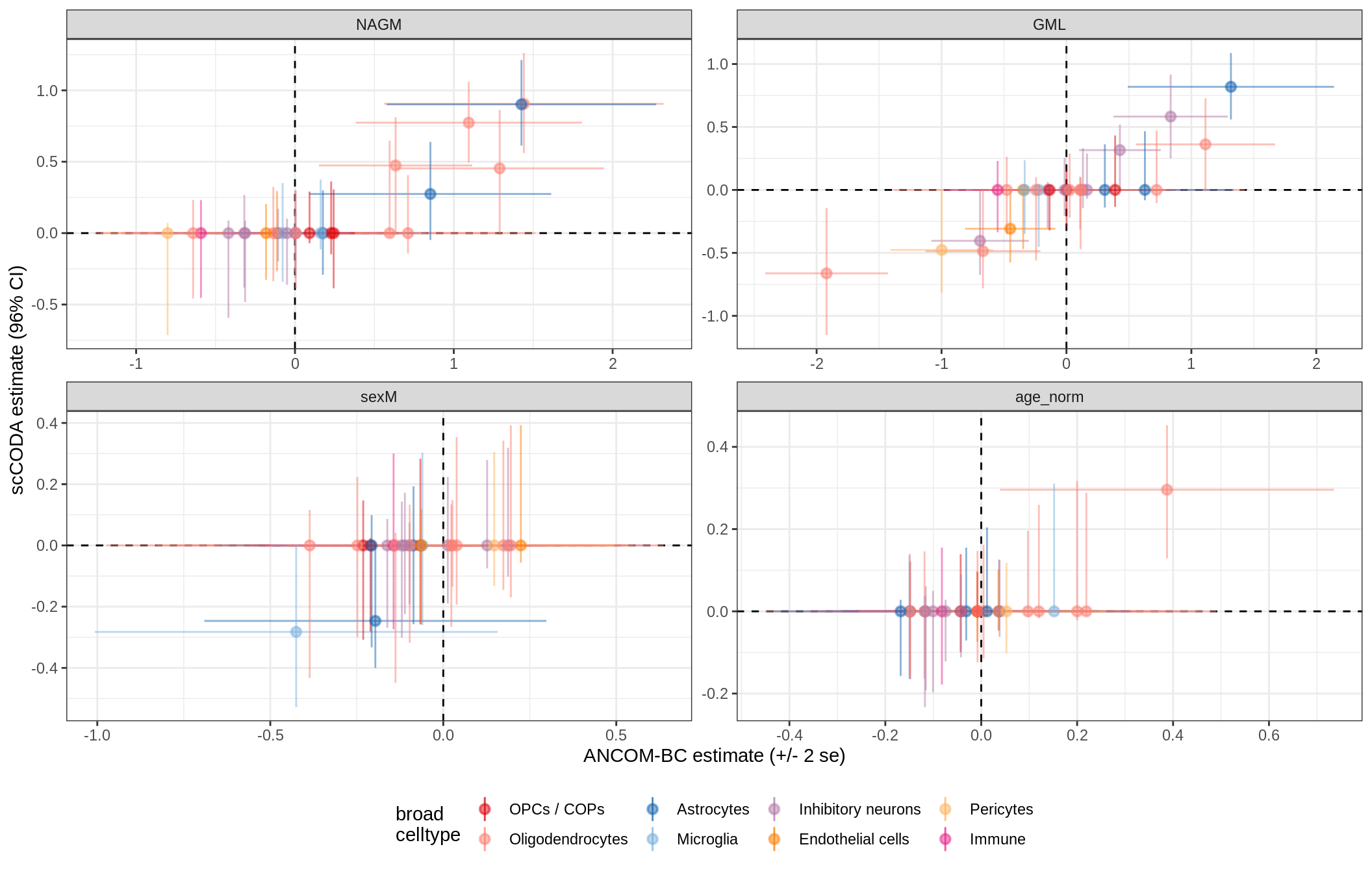
GM_vs_WM
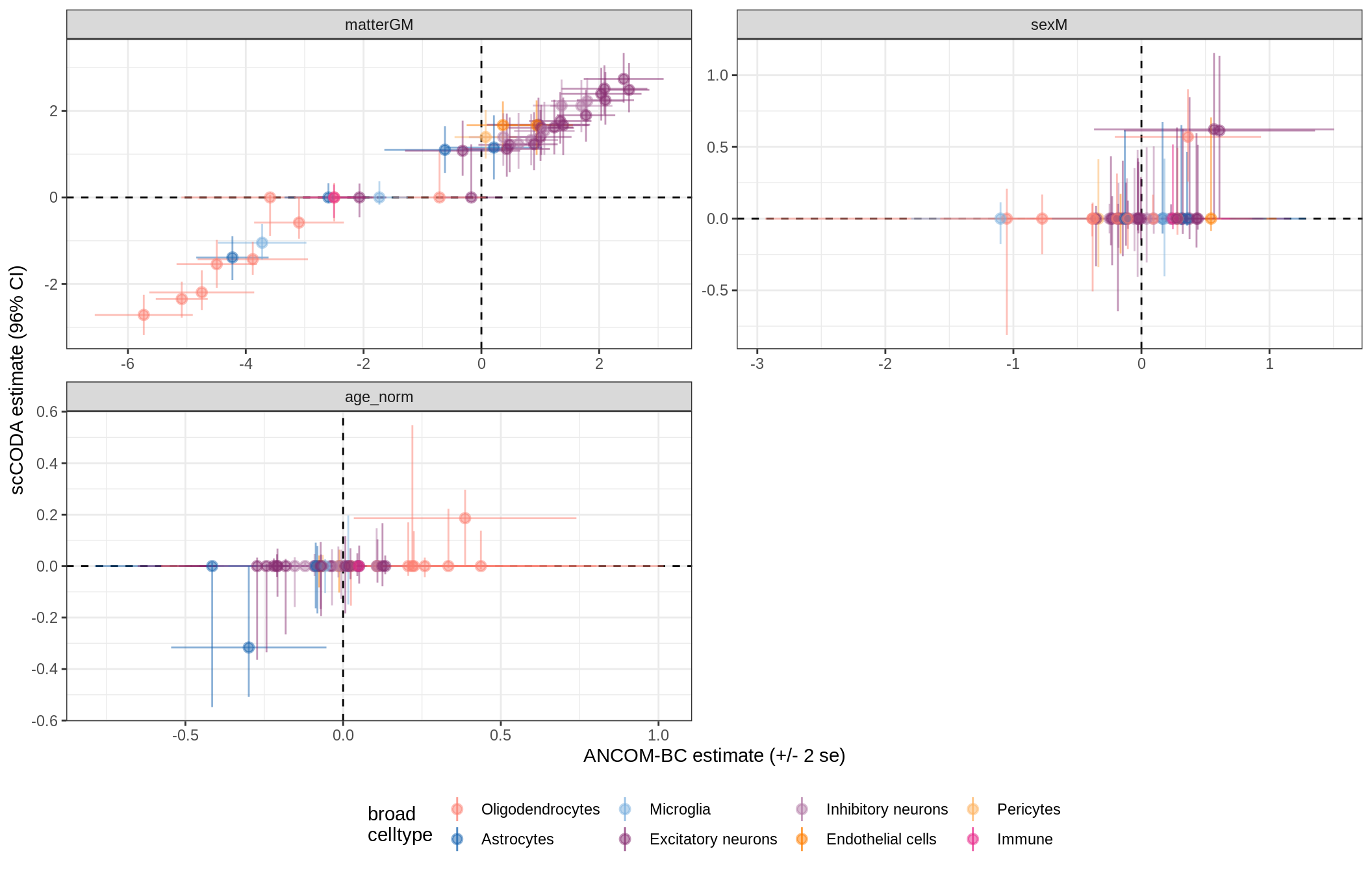
lesions_WM_oligos
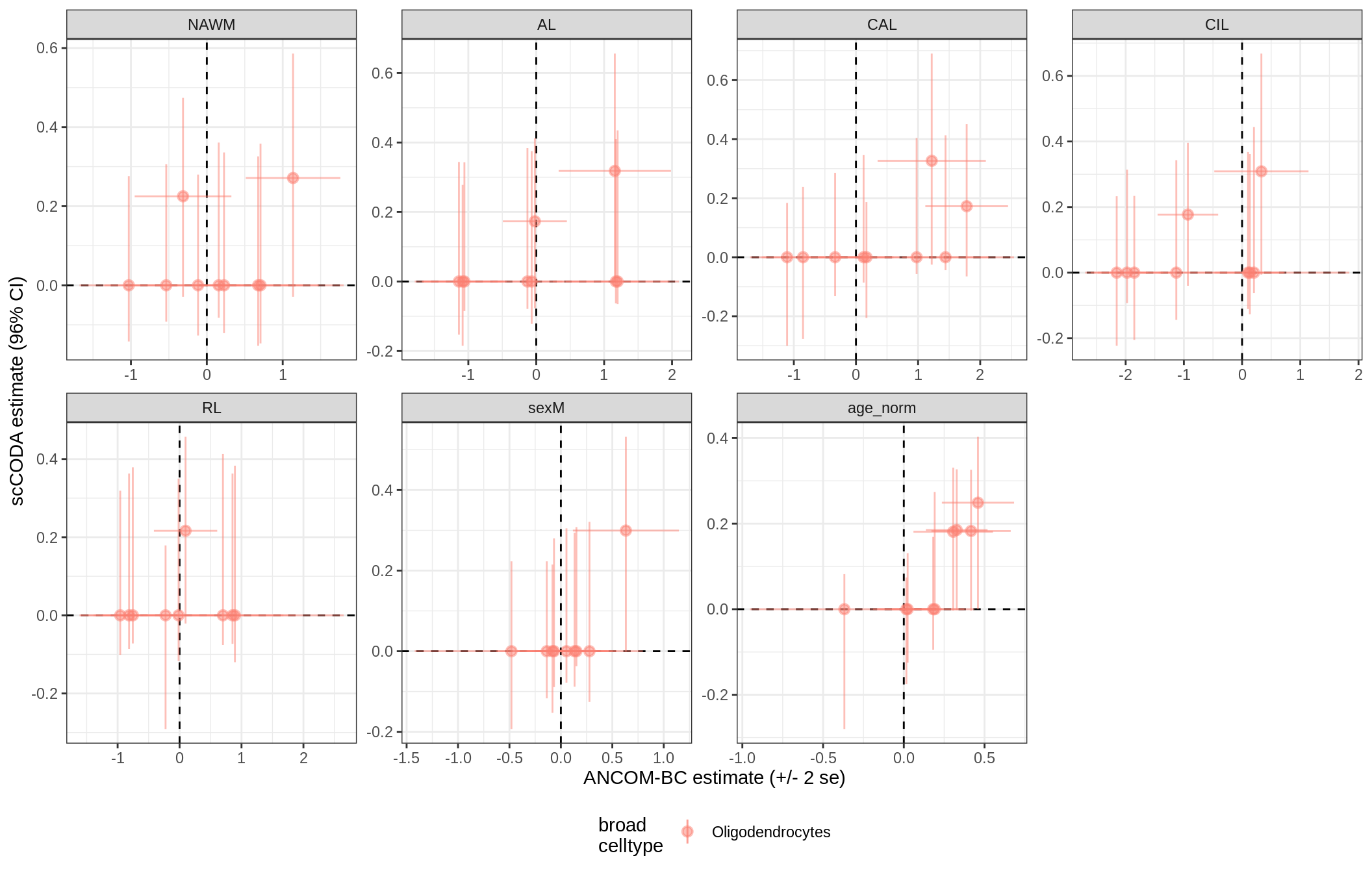
lesions_GM_oligos
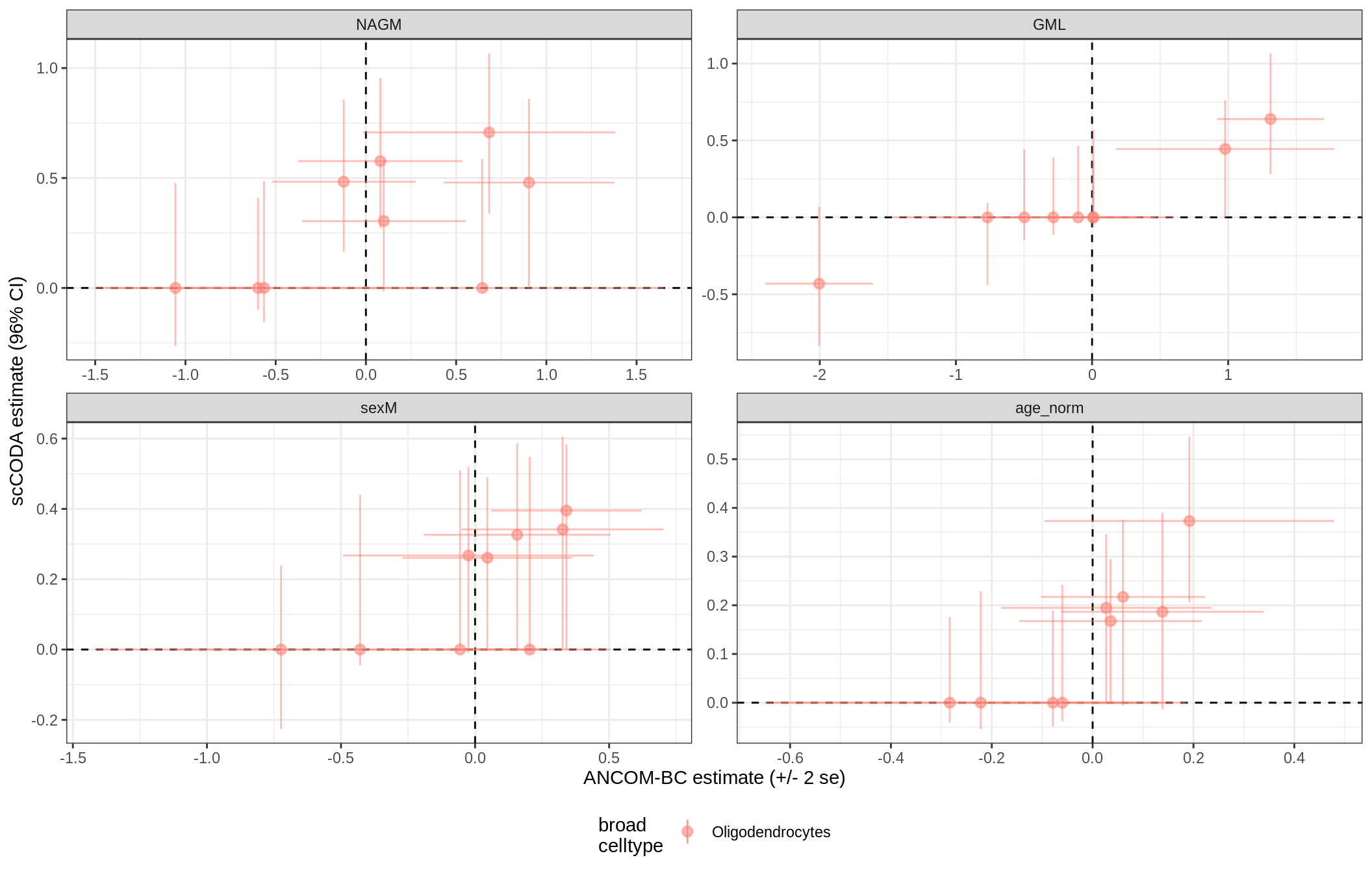
GM_vs_WM_oligos
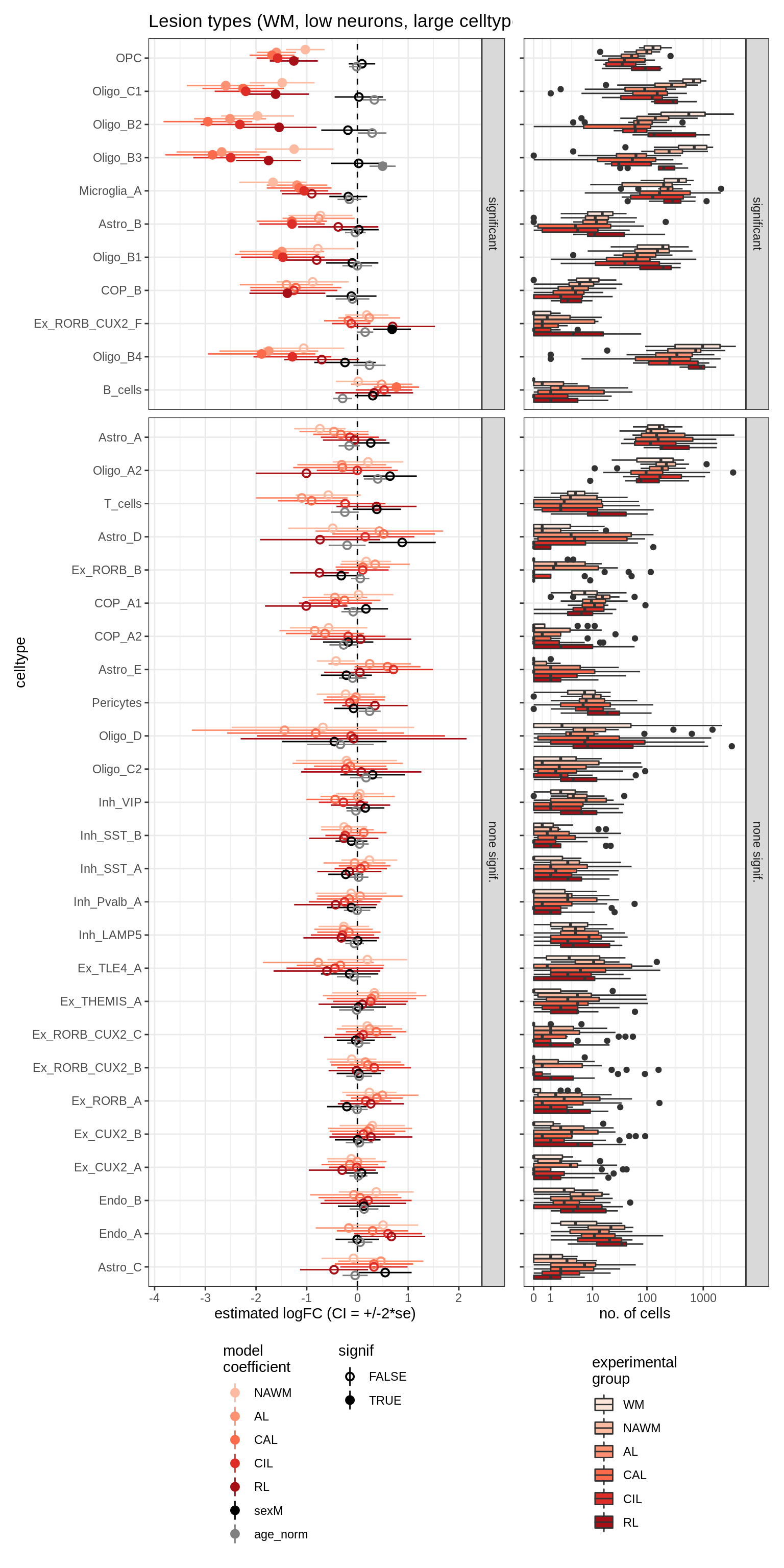
ANCOM CIs (all)
for (nn in model_names) {
# extract this model
cat('### ', nn, '\n')
print(plot_ancombc_ci(ancom_list[[nn]], counts_wide, names_list[[nn]],
whatplot = whatplot_list[[nn]], reported_only = FALSE))
cat('\n\n')
}lesions_WM_mad
lesions_WM_big_mad
lesions_GM_mad
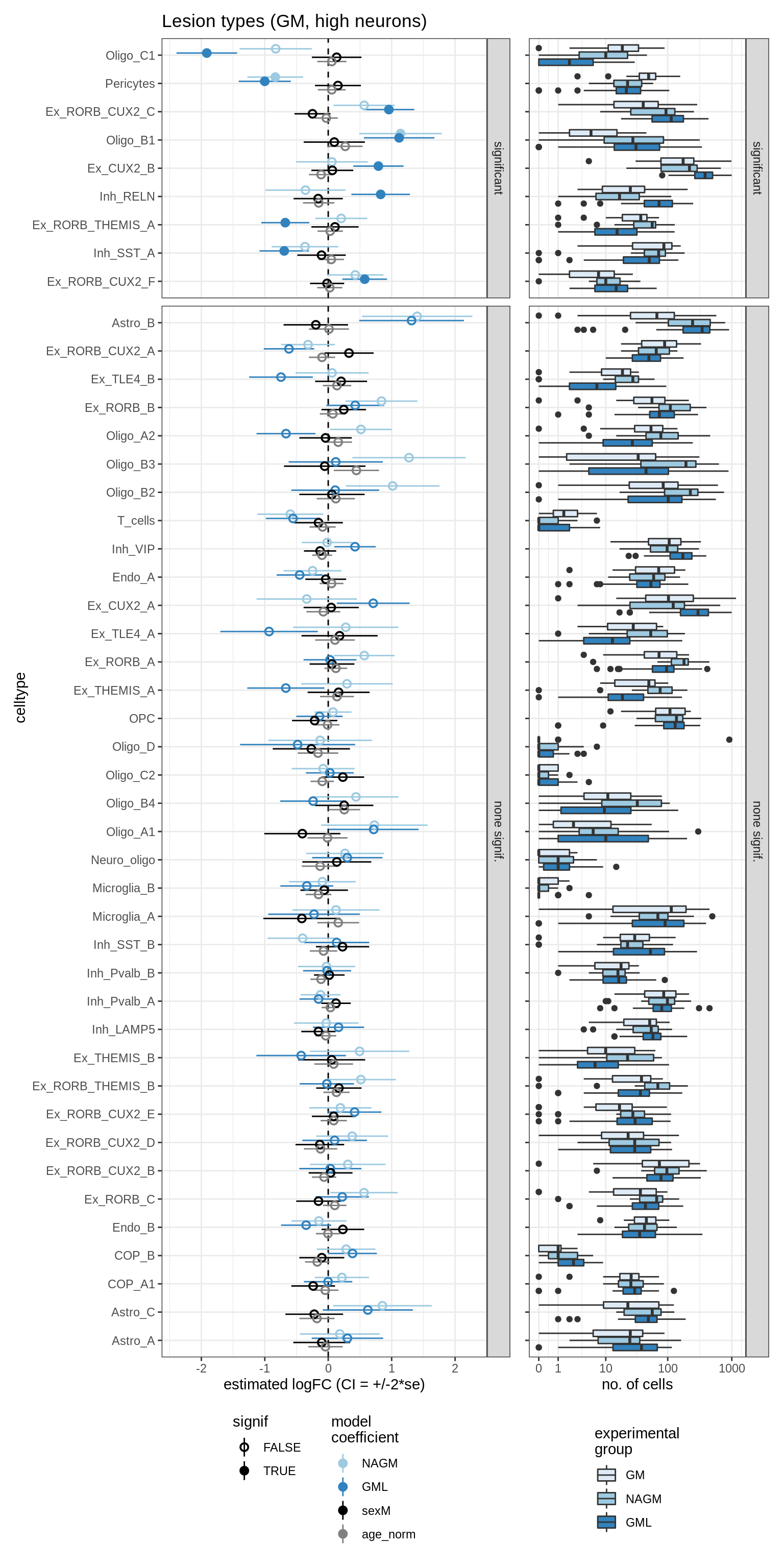
lesions_GM_big_mad
lesions_WM_no_neuro
lesions_GM_neuro
lesions_WM_all
lesions_GM_all
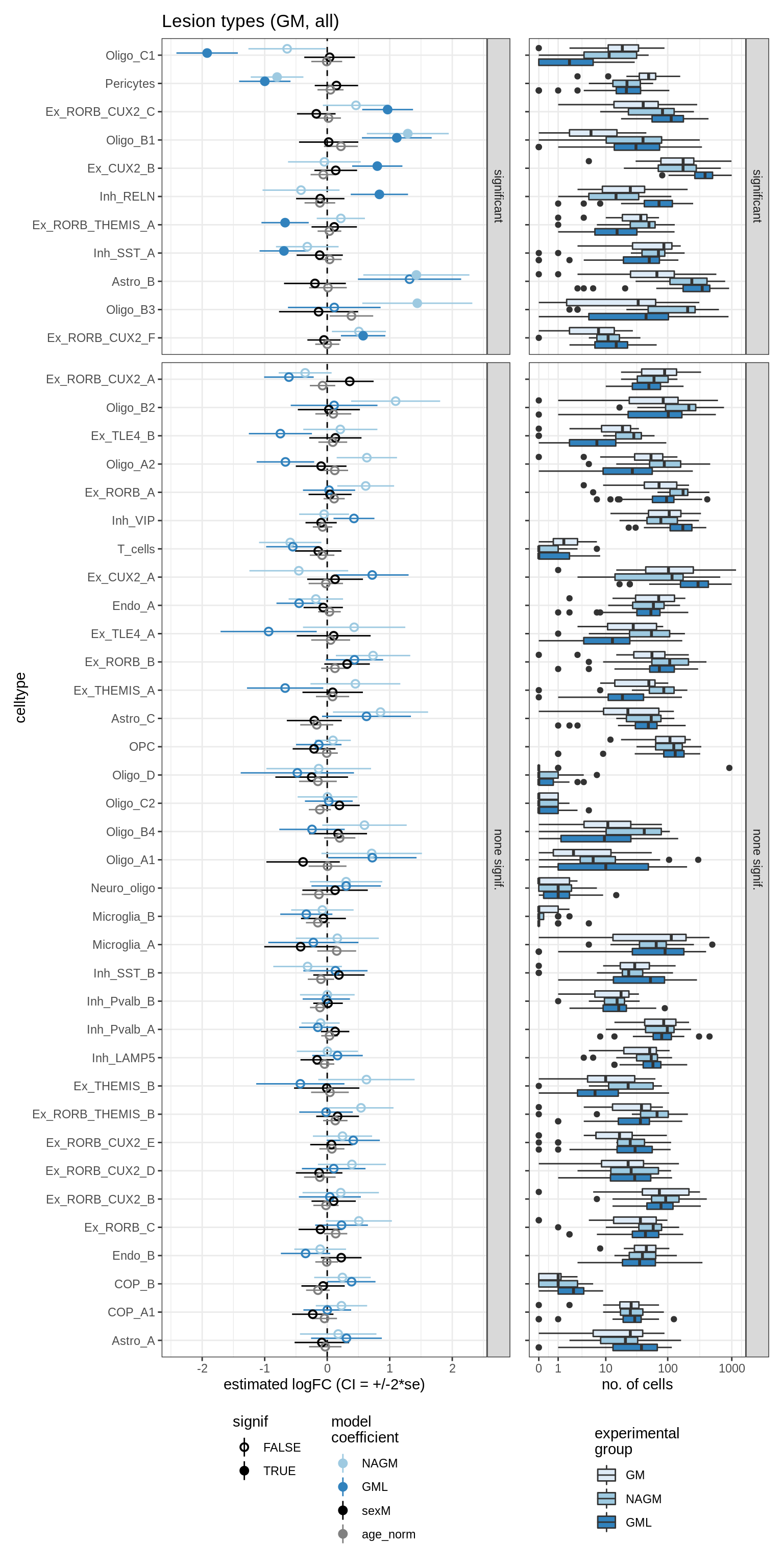
GM_vs_WM
lesions_WM_oligos
lesions_GM_oligos
GM_vs_WM_oligos
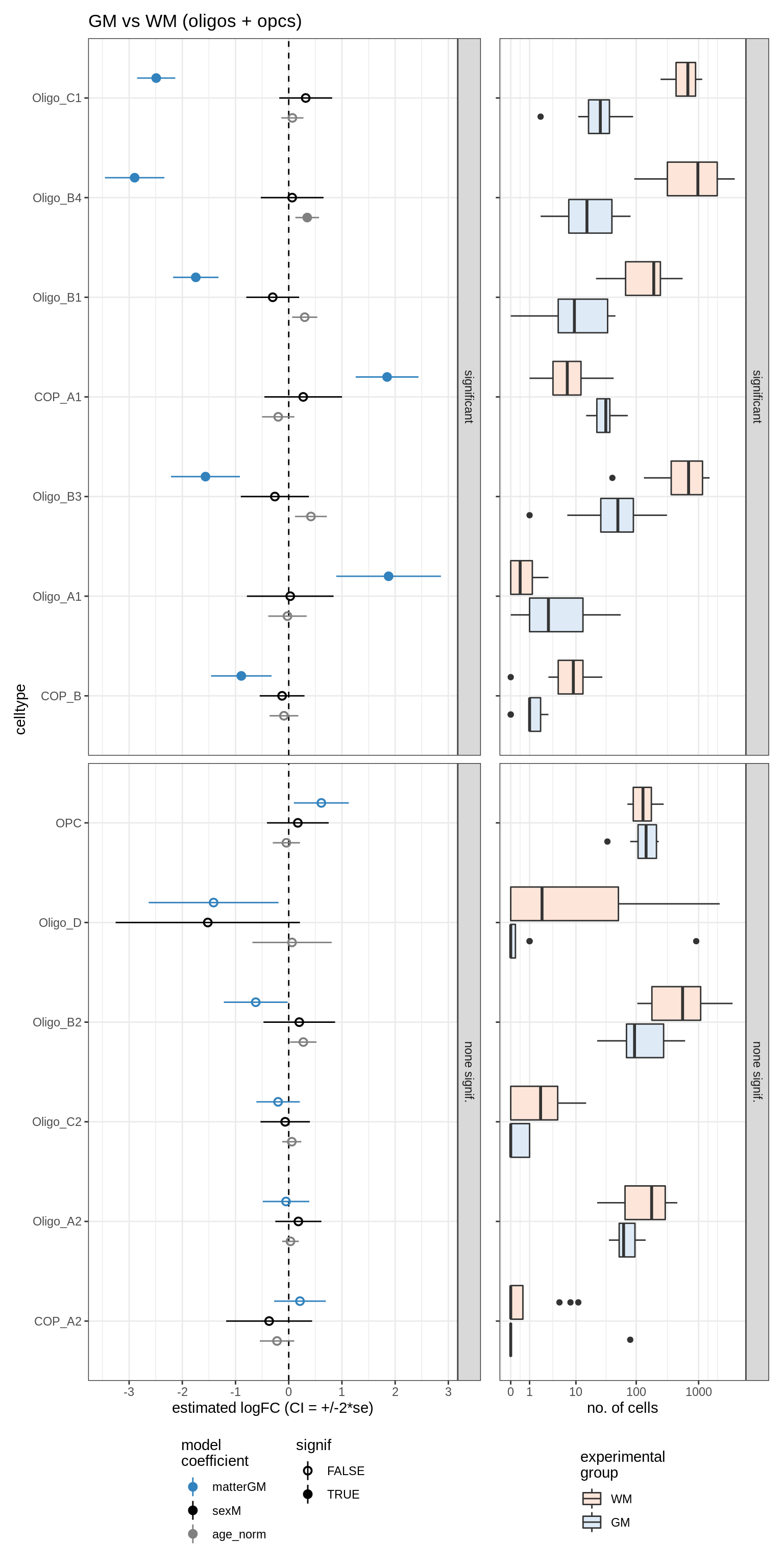
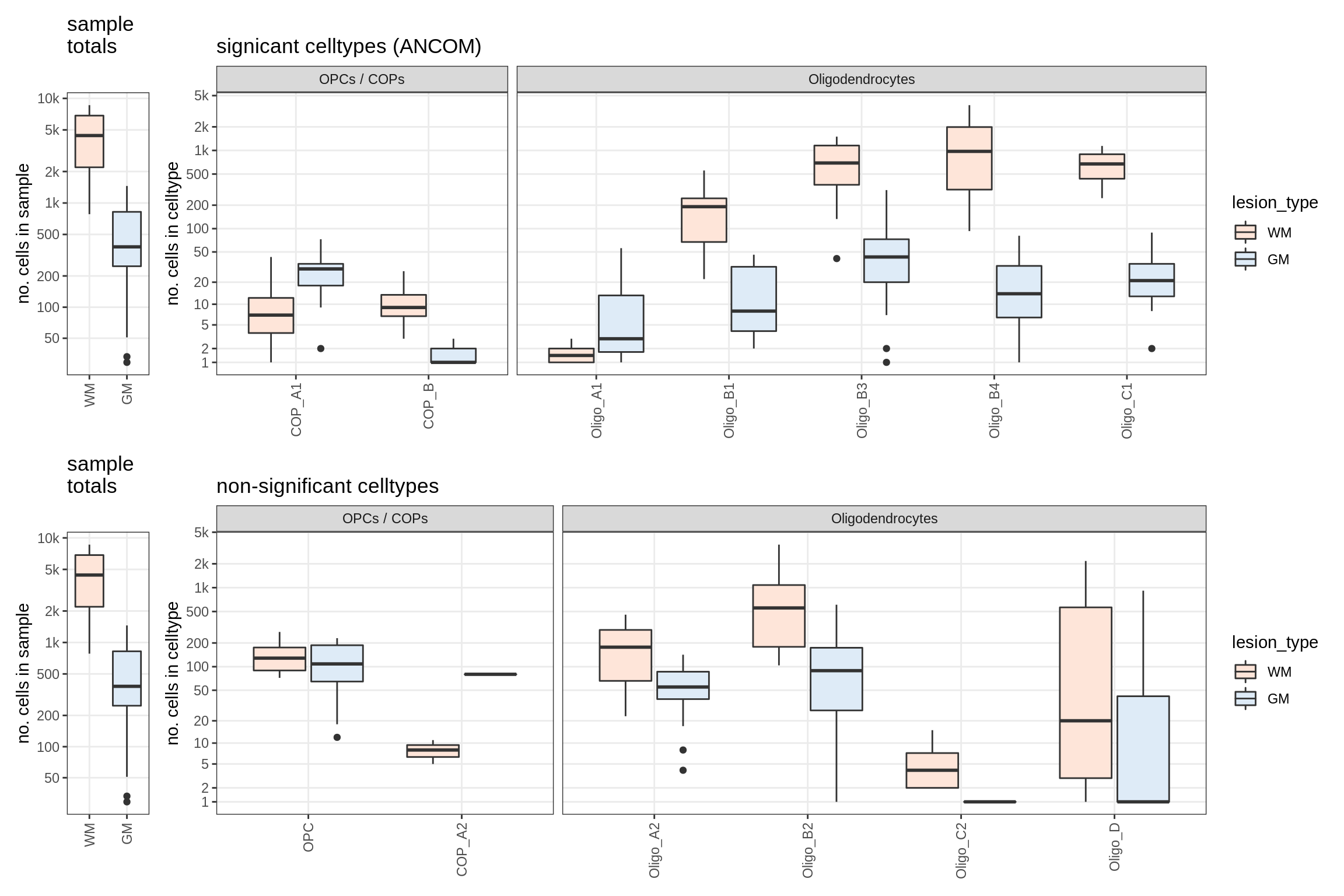
Raw counts across conditions
for (nn in model_names) {
# extract this model
cat('### ', nn, '\n')
print(plot_raw_counts(conos_dt, subset_list[[nn]]$type_spec,
subset_list[[nn]]$subset_spec, subset_list[[nn]]$size_spec,
what = 'boxplot', ancom_list[[nn]]))
cat('\n\n')
}lesions_WM_mad
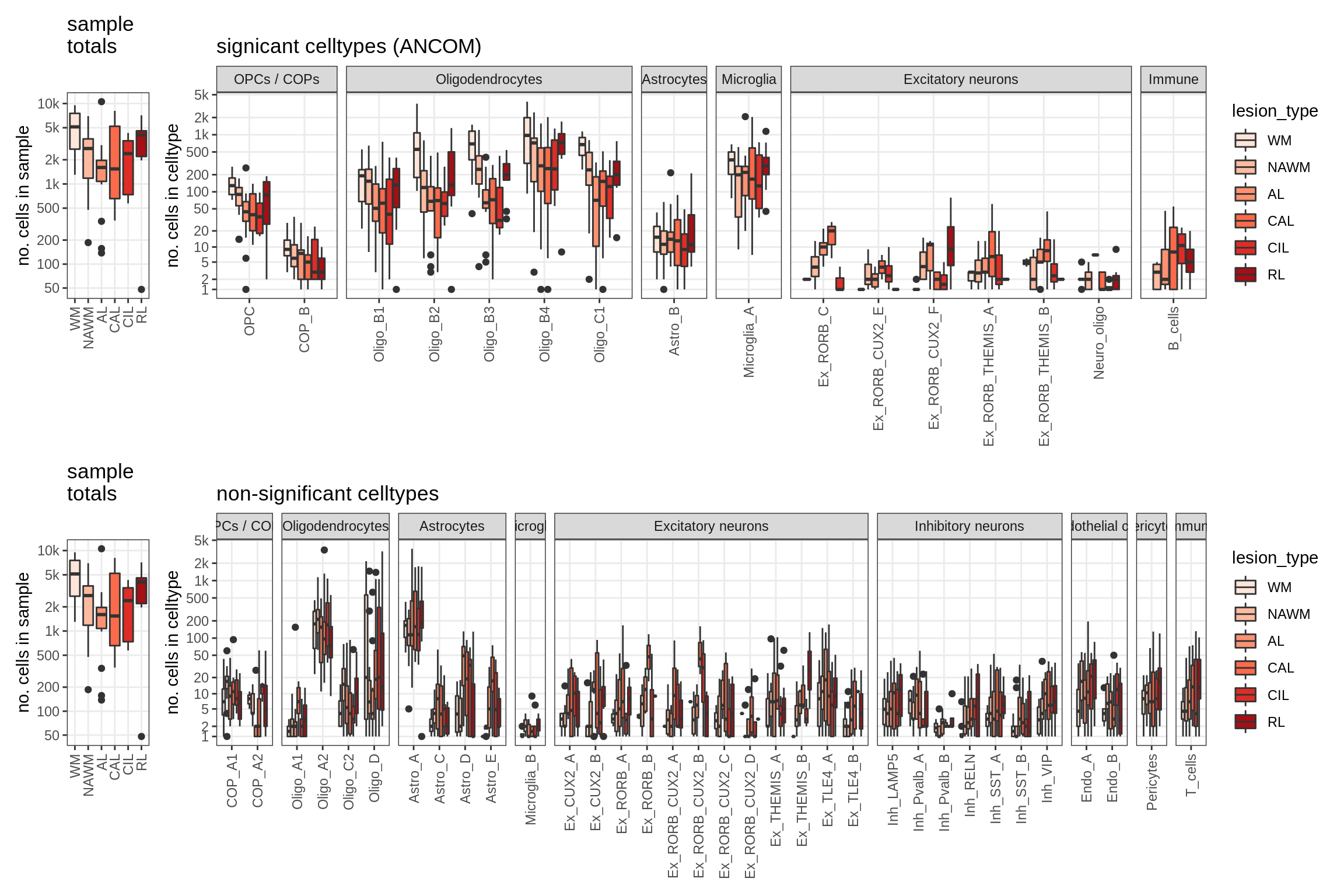
lesions_WM_big_mad
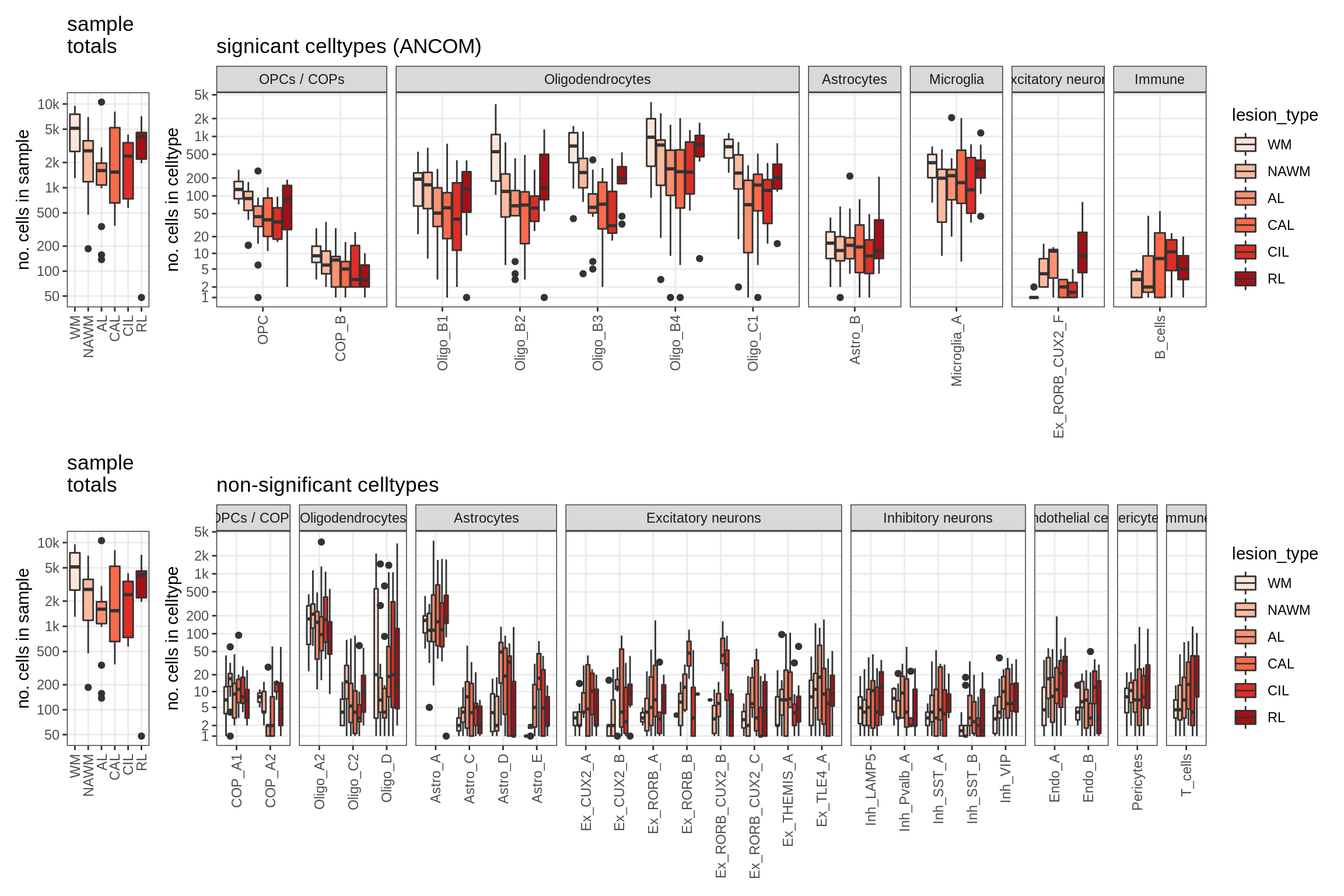
lesions_GM_mad
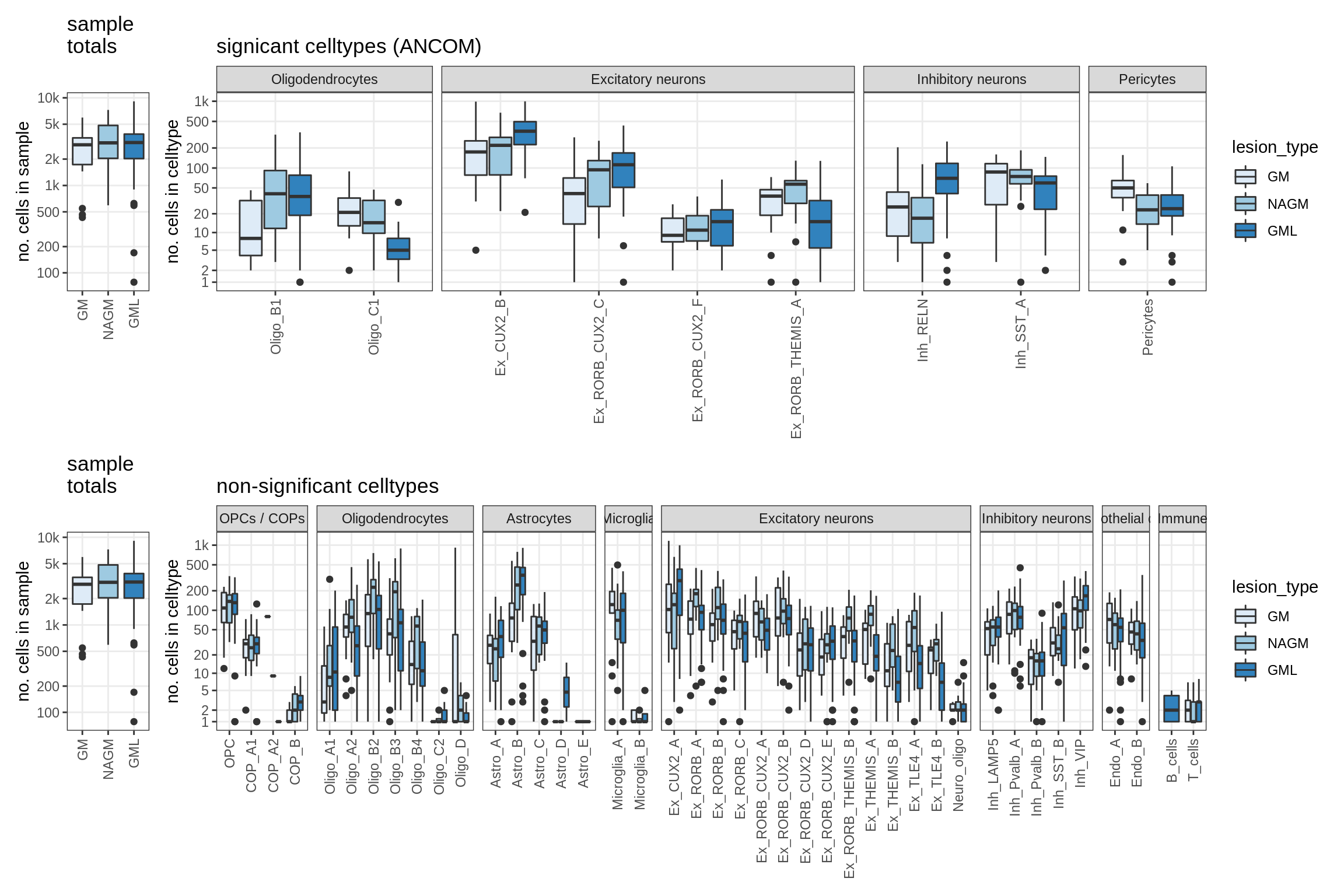
lesions_GM_big_mad
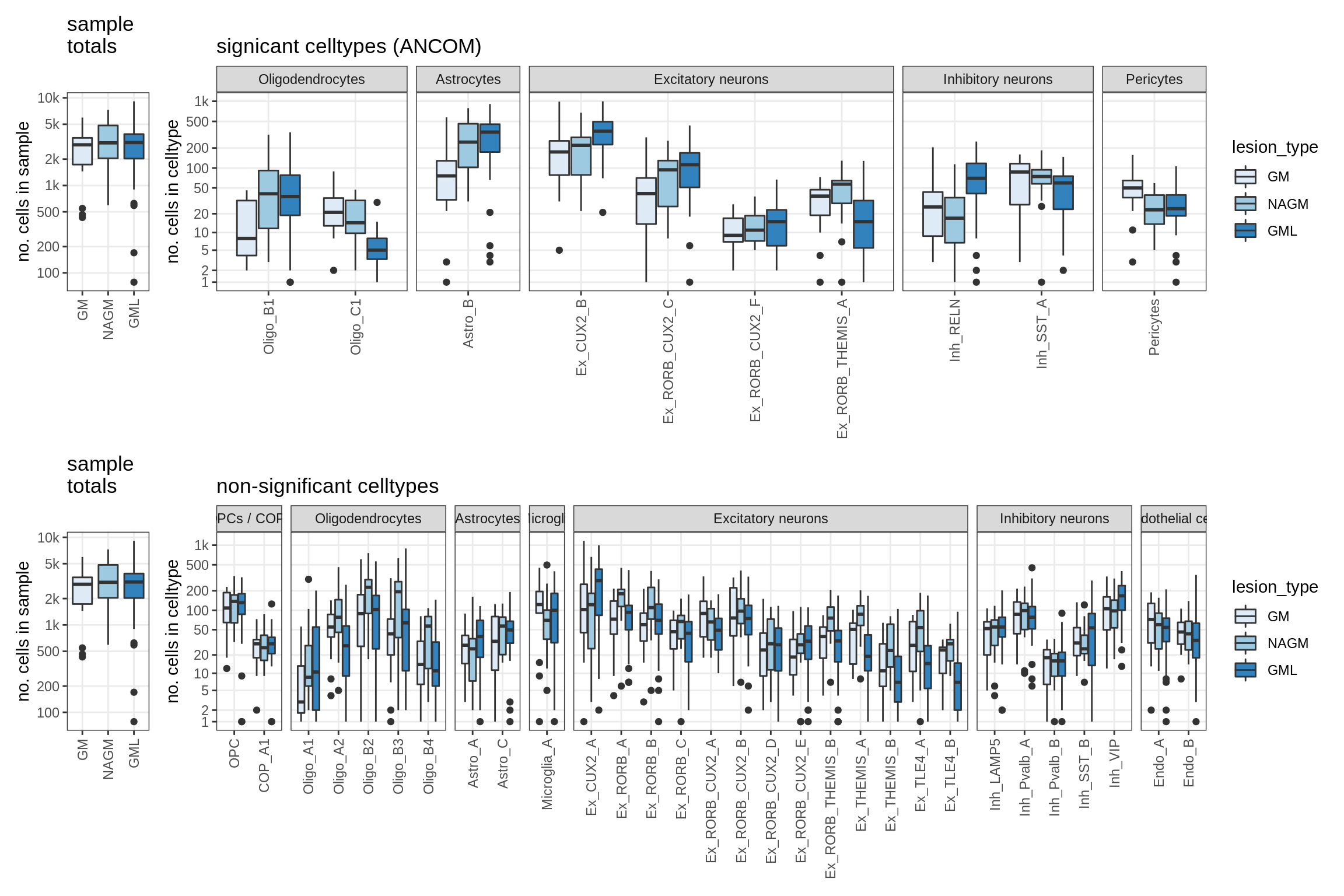
lesions_WM_no_neuro
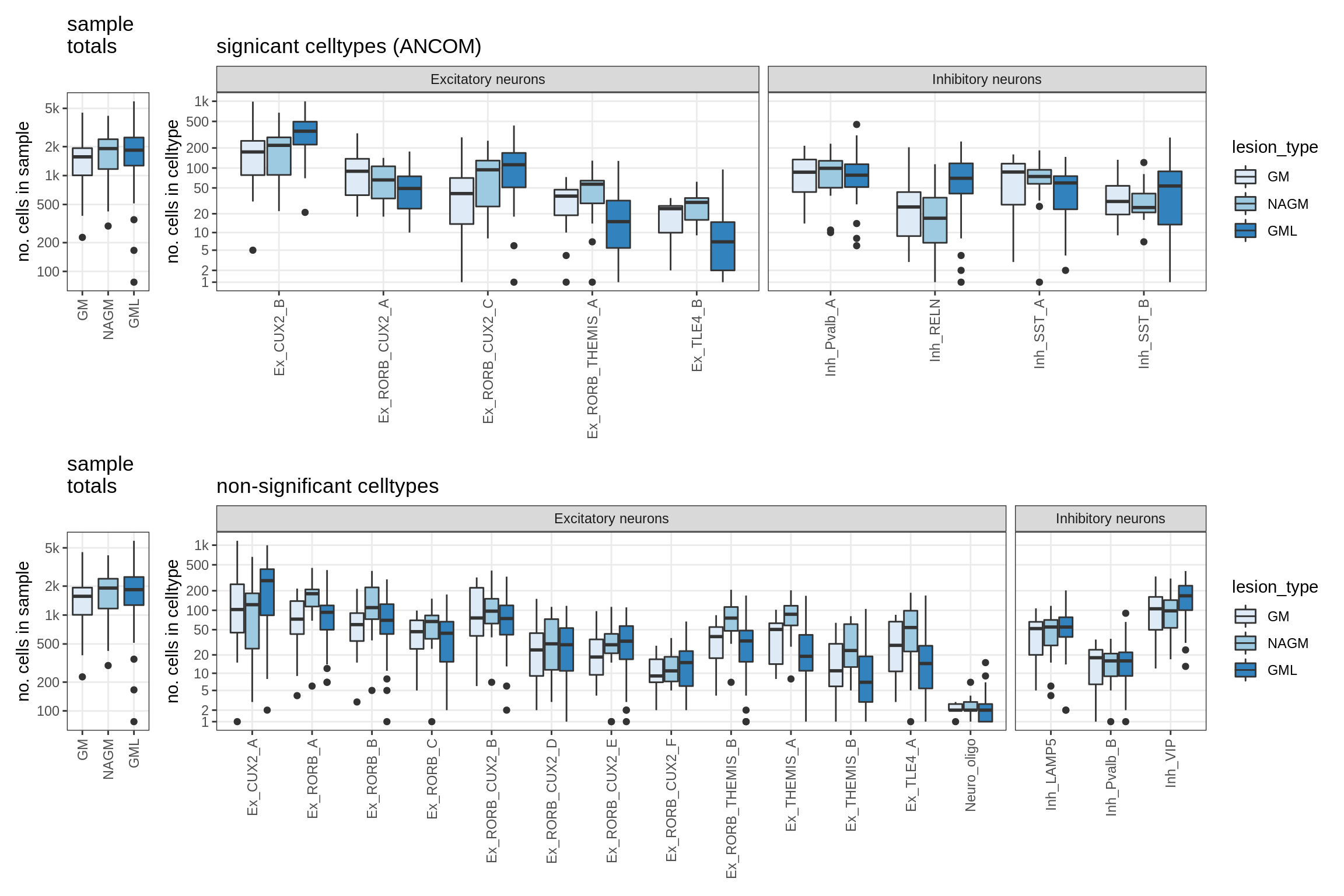
lesions_GM_neuro
lesions_WM_all
lesions_GM_all
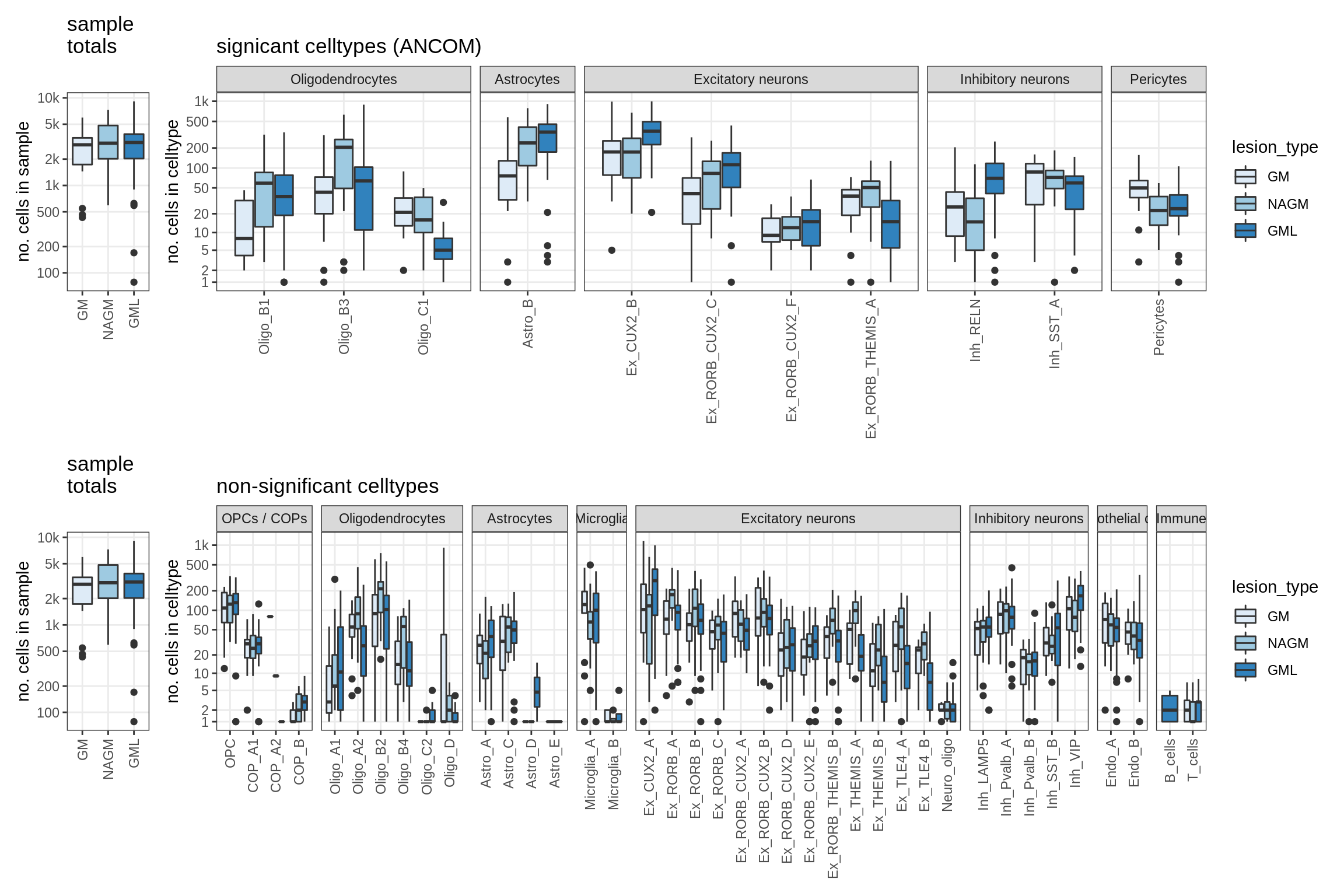
GM_vs_WM
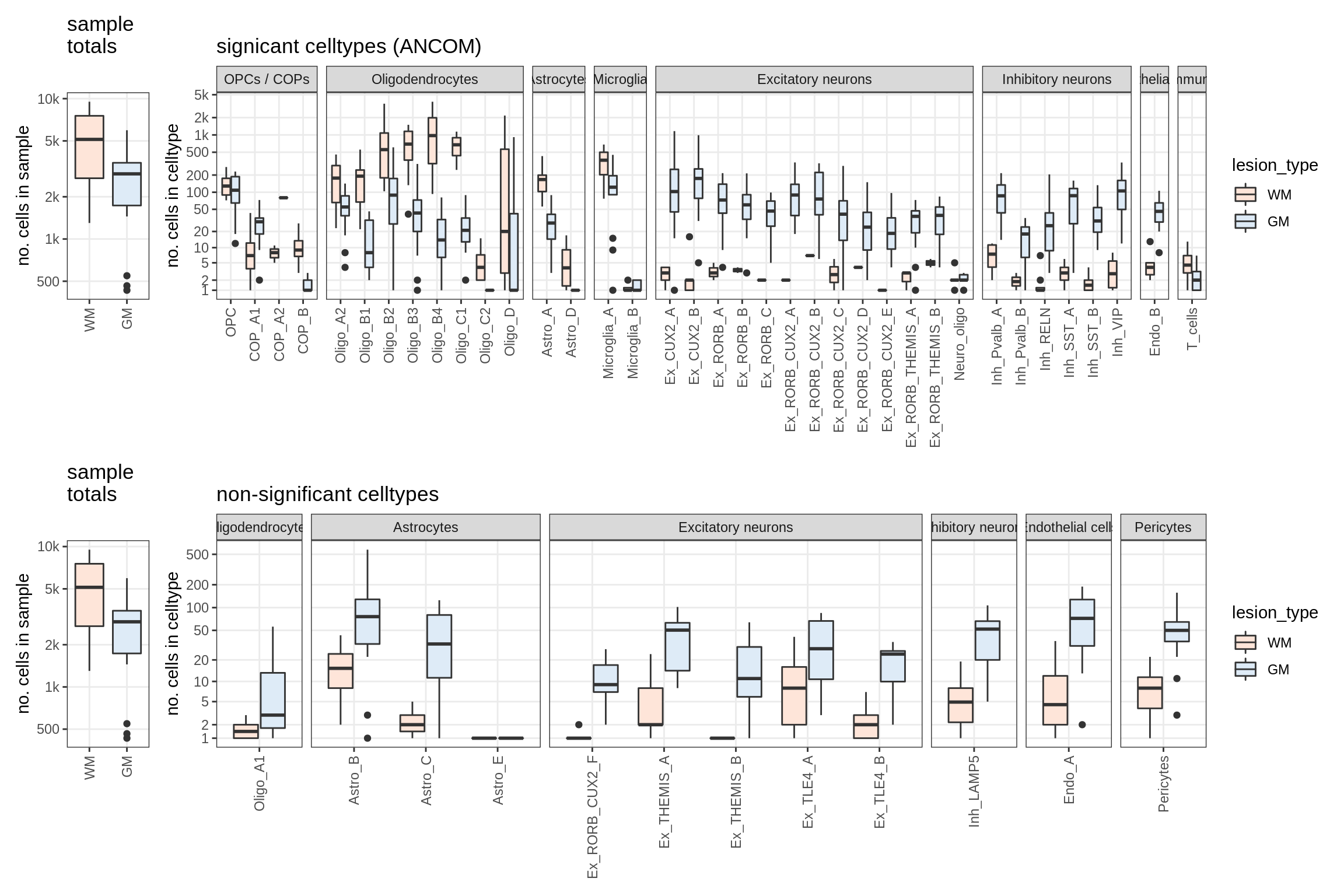
lesions_WM_oligos
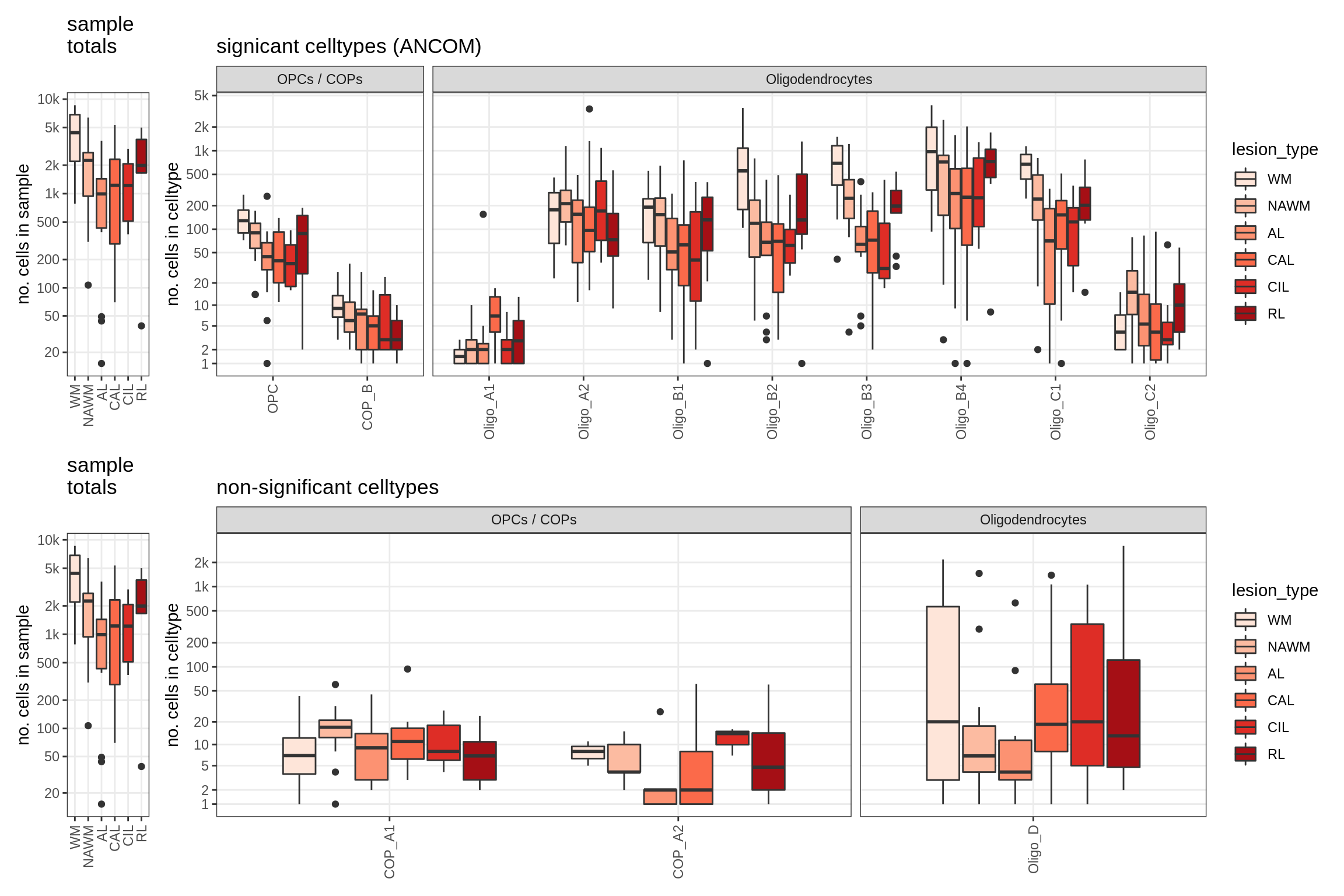
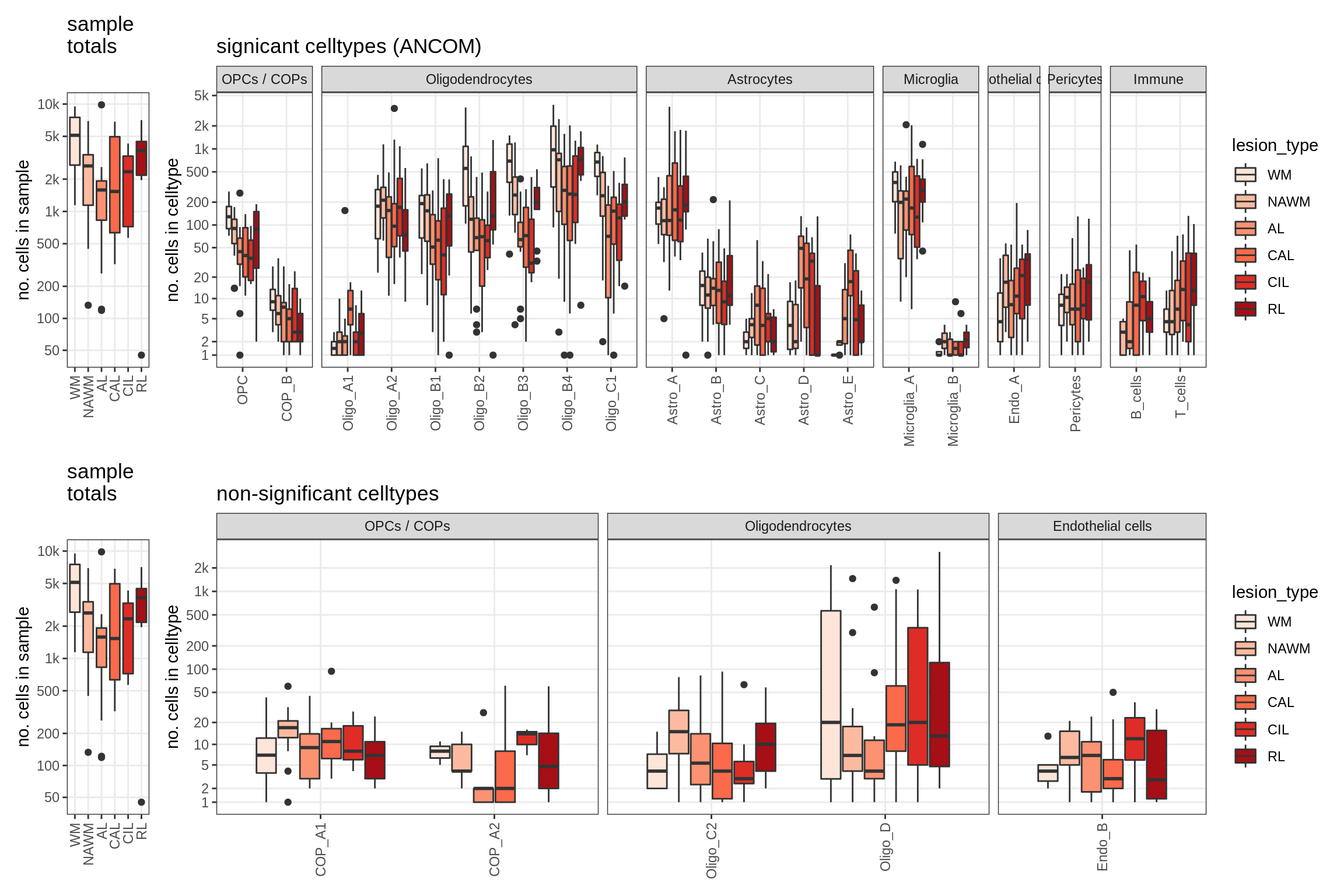
lesions_GM_oligos
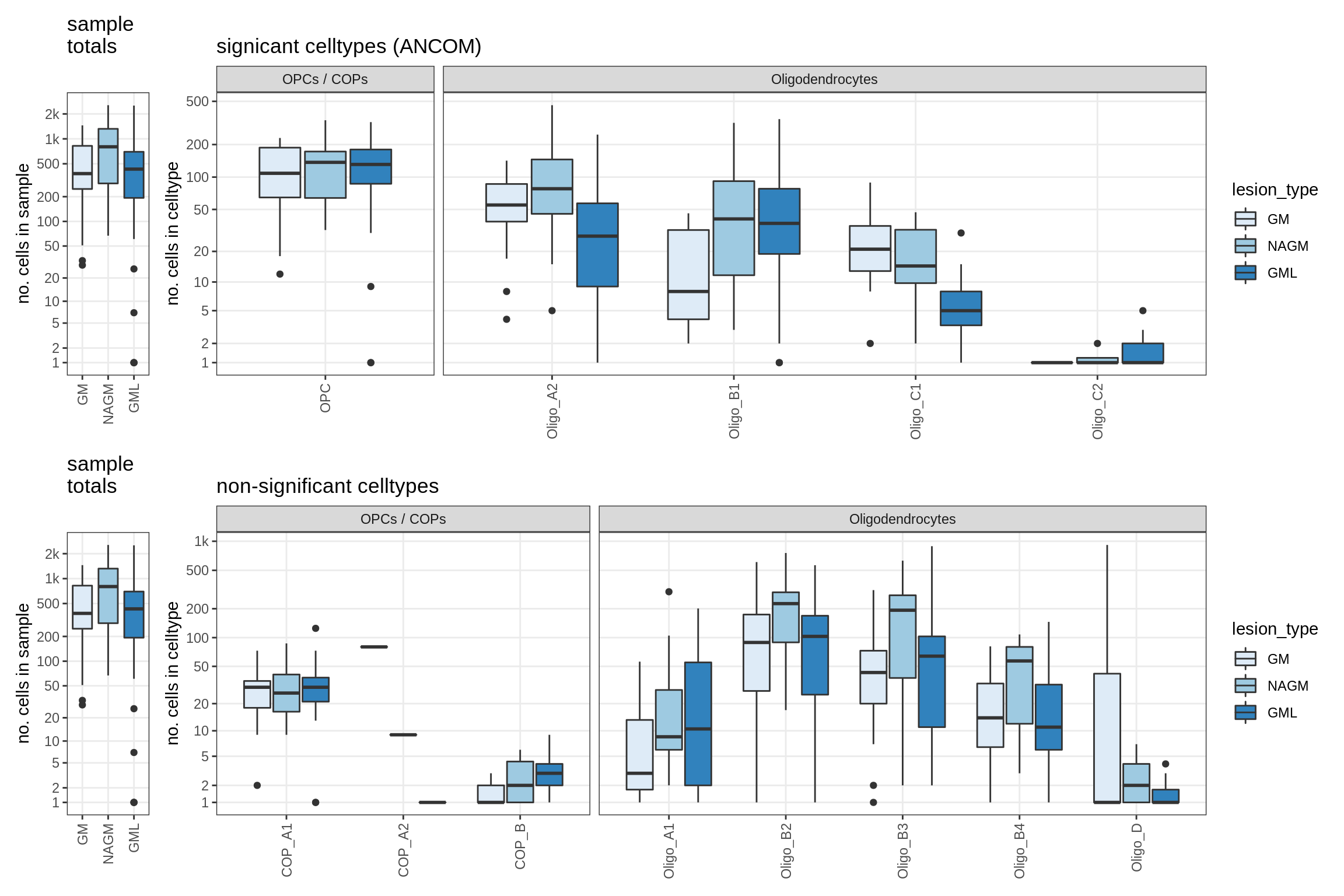
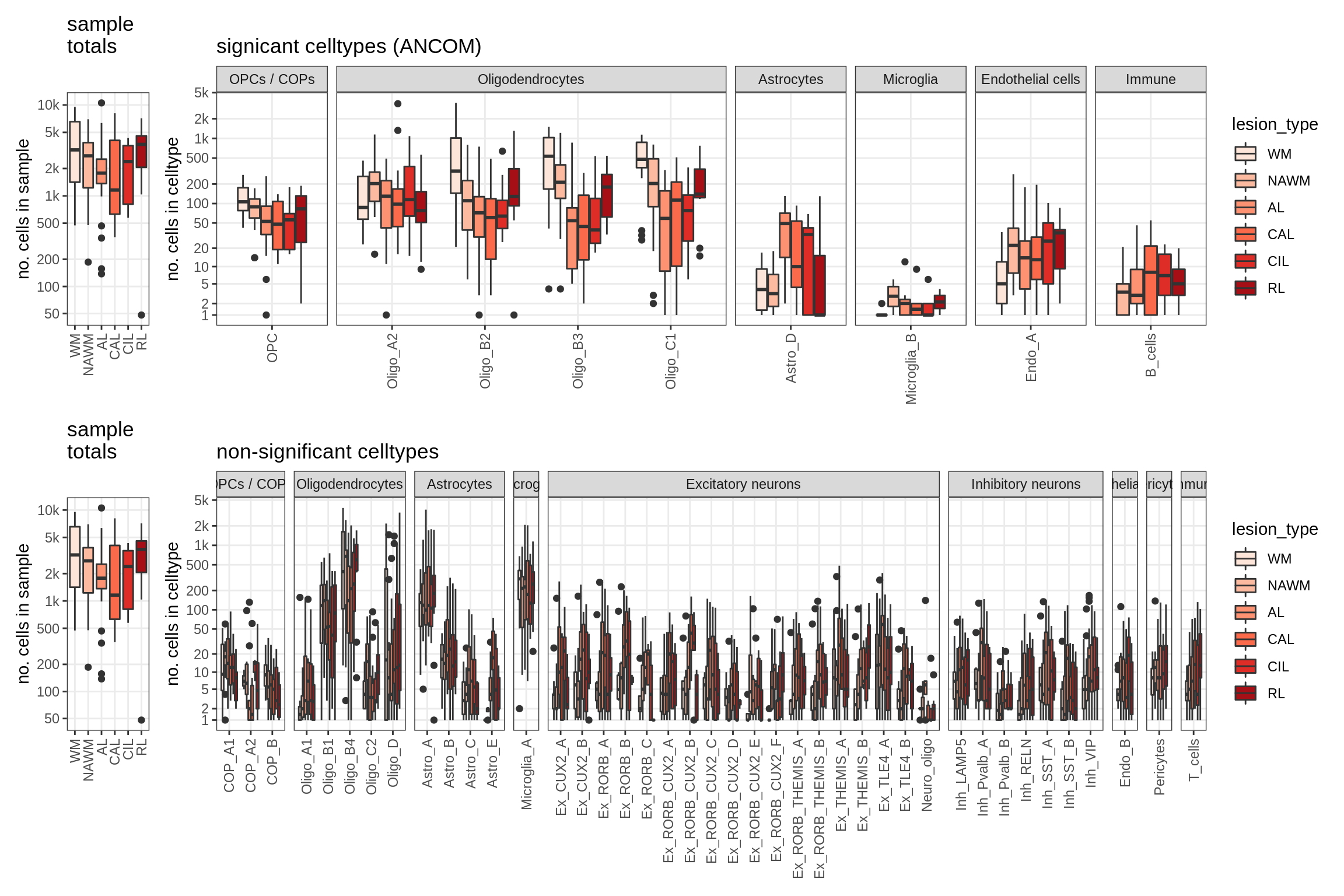
