Cell-level QC with SampleQC
Will Macnair
Institute for Molecular Life Sciences, University of Zurich, SwitzerlandSwiss Institute of Bioinformatics (SIB), University of Zurich, SwitzerlandApril 28, 2021
Last updated: 2021-04-28
Checks: 5 2
Knit directory: MS_lesions/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
The global environment had objects present when the code in the R Markdown file was run. These objects can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment. Use wflow_publish or wflow_build to ensure that the code is always run in an empty environment.
The following objects were defined in the global environment when these results were created:
| Name | Class | Size |
|---|---|---|
| q | function | 1008 bytes |
The command set.seed(20210118) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version a6bdc98. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rprofile
Ignored: .Rproj.user/
Ignored: ._.DS_Store
Ignored: ._MS_lesions.sublime-project
Ignored: MS_lesions.sublime-project
Ignored: MS_lesions.sublime-workspace
Ignored: analysis/.__site.yml
Ignored: analysis/ms02_doublet_id_cache/
Ignored: analysis/ms07_soup_cache/
Ignored: analysis/ms08_modules_cache/
Ignored: analysis/ms10_muscat_run01_cache/
Ignored: analysis/ms10_muscat_run02_cache/
Ignored: analysis/ms10_muscat_template_cache/
Ignored: analysis/supp10_muscat_cache/
Ignored: data/
Ignored: output/
Untracked files:
Untracked: analysis/ms10_muscat_run01.Rmd
Untracked: analysis/ms10_muscat_run02.Rmd
Untracked: analysis/ms10_muscat_template.Rmd
Untracked: code/ms10_muscat_fns.R
Untracked: code/ms10_muscat_runs.R
Untracked: code/muscat_plan.txt
Untracked: code/plot_dotplot.R
Untracked: code/supp10_muscat.R
Unstaged changes:
Modified: analysis/index.Rmd
Modified: analysis/ms03_SampleQC.Rmd
Modified: analysis/ms07_soup.Rmd