GM_vs_WM_oligos
Outputs
devtools::session_info()Registered S3 method overwritten by 'cli':
method from
print.boxx spatstat.geom- Session info ---------------------------------------------------------------
setting value
version R version 4.0.3 (2020-10-10)
os CentOS Linux 7 (Core)
system x86_64, linux-gnu
ui X11
language (EN)
collate en_US.UTF-8
ctype C
tz Europe/Zurich
date 2021-06-04
- Packages -------------------------------------------------------------------
package * version date lib
abind 1.4-5 2016-07-21 [2]
ade4 1.7-16 2020-10-28 [1]
ANCOMBC * 1.0.5 2021-03-09 [1]
ape 5.5 2021-04-25 [1]
assertthat * 0.2.1 2019-03-21 [2]
beachmat 2.6.4 2020-12-20 [1]
beeswarm 0.3.1 2021-03-07 [1]
Biobase * 2.50.0 2020-10-27 [1]
BiocGenerics * 0.36.1 2021-04-16 [1]
BiocManager 1.30.15 2021-05-11 [1]
BiocNeighbors 1.8.2 2020-12-07 [1]
BiocParallel * 1.24.1 2020-11-06 [1]
BiocSingular 1.6.0 2020-10-27 [1]
BiocStyle * 2.18.1 2020-11-24 [1]
biomformat 1.18.0 2020-10-27 [1]
Biostrings 2.58.0 2020-10-27 [1]
bitops 1.0-7 2021-04-24 [2]
bluster 1.0.0 2020-10-27 [1]
bslib 0.2.5 2021-05-12 [2]
cachem 1.0.5 2021-05-15 [2]
Cairo 1.5-12.2 2020-07-07 [2]
callr 3.7.0 2021-04-20 [2]
circlize * 0.4.12 2021-01-08 [1]
cli 2.5.0 2021-04-26 [2]
clue 0.3-59 2021-04-16 [1]
cluster 2.1.2 2021-04-17 [2]
codetools 0.2-18 2020-11-04 [2]
colorout * 1.2-2 2021-04-15 [1]
colorspace 2.0-1 2021-05-04 [2]
ComplexHeatmap * 2.6.2 2020-11-12 [1]
conos * 1.4.1 2021-05-15 [1]
cowplot 1.1.1 2020-12-30 [2]
crayon 1.4.1 2021-02-08 [2]
data.table * 1.14.0 2021-02-21 [2]
DBI 1.1.1 2021-01-15 [2]
DelayedArray 0.16.3 2021-03-24 [1]
DelayedMatrixStats 1.12.3 2021-02-03 [1]
deldir 0.2-10 2021-02-16 [2]
desc 1.3.0 2021-03-05 [2]
devtools 2.4.1 2021-05-05 [1]
digest 0.6.27 2020-10-24 [2]
dplyr 1.0.6 2021-05-05 [2]
dqrng 0.3.0 2021-05-01 [2]
edgeR 3.32.1 2021-01-14 [1]
ellipsis 0.3.2 2021-04-29 [2]
evaluate 0.14 2019-05-28 [2]
fansi 0.4.2 2021-01-15 [2]
farver 2.1.0 2021-02-28 [2]
fastmap 1.1.0 2021-01-25 [2]
fitdistrplus 1.1-3 2020-12-05 [2]
forcats * 0.5.1 2021-01-27 [2]
foreach 1.5.1 2020-10-15 [2]
fs 1.5.0 2020-07-31 [2]
future 1.21.0 2020-12-10 [2]
future.apply 1.7.0 2021-01-04 [2]
generics 0.1.0 2020-10-31 [2]
GenomeInfoDb * 1.26.7 2021-04-08 [1]
GenomeInfoDbData 1.2.4 2021-04-15 [1]
GenomicRanges * 1.42.0 2020-10-27 [1]
GetoptLong 1.0.5 2020-12-15 [1]
ggbeeswarm 0.6.0 2017-08-07 [1]
ggplot.multistats * 1.0.0 2019-10-28 [1]
ggplot2 * 3.3.3 2020-12-30 [2]
ggrepel * 0.9.1 2021-01-15 [2]
ggridges 0.5.3 2021-01-08 [2]
git2r 0.28.0 2021-01-10 [1]
GlobalOptions 0.1.2 2020-06-10 [1]
globals 0.14.0 2020-11-22 [2]
glue 1.4.2 2020-08-27 [2]
goftest 1.2-2 2019-12-02 [2]
gridExtra 2.3 2017-09-09 [2]
grr 0.9.5 2016-08-26 [1]
gtable 0.3.0 2019-03-25 [2]
hexbin 1.28.2 2021-01-08 [2]
highr 0.9 2021-04-16 [2]
hms 1.1.0 2021-05-17 [2]
htmltools 0.5.1.1 2021-01-22 [2]
htmlwidgets 1.5.3 2020-12-10 [2]
httpuv 1.6.1 2021-05-07 [2]
httr 1.4.2 2020-07-20 [2]
ica 1.0-2 2018-05-24 [2]
igraph * 1.2.6 2020-10-06 [2]
IRanges * 2.24.1 2020-12-12 [1]
irlba 2.3.3 2019-02-05 [2]
iterators 1.0.13 2020-10-15 [2]
janitor 2.1.0 2021-01-05 [1]
jquerylib 0.1.4 2021-04-26 [2]
jsonlite 1.7.2 2020-12-09 [2]
KernSmooth 2.23-20 2021-05-03 [2]
knitr 1.33 2021-04-24 [1]
labeling 0.4.2 2020-10-20 [2]
later 1.2.0 2021-04-23 [2]
lattice 0.20-44 2021-05-02 [2]
lazyeval 0.2.2 2019-03-15 [2]
leiden 0.3.7 2021-01-26 [2]
leidenAlg 0.1.1 2021-03-03 [1]
lifecycle 1.0.0 2021-02-15 [2]
limma 3.46.0 2020-10-27 [1]