Modified: analysis/ms08_modules.Rmd
Modified: analysis/supp10_muscat.Rmd
Modified: code/ms00_utils.R
Modified: code/ms03_SampleQC.R
Modified: code/ms07_soup.R
Deleted: code/ms10_muscat.R
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/ms03_SampleQC.Rmd) and HTML (docs/ms03_SampleQC.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 3f50452 | wmacnair | 2021-02-10 | Running and checking SampleQC |
| Rmd | c7a8e50 | wmacnair | 2021-01-19 | Added SampleQC splicing analysis |
Setup / definitions
Libraries
Helper functions
source('code/ms00_utils.R')
source('code/ms03_SampleQC.R')Inputs
loom_dir = 'data/loom_files'
sce_f = 'data/sce_raw/ms_sce.rds'
dbl_f = 'output/ms02_doublet_id/scDblFinder_outputs.txt'
meta_f = 'data/metadata/metadata_updated_20201127.txt'Outputs
save_dir = 'output/ms03_SampleQC'
date_tag = '2021-02-10'
if (!dir.exists(save_dir))
dir.create(save_dir)
qc_f = file.path(save_dir, 'ms_qc_dt.txt')
cols_f = file.path(save_dir, 'cols_dt.txt')
# hard threshold for min counts
min_feats = 300
min_counts = 500
min_cells = 500
# specifications for SampleQC
# qc_names = c('log_counts', 'log_feats', 'logit_mito', 'splice_ratio')
qc_names = c('log_counts', 'logit_mito', 'splice_ratio')
annots_disc = c('patient_id', 'lesion_type', 'seq_pool', 'source', 'disease_status', 'sex')
annots_cont = c('age', 'post_mortem_m', 'med_splice')
# files to save
sampleqc_f = sprintf('%s/sampleQC_obj_%s.rds', save_dir, date_tag)
proj_name = sprintf('MS_lesions_%s', date_tag)
report_f = sprintf('%s/SampleQC_report_%s.html', save_dir, proj_name)
outliers_f = sprintf('%s/outliers_dt_%s.txt', save_dir, date_tag)
keep_f = sprintf('%s/keep_dt_%s.txt', save_dir, date_tag)
# one sample is a massive outlier
outlier_s = 'EU042'
# we want to exclude cells with high splice ratios
splice_cut = 3Load inputs
qc_dt = make_qc_dt_file(sce_f, qc_f) %>%