listenv 0.8.0 2019-12-05 [2]
lmtest 0.9-38 2020-09-09 [2]
locfit 1.5-9.4 2020-03-25 [1]
lubridate 1.7.10 2021-02-26 [2]
magrittr * 2.0.1 2020-11-17 [1]
MASS * 7.3-54 2021-05-03 [2]
Matrix * 1.3-3 2021-05-04 [2]
Matrix.utils 0.9.8 2020-02-26 [1]
MatrixGenerics * 1.2.1 2021-01-30 [1]
matrixStats * 0.58.0 2021-01-29 [2]
memoise 2.0.0 2021-01-26 [1]
mgcv 1.8-35 2021-04-18 [2]
microbiome 1.12.0 2020-10-27 [1]
mime 0.10 2021-02-13 [2]
miniUI 0.1.1.1 2018-05-18 [2]
multtest 2.46.0 2020-10-27 [1]
munsell 0.5.0 2018-06-12 [2]
nlme 3.1-152 2021-02-04 [2]
nloptr 1.2.2.2 2020-07-02 [1]
parallelly 1.25.0 2021-04-30 [2]
patchwork * 1.1.1 2020-12-17 [2]
pbapply 1.4-3 2020-08-18 [2]
permute 0.9-5 2019-03-12 [1]
phyloseq * 1.34.0 2020-10-27 [1]
pillar 1.6.1 2021-05-16 [2]
pkgbuild 1.2.0 2020-12-15 [1]
pkgconfig 2.0.3 2019-09-22 [2]
pkgload 1.2.1 2021-04-06 [2]
plotly 4.9.3 2021-01-10 [2]
plyr 1.8.6 2020-03-03 [2]
png 0.1-7 2013-12-03 [2]
polyclip 1.10-0 2019-03-14 [2]
prettyunits 1.1.1 2020-01-24 [2]
processx 3.5.2 2021-04-30 [2]
progress 1.2.2 2019-05-16 [2]
promises 1.2.0.1 2021-02-11 [2]
ps 1.6.0 2021-02-28 [2]
purrr * 0.3.4 2020-04-17 [2]
R.methodsS3 1.8.1 2020-08-26 [1]
R.oo 1.24.0 2020-08-26 [1]
R.utils 2.10.1 2020-08-26 [1]
R6 2.5.0 2020-10-28 [2]
RANN 2.6.1 2019-01-08 [2]
rappdirs 0.3.3 2021-01-31 [2]
rbibutils 2.1.1 2021-04-28 [1]
RColorBrewer * 1.1-2 2014-12-07 [2]
Rcpp 1.0.6 2021-01-15 [2]
RcppAnnoy 0.0.18 2020-12-15 [2]
RCurl 1.98-1.3 2021-03-16 [1]
Rdpack 2.1.1 2021-02-23 [1]
registry 0.5-1 2019-03-05 [1]
remotes 2.3.0 2021-04-01 [1]
reshape2 1.4.4 2020-04-09 [2]
reticulate * 1.20 2021-05-03 [2]
rhdf5 2.34.0 2020-10-27 [1]
rhdf5filters 1.2.1 2021-05-03 [1]
Rhdf5lib 1.12.1 2021-01-26 [1]
rjson 0.2.20 2018-06-08 [1]
rlang 0.4.11 2021-04-30 [2]
rmarkdown 2.8 2021-05-07 [2]
ROCR 1.0-11 2020-05-02 [2]
rpart 4.1-15 2019-04-12 [2]
rprojroot 2.0.2 2020-11-15 [2]
rsvd 1.0.5 2021-04-16 [1]
Rtsne 0.15 2018-11-10 [2]
S4Vectors * 0.28.1 2020-12-09 [1]
sass 0.4.0 2021-05-12 [2]
scales * 1.1.1 2020-05-11 [2]
scater * 1.18.6 2021-02-26 [1]
scattermore 0.7 2020-11-24 [2]
sccore 0.1.3 2021-05-05 [1]
scran * 1.18.7 2021-04-16 [1]
sctransform 0.3.2 2020-12-16 [2]
scuttle 1.0.4 2020-12-17 [1]
seriation * 1.2-9 2020-10-01 [1]
sessioninfo 1.1.1 2018-11-05 [1]
Seurat * 4.0.1 2021-03-18 [2]
SeuratObject * 4.0.1 2021-05-08 [2]
shape 1.4.5 2020-09-13 [2]
shiny 1.6.0 2021-01-25 [2]
SingleCellExperiment * 1.12.0 2020-10-27 [1]
snakecase 0.11.0 2019-05-25 [1]
sparseMatrixStats 1.2.1 2021-02-02 [1]
spatstat.core 2.1-2 2021-04-18 [2]
spatstat.data 2.1-0 2021-03-21 [2]
spatstat.geom 2.1-0 2021-04-15 [2]
spatstat.sparse 2.0-0 2021-03-16 [2]
spatstat.utils 2.1-0 2021-03-15 [2]
statmod 1.4.36 2021-05-10 [1]
stringi 1.6.2 2021-05-17 [2]
stringr * 1.4.0 2019-02-10 [2]
SummarizedExperiment * 1.20.0 2020-10-27 [1]
survival 3.2-11 2021-04-26 [2]
tensor 1.5 2012-05-05 [2]
testthat 3.0.2 2021-02-14 [2]
tibble 3.1.2 2021-05-16 [2]
tidyr 1.1.3 2021-03-03 [2]
tidyselect 1.1.1 2021-04-30 [2]
TSP 1.1-10 2020-04-17 [1]
usethis 2.0.1 2021-02-10 [1]
utf8 1.2.1 2021-03-12 [2]
uwot * 0.1.10 2020-12-15 [2]
vctrs 0.3.8 2021-04-29 [2]
vegan 2.5-7 2020-11-28 [1]
vipor 0.4.5 2017-03-22 [1]
viridis * 0.6.1 2021-05-11 [1]
viridisLite * 0.4.0 2021-04-13 [2]
whisker 0.4 2019-08-28 [1]
withr 2.4.2 2021-04-18 [2]
workflowr * 1.6.2 2020-04-30 [1]
xfun 0.23 2021-05-15 [1]
xtable 1.8-4 2019-04-21 [2]
XVector 0.30.0 2020-10-27 [1]
yaml 2.2.1 2020-02-01 [2]
zlibbioc 1.36.0 2020-10-27 [1]