.[, .(cell_id,
sample_id = str_match(cell_id, '^((EU|WM)[0-9]{3})')[,2],
log_counts = log10(all_counts),
log_feats = log10(all_feats),
logit_mito = qlogis((mito_counts+1) / (all_counts + 2))
)] %>%
setkey('cell_id')extract_splice_ratios(loom_dir, save_dir)extracting spliced, unspliced dataspliced_dt = get_spliced_dt(save_dir)
assert_that( all(sort(qc_dt$cell_id) == sort(spliced_dt$cell_id)))[1] TRUEqc_dt = merge(qc_dt, spliced_dt[, -c('matter', 'barcode')], by=c('cell_id', 'sample_id'))dbl_dt = dbl_f %>% fread %>%
.[, .(cell_id, sample_id=sample, dbl_class=class)]
assert_that( all(sort(qc_dt$cell_id) == sort(dbl_dt$cell_id)))[1] TRUEcols_dt = get_cols_dt(sce_f, cols_f) %>%
setnames('library_id', 'sample_id')
patients_dt = cols_dt[, .(sample_id, patient_id)] %>% unique %>%
.[, N:=.N, by=patient_id] %>%
.[ N < 3, patient_id := NA ] %>%
.[, N := NULL]
cols_dt = merge(cols_dt[, -'patient_id'], patients_dt, by='sample_id')