zoo 1.8-9 2021-03-09 [2]
source
CRAN (R 4.0.0)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.3)
Bioconductor
Bioconductor
CRAN (R 4.0.3)
Bioconductor
Bioconductor
Bioconductor
Bioconductor
Bioconductor
Bioconductor
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.2)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Github (jalvesaq/colorout@79931fd)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Bioconductor
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.2)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Bioconductor
Bioconductor
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.2)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.2)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
Bioconductor
Bioconductor
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.2)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.3)
[1] /pstore/home/macnairw/lib/conda_r3.12
[2] /pstore/home/macnairw/.conda/envs/r_4.0.3/lib/R/library
sessionInfo()R version 4.0.3 (2020-10-10)
Platform: x86_64-conda-linux-gnu (64-bit)
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS/LAPACK: /pstore/home/macnairw/.conda/envs/r_4.0.3/lib/libopenblasp-r0.3.12.so
locale:
[1] LC_CTYPE=C LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats4 grid stats graphics grDevices utils
[8] datasets methods base
other attached packages:
[1] ggrepel_0.9.1 reticulate_1.20
[3] MASS_7.3-54 phyloseq_1.34.0
[5] ANCOMBC_1.0.5 purrr_0.3.4
[7] scran_1.18.7 uwot_0.1.10
[9] scater_1.18.6 SingleCellExperiment_1.12.0
[11] SummarizedExperiment_1.20.0 Biobase_2.50.0
[13] GenomicRanges_1.42.0 GenomeInfoDb_1.26.7
[15] IRanges_2.24.1 S4Vectors_0.28.1
[17] BiocGenerics_0.36.1 MatrixGenerics_1.2.1
[19] matrixStats_0.58.0 BiocParallel_1.24.1
[21] ggplot.multistats_1.0.0 patchwork_1.1.1
[23] seriation_1.2-9 ComplexHeatmap_2.6.2
[25] SeuratObject_4.0.1 Seurat_4.0.1
[27] conos_1.4.1 igraph_1.2.6
[29] Matrix_1.3-3 forcats_0.5.1
[31] ggplot2_3.3.3 scales_1.1.1
[33] viridis_0.6.1 viridisLite_0.4.0
[35] assertthat_0.2.1 stringr_1.4.0
[37] data.table_1.14.0 magrittr_2.0.1
[39] circlize_0.4.12 RColorBrewer_1.1-2
[41] BiocStyle_2.18.1 colorout_1.2-2
[43] workflowr_1.6.2
loaded via a namespace (and not attached):
[1] rappdirs_0.3.3 scattermore_0.7
[3] R.methodsS3_1.8.1 tidyr_1.1.3
[5] knitr_1.33 R.utils_2.10.1
[7] irlba_2.3.3 DelayedArray_0.16.3
[9] rpart_4.1-15 RCurl_1.98-1.3
[11] generics_0.1.0 callr_3.7.0
[13] cowplot_1.1.1 microbiome_1.12.0
[15] usethis_2.0.1 RANN_2.6.1
[17] future_1.21.0 lubridate_1.7.10
[19] spatstat.data_2.1-0 httpuv_1.6.1
[21] xfun_0.23 hms_1.1.0
[23] jquerylib_0.1.4 evaluate_0.14
[25] promises_1.2.0.1 TSP_1.1-10
[27] fansi_0.4.2 progress_1.2.2
[29] DBI_1.1.1 htmlwidgets_1.5.3
[31] spatstat.geom_2.1-0 ellipsis_0.3.2
[33] dplyr_1.0.6 permute_0.9-5
[35] deldir_0.2-10 sparseMatrixStats_1.2.1
[37] vctrs_0.3.8 remotes_2.3.0
[39] Cairo_1.5-12.2 ROCR_1.0-11
[41] abind_1.4-5 cachem_1.0.5
[43] withr_2.4.2 grr_0.9.5
[45] sctransform_0.3.2 vegan_2.5-7
[47] prettyunits_1.1.1 goftest_1.2-2
[49] cluster_2.1.2 ape_5.5
[51] lazyeval_0.2.2 crayon_1.4.1
[53] labeling_0.4.2 edgeR_3.32.1
[55] pkgconfig_2.0.3 pkgload_1.2.1
[57] nlme_3.1-152 vipor_0.4.5
[59] devtools_2.4.1 rlang_0.4.11
[61] globals_0.14.0 lifecycle_1.0.0
[63] miniUI_0.1.1.1 registry_0.5-1
[65] rsvd_1.0.5 rprojroot_2.0.2
[67] polyclip_1.10-0 lmtest_0.9-38
[69] Rhdf5lib_1.12.1 zoo_1.8-9
[71] Matrix.utils_0.9.8 beeswarm_0.3.1
[73] processx_3.5.2 whisker_0.4
[75] ggridges_0.5.3 GlobalOptions_0.1.2