assert_that( all(sort(qc_dt$cell_id) == sort(cols_dt$cell_id)))[1] TRUEqc_dt = qc_dt %>% merge(cols_dt, by=c('cell_id', 'sample_id'))Processing / calculations
keep_ids = dbl_dt[ dbl_class == 'singlet' ]$cell_id
qc_dt = qc_dt[ cell_id %in% keep_ids ]qc_dt = qc_dt[(log_counts >= log10(min_counts)) &
(log_feats >= log10(min_feats))]
n_dt = qc_dt[, .N, by=sample_id]
keep_s = n_dt[N >= min_cells]$sample_id
qc_dt = qc_dt[sample_id %in% keep_s]qc_dt[, med_splice := median(splice_ratio), by=sample_id]qc_dt = make_qc_dt(qc_dt, qc_names=qc_names)# define number of components
K_list = c(4, 4, 3, 2, 1, 3, 4, 4, 4)
if (!file.exists(sampleqc_f)) {
# calculate sample-sample distances
set.seed(20210210)
qc_obj = calculate_sample_to_sample_MMDs(qc_dt, qc_names,
annots_disc = annots_disc, annots_cont = annots_cont,
n_cores = 16)
print(table(colData(qc_obj)$group_id))
# fit models
set.seed(20210210)
qc_obj = fit_sampleQC(qc_obj, K_list = K_list)
# save results
saveRDS(qc_obj, file=sampleqc_f)
} else {
# load results
qc_obj = readRDS(sampleqc_f)
}# extract component means
group_ids = levels(colData(qc_obj)$group_id)
beta_ks = group_ids %>%
lapply(function(g) {
beta_k = metadata(qc_obj)$fit_list[[g]]$beta_k
mu_0 = metadata(qc_obj)$fit_list[[g]]$mu_0
beta_k = sweep(beta_k, 2, mu_0, '+')
assert_that( all(abs(colMeans(beta_k) - mu_0) < 1e-10) )
return(beta_k)
})
# get clusters with positive splice ratio
splice_idx = qc_names == 'splice_ratio'
exc_clusters = lapply(beta_ks,
function(beta_k_mat) which(beta_k_mat[, splice_idx] > splice_cut)
) %>% setNames(group_ids)
exc_clusters = exc_clusters[ sapply(exc_clusters, function(x) length(x) > 0) ]
assert_that(all(sapply(exc_clusters, length) == 1))[1] TRUE# get outliers
outliers_dt = get_outliers(qc_obj, exc_clusters=exc_clusters)
outliers_dt[ sample_id == outlier_s, outlier := TRUE ]
# check no samples very small
n_kept_dt = outliers_dt[outlier == FALSE, .N, by=sample_id]
assert_that( all(n_kept_dt$N > min_cells) )[1] TRUEkeep_dt = outliers_dt[outlier == FALSE, .(sample_id, cell_id)]Analysis
(plot_spliced_distns(spliced_dt))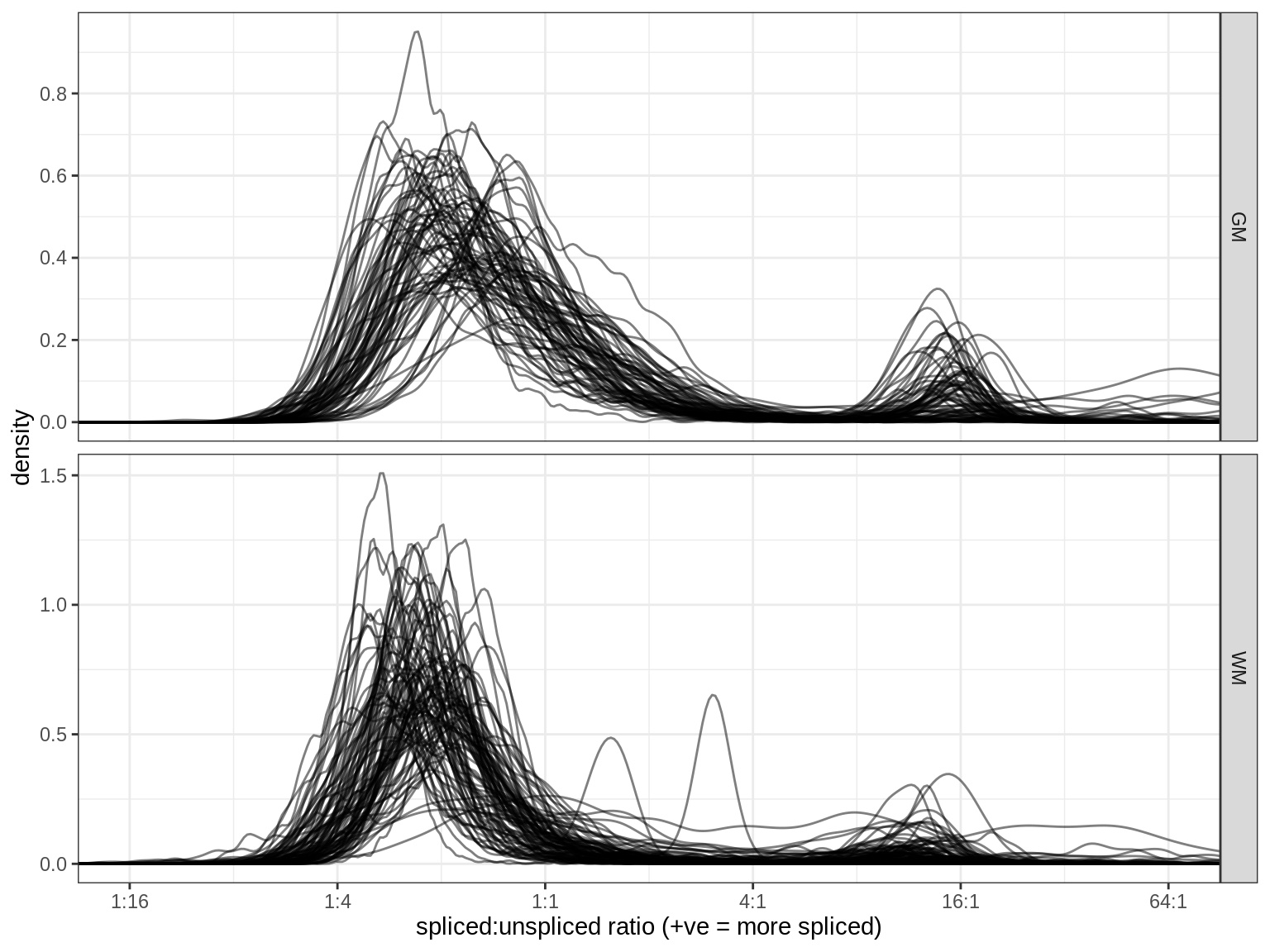
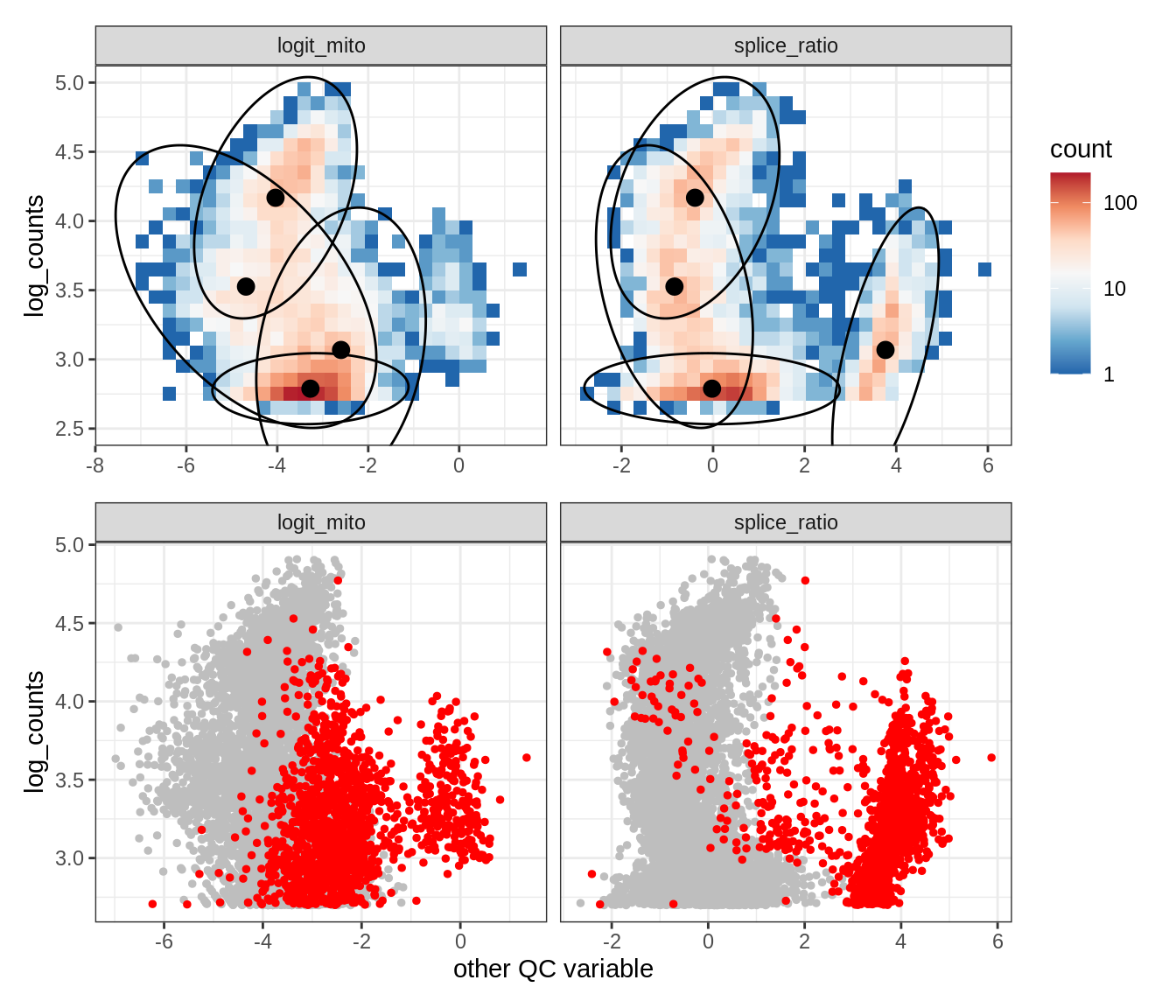
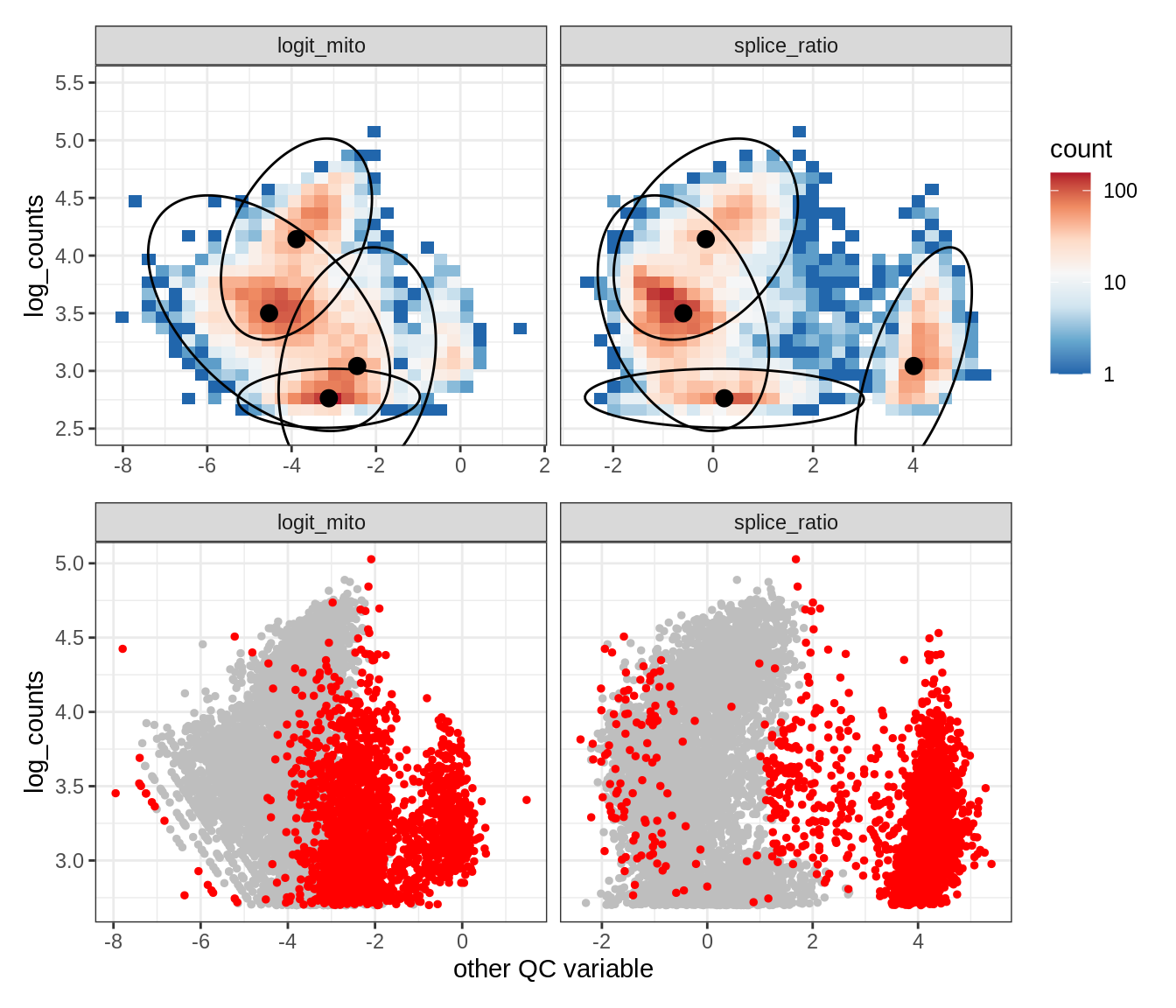
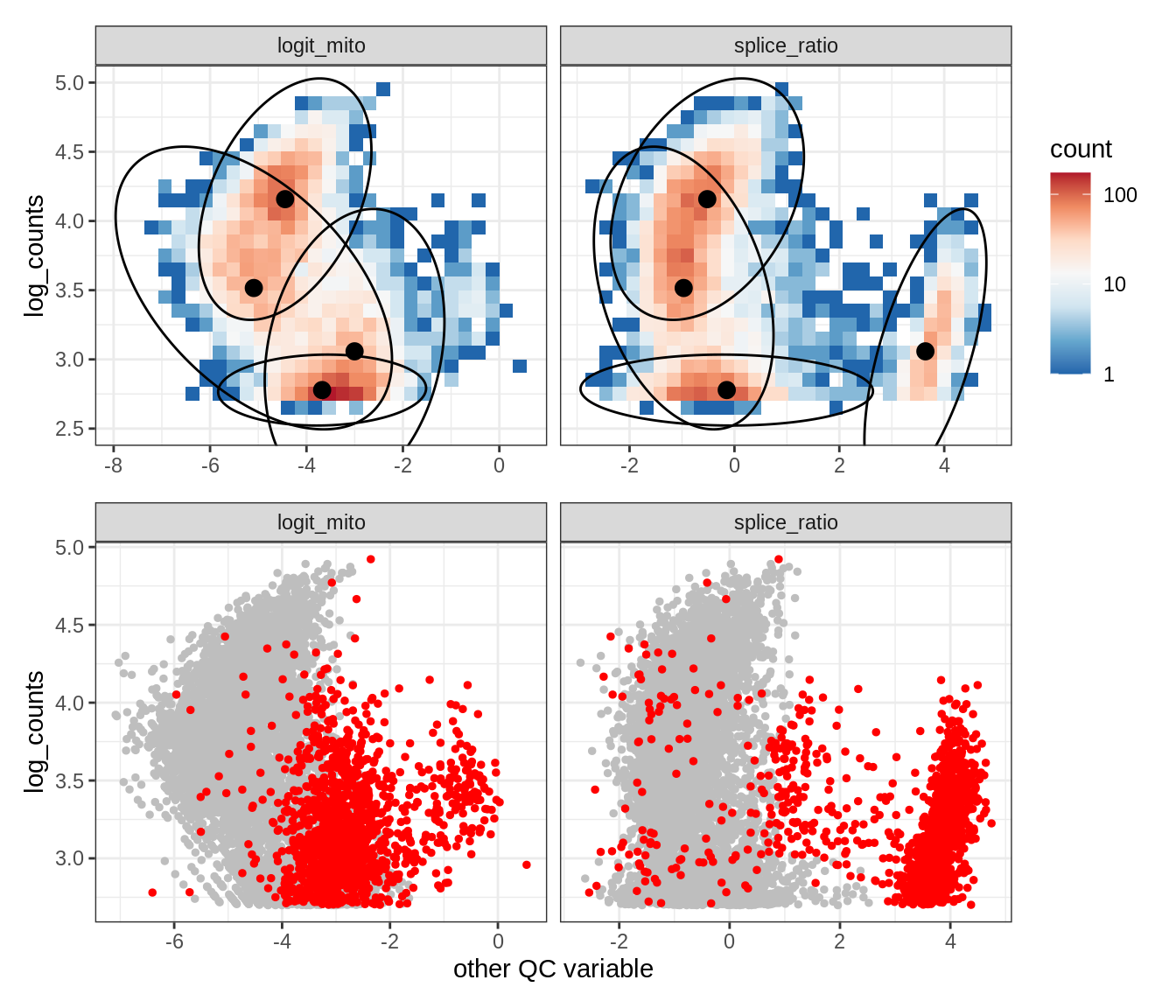
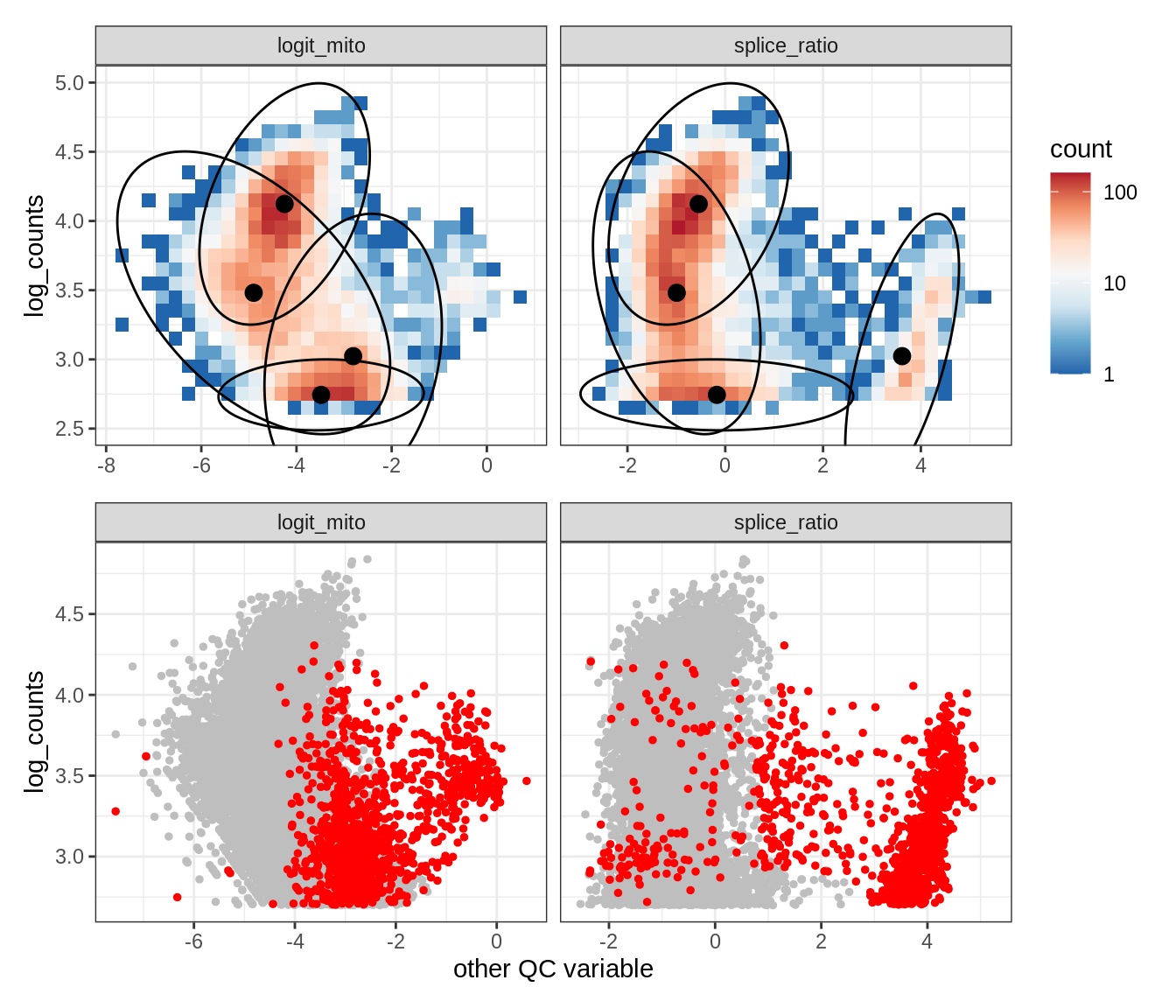
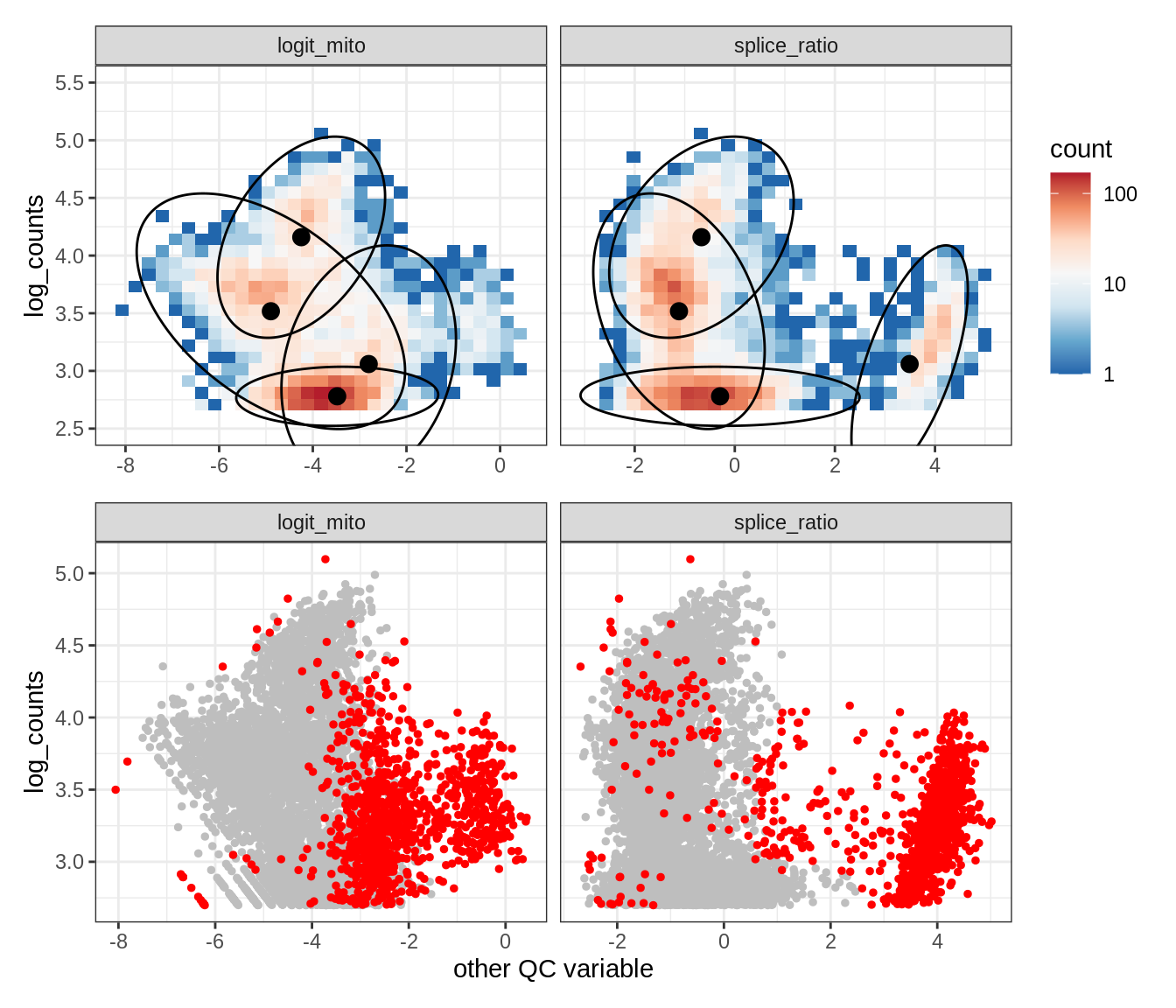
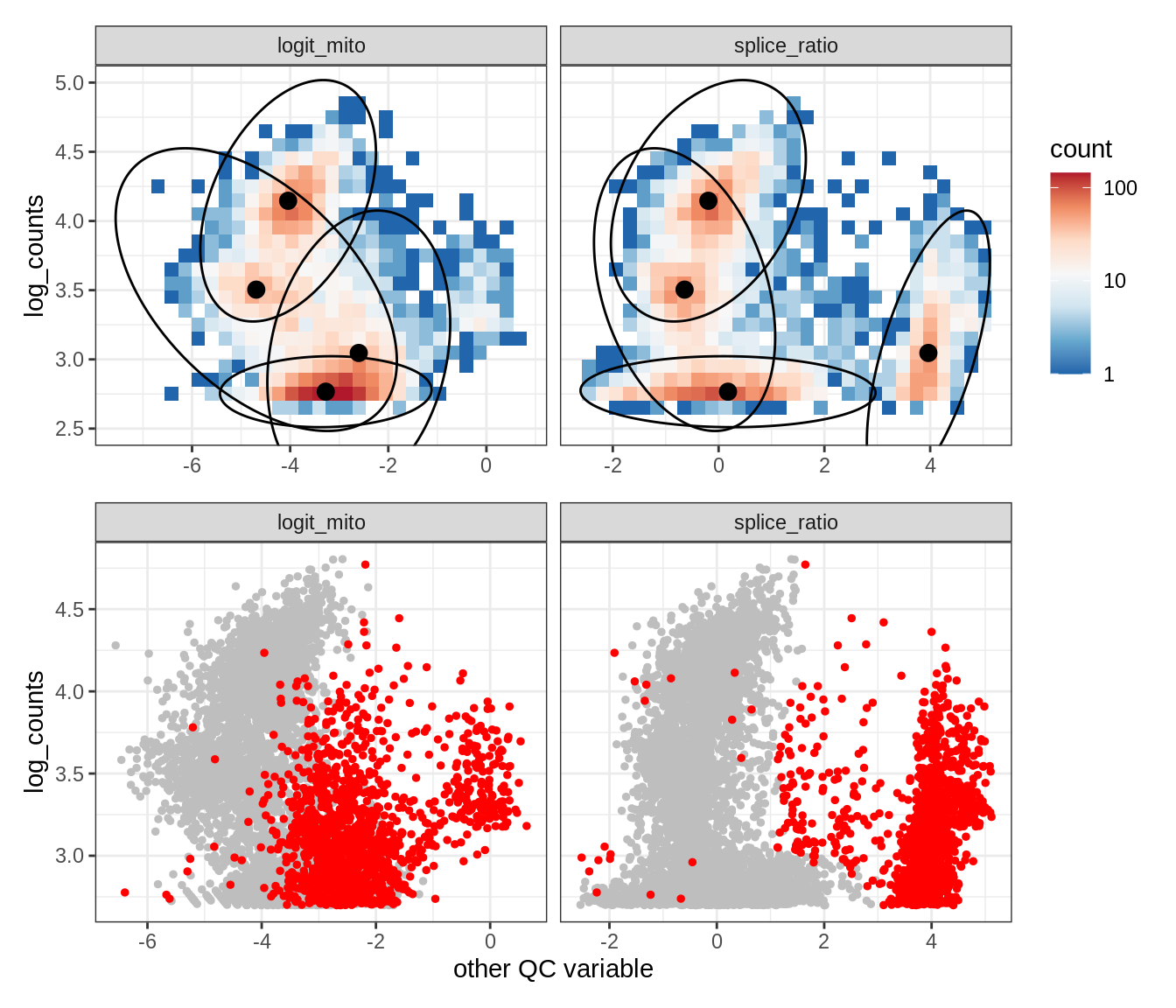
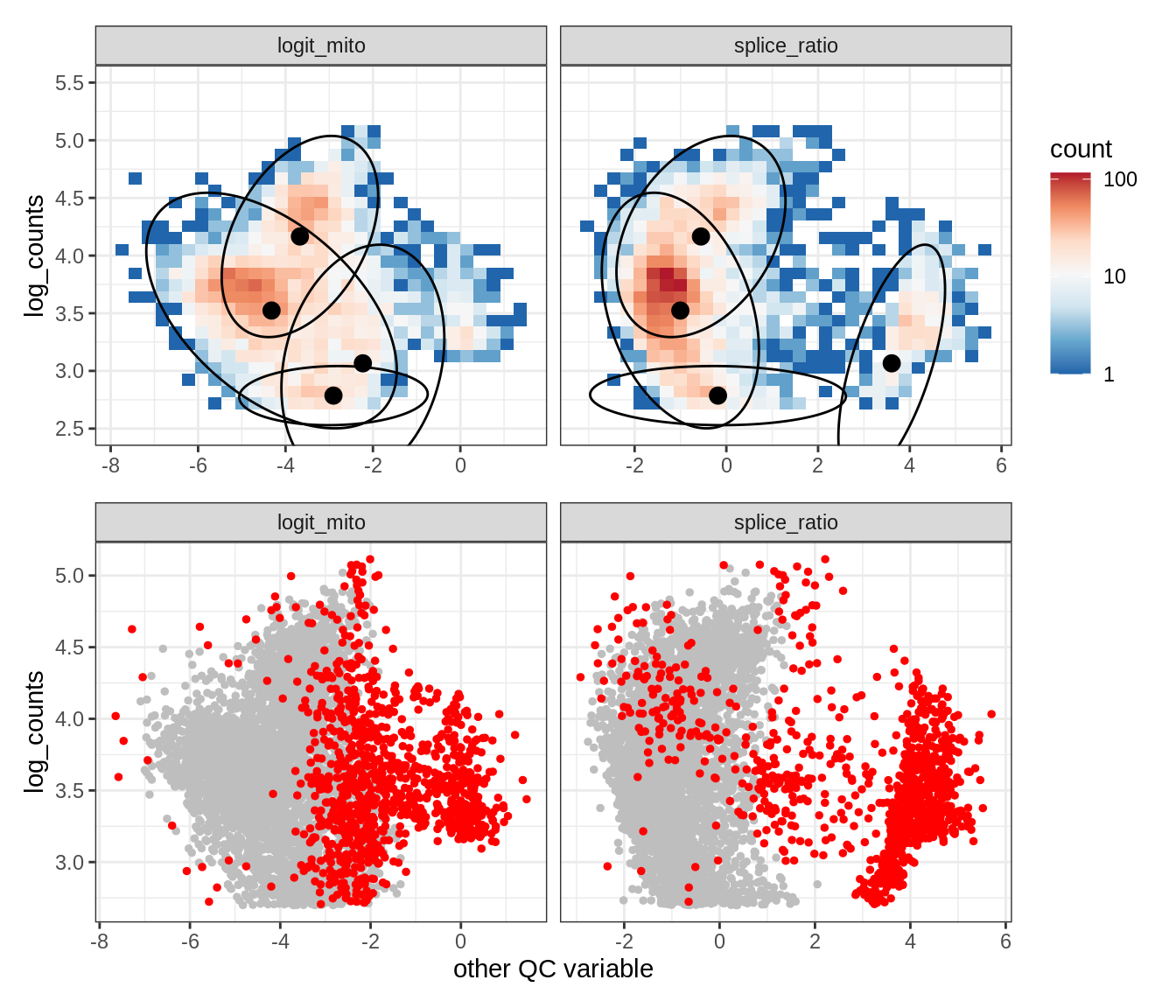
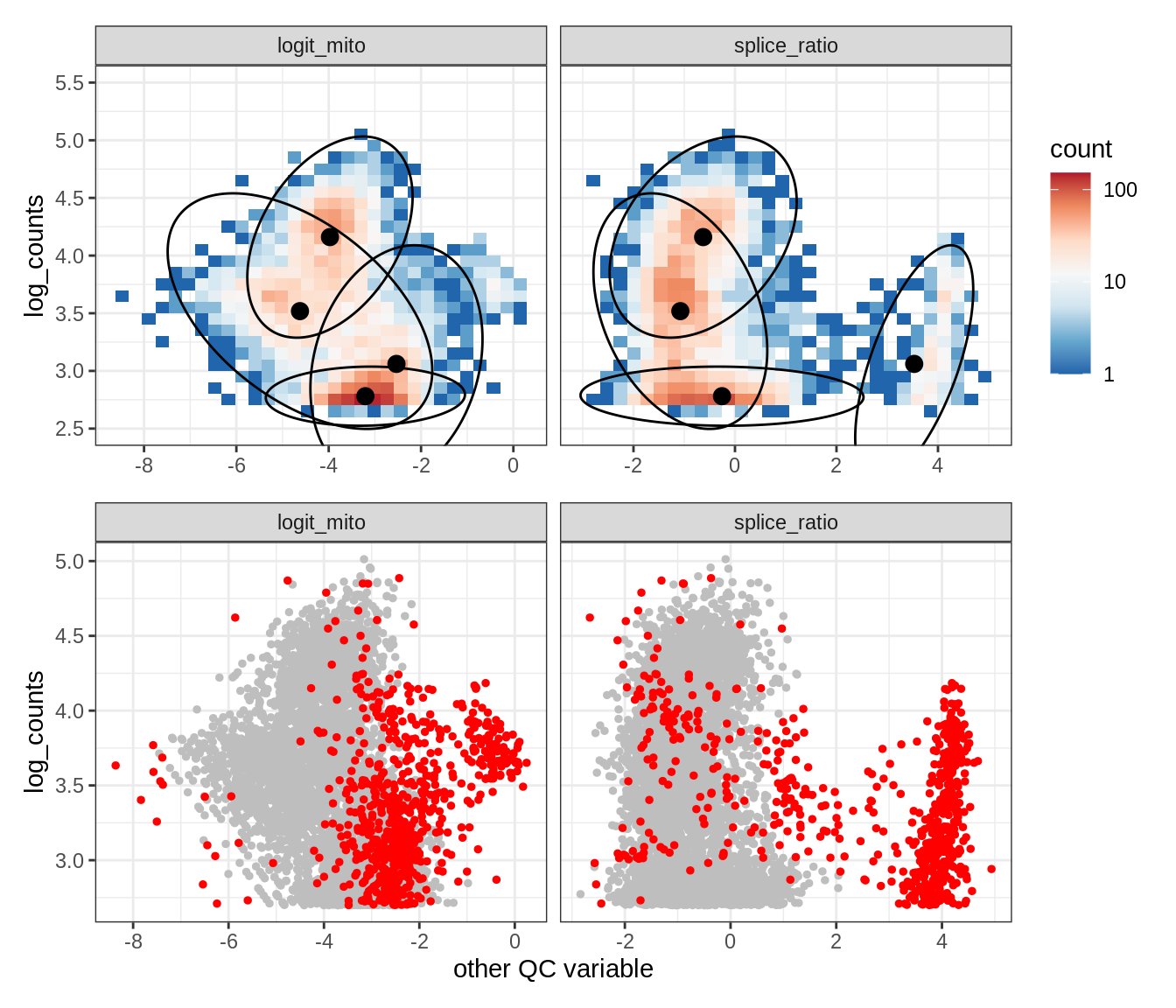
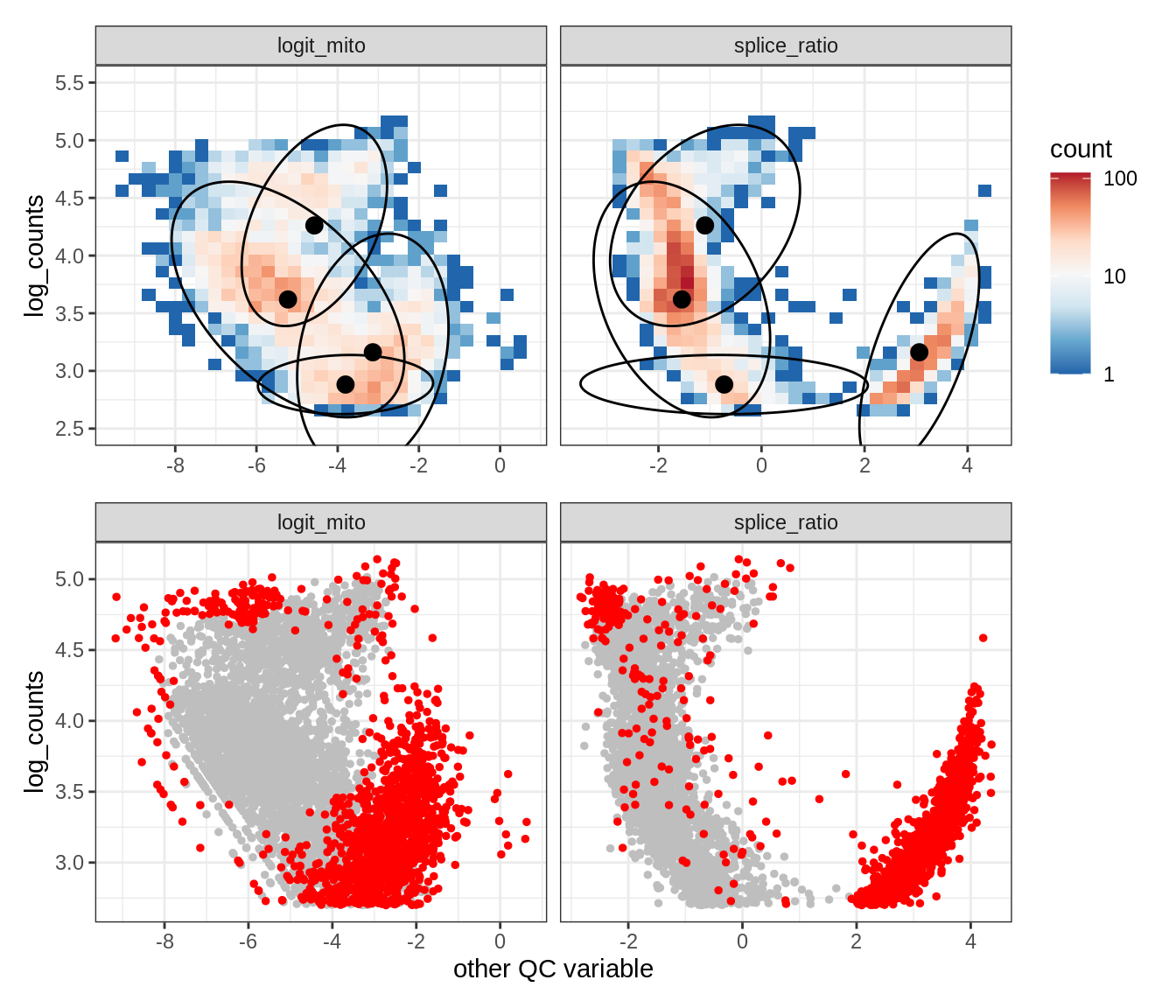
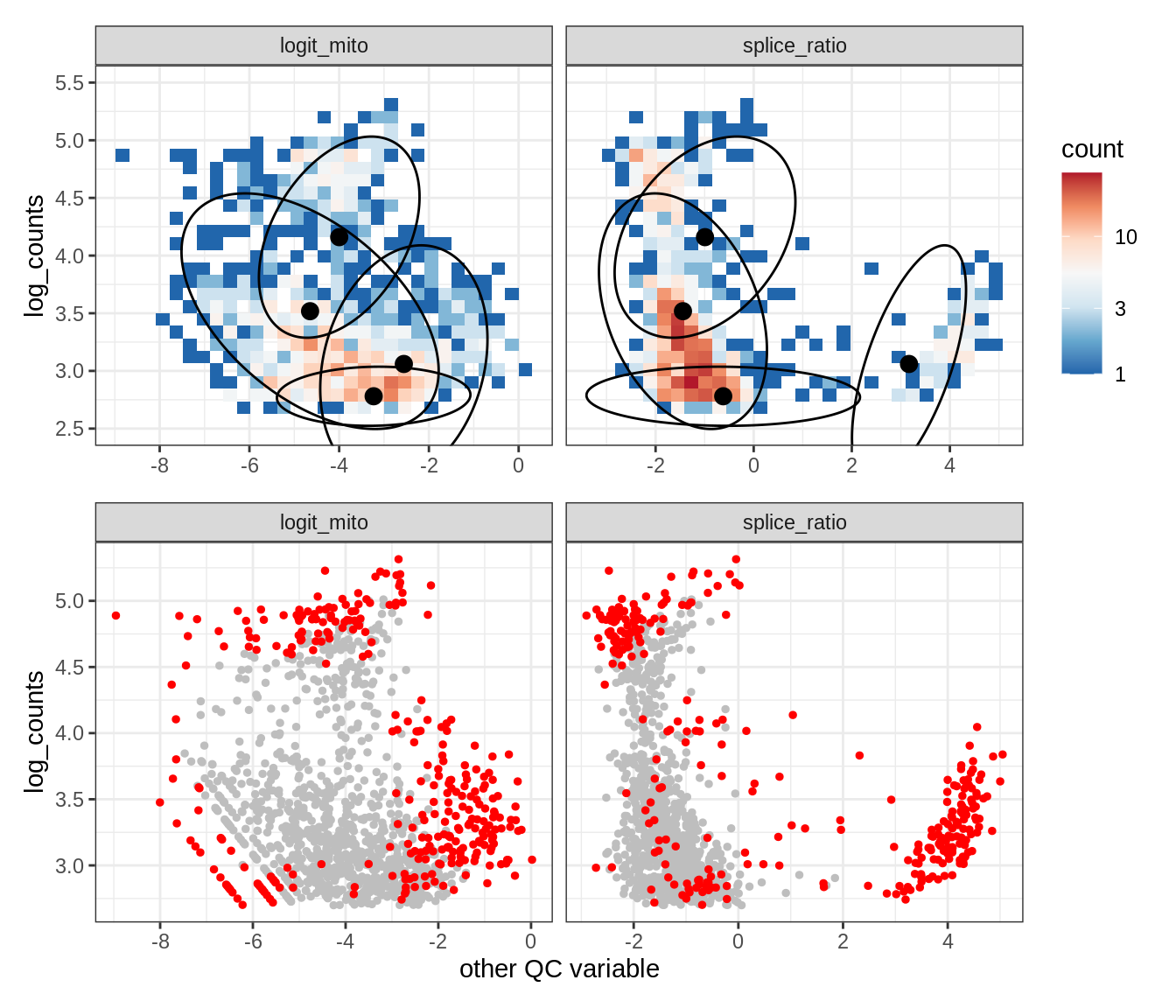
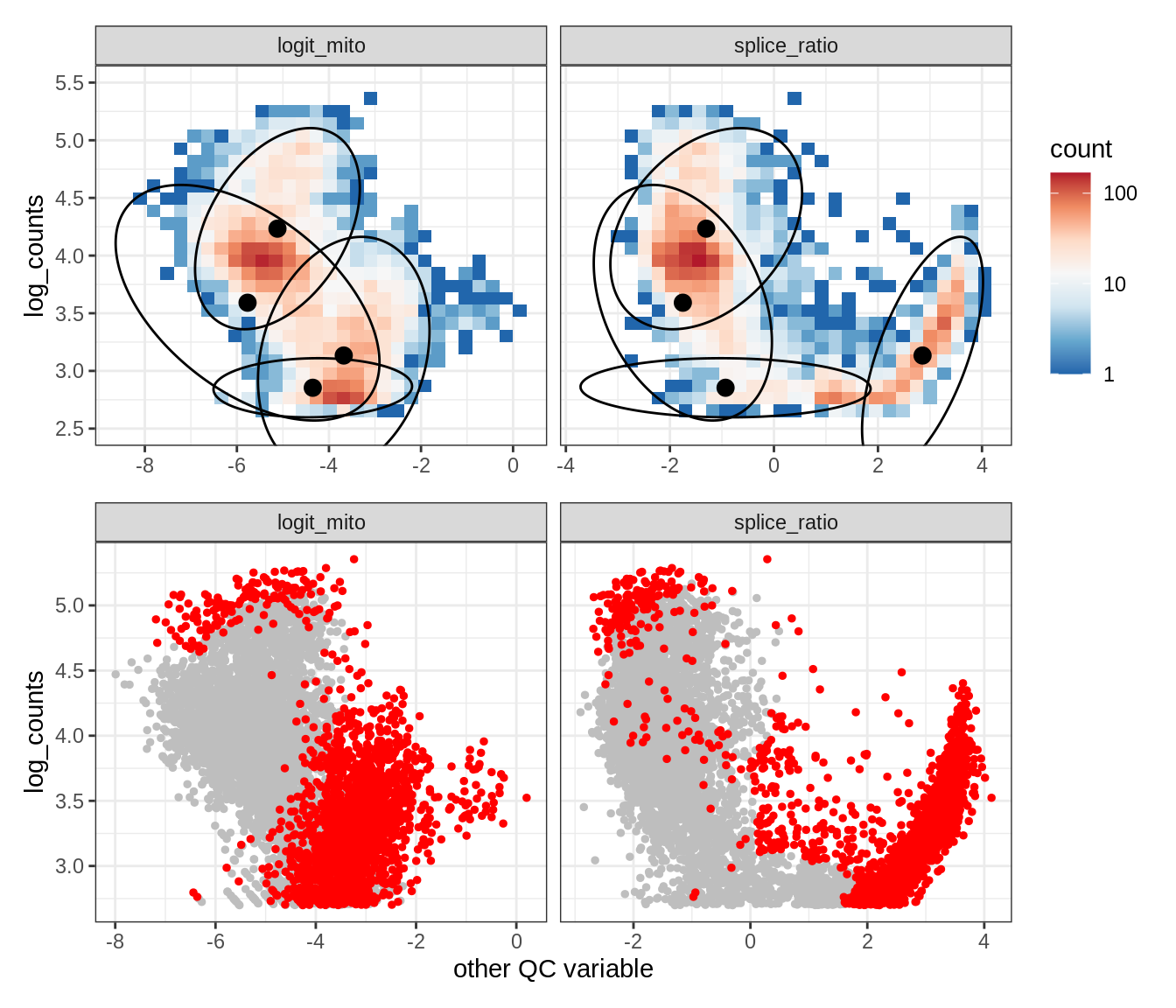
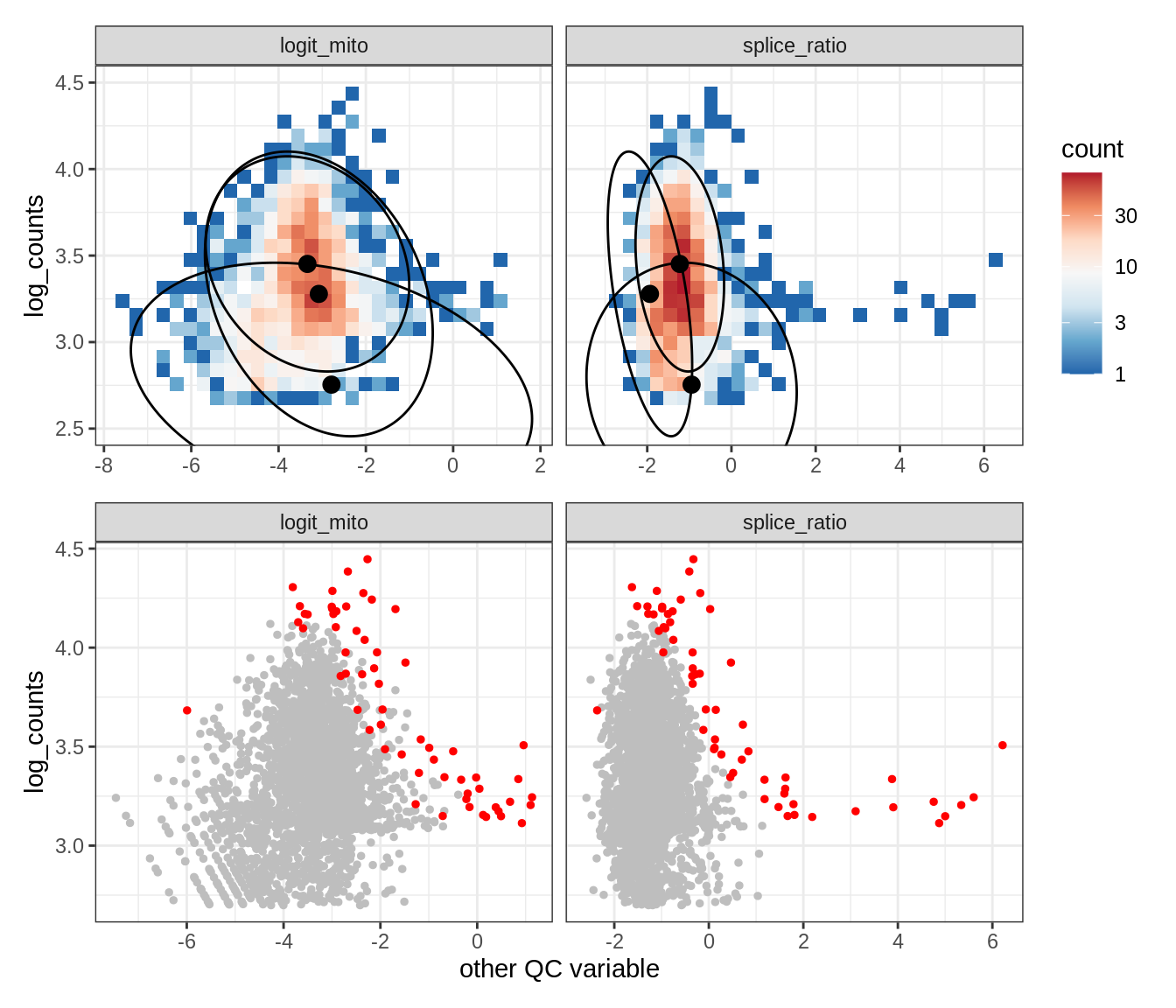
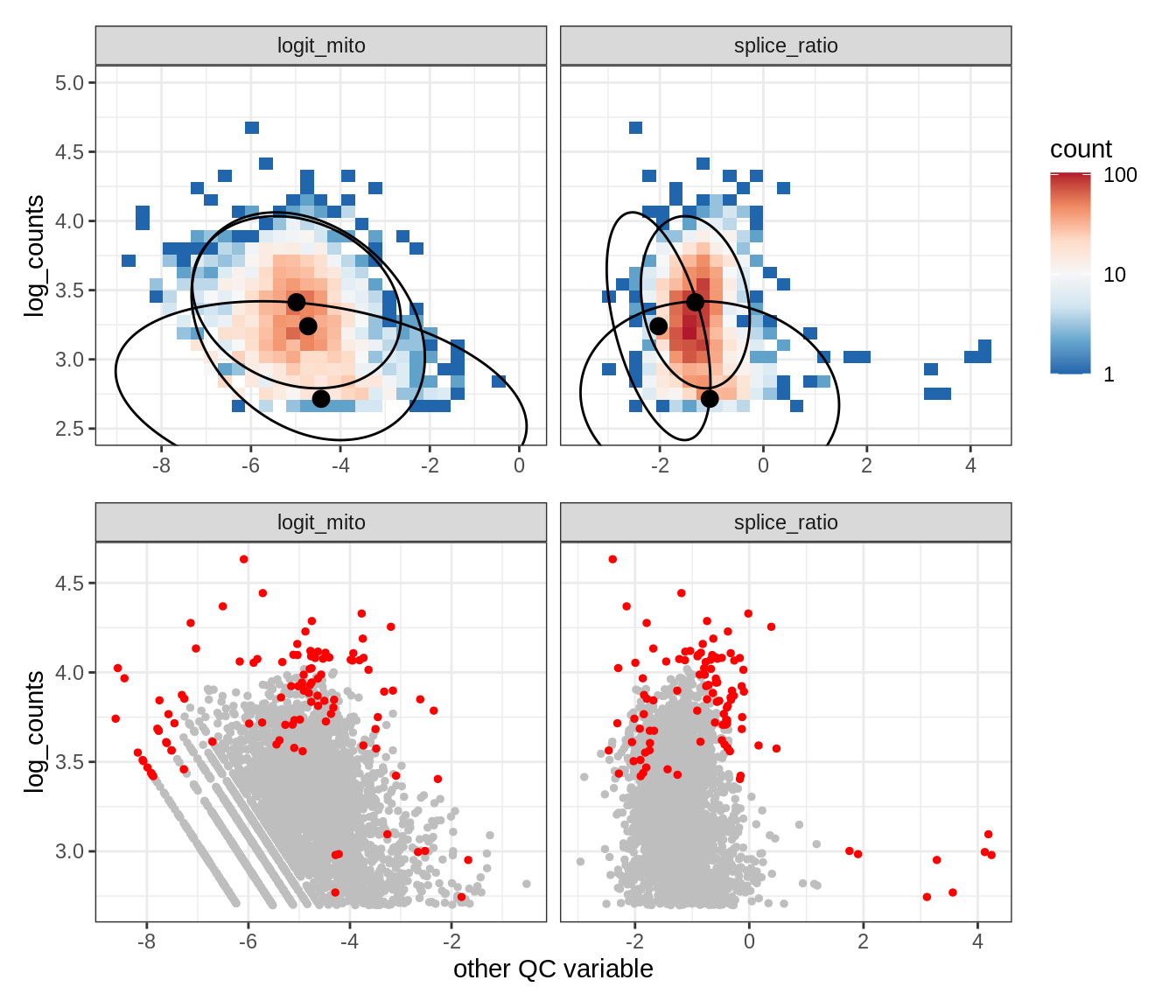
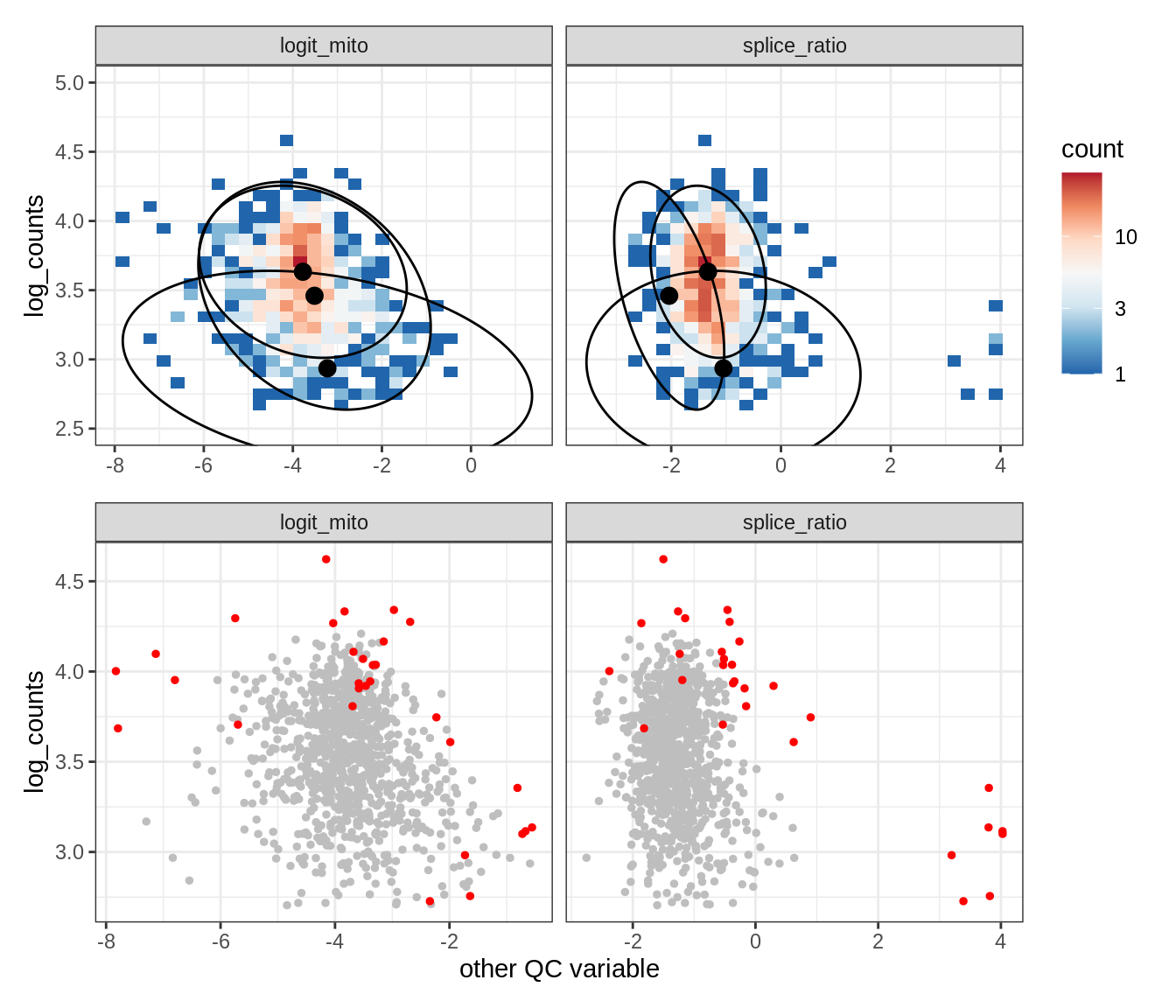
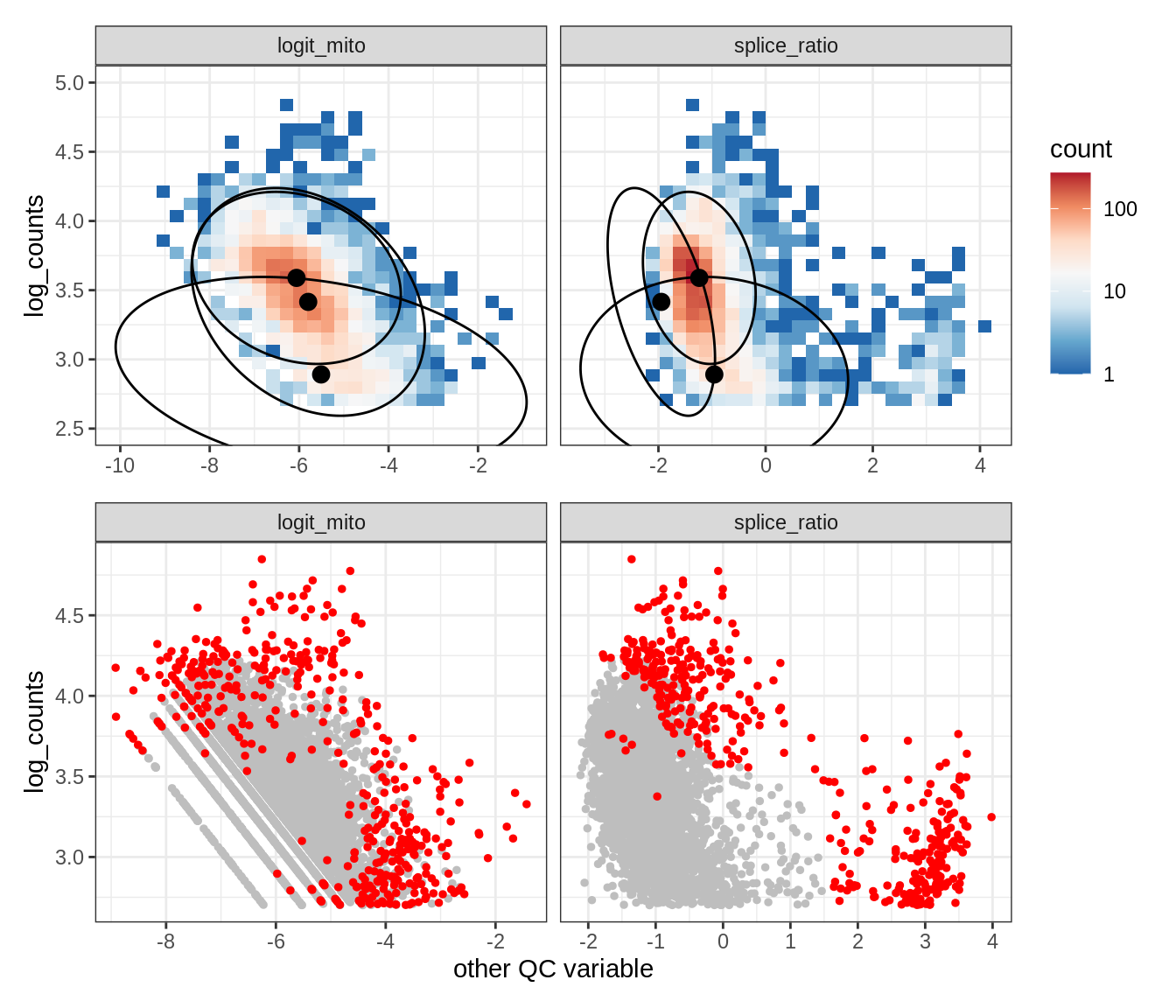