[77] png_0.1-7 rjson_0.2.20
[79] bitops_1.0-7 R.oo_1.24.0
[81] KernSmooth_2.23-20 rhdf5filters_1.2.1
[83] Biostrings_2.58.0 DelayedMatrixStats_1.12.3
[85] shape_1.4.5 parallelly_1.25.0
[87] sccore_0.1.3 beachmat_2.6.4
[89] memoise_2.0.0 plyr_1.8.6
[91] hexbin_1.28.2 ica_1.0-2
[93] zlibbioc_1.36.0 compiler_4.0.3
[95] dqrng_0.3.0 clue_0.3-59
[97] fitdistrplus_1.1-3 cli_2.5.0
[99] snakecase_0.11.0 ade4_1.7-16
[101] XVector_0.30.0 listenv_0.8.0
[103] ps_1.6.0 pbapply_1.4-3
[105] mgcv_1.8-35 tidyselect_1.1.1
[107] stringi_1.6.2 highr_0.9
[109] yaml_2.2.1 BiocSingular_1.6.0
[111] locfit_1.5-9.4 sass_0.4.0
[113] tools_4.0.3 future.apply_1.7.0
[115] rstudioapi_0.13 bluster_1.0.0
[117] foreach_1.5.1 git2r_0.28.0
[119] janitor_2.1.0 gridExtra_2.3
[121] farver_2.1.0 Rtsne_0.15
[123] digest_0.6.27 BiocManager_1.30.15
[125] shiny_1.6.0 Rcpp_1.0.6
[127] scuttle_1.0.4 later_1.2.0
[129] RcppAnnoy_0.0.18 httr_1.4.2
[131] Rdpack_2.1.1 colorspace_2.0-1
[133] fs_1.5.0 tensor_1.5
[135] splines_4.0.3 statmod_1.4.36
[137] spatstat.utils_2.1-0 multtest_2.46.0
[139] sessioninfo_1.1.1 plotly_4.9.3
[141] xtable_1.8-4 jsonlite_1.7.2
[143] nloptr_1.2.2.2 leidenAlg_0.1.1
[145] testthat_3.0.2 R6_2.5.0
[147] pillar_1.6.1 htmltools_0.5.1.1
[149] mime_0.10 glue_1.4.2
[151] fastmap_1.1.0 BiocNeighbors_1.8.2
[153] codetools_0.2-18 pkgbuild_1.2.0
[155] utf8_1.2.1 lattice_0.20-44
[157] bslib_0.2.5 spatstat.sparse_2.0-0
[159] tibble_3.1.2 ggbeeswarm_0.6.0
[161] leiden_0.3.7 survival_3.2-11
[163] limma_3.46.0 rmarkdown_2.8
[165] desc_1.3.0 biomformat_1.18.0
[167] munsell_0.5.0 GetoptLong_1.0.5
[169] rhdf5_2.34.0 GenomeInfoDbData_1.2.4
[171] iterators_1.0.13 reshape2_1.4.4
[173] gtable_0.3.0 rbibutils_2.1.1
[175] spatstat.core_2.1-2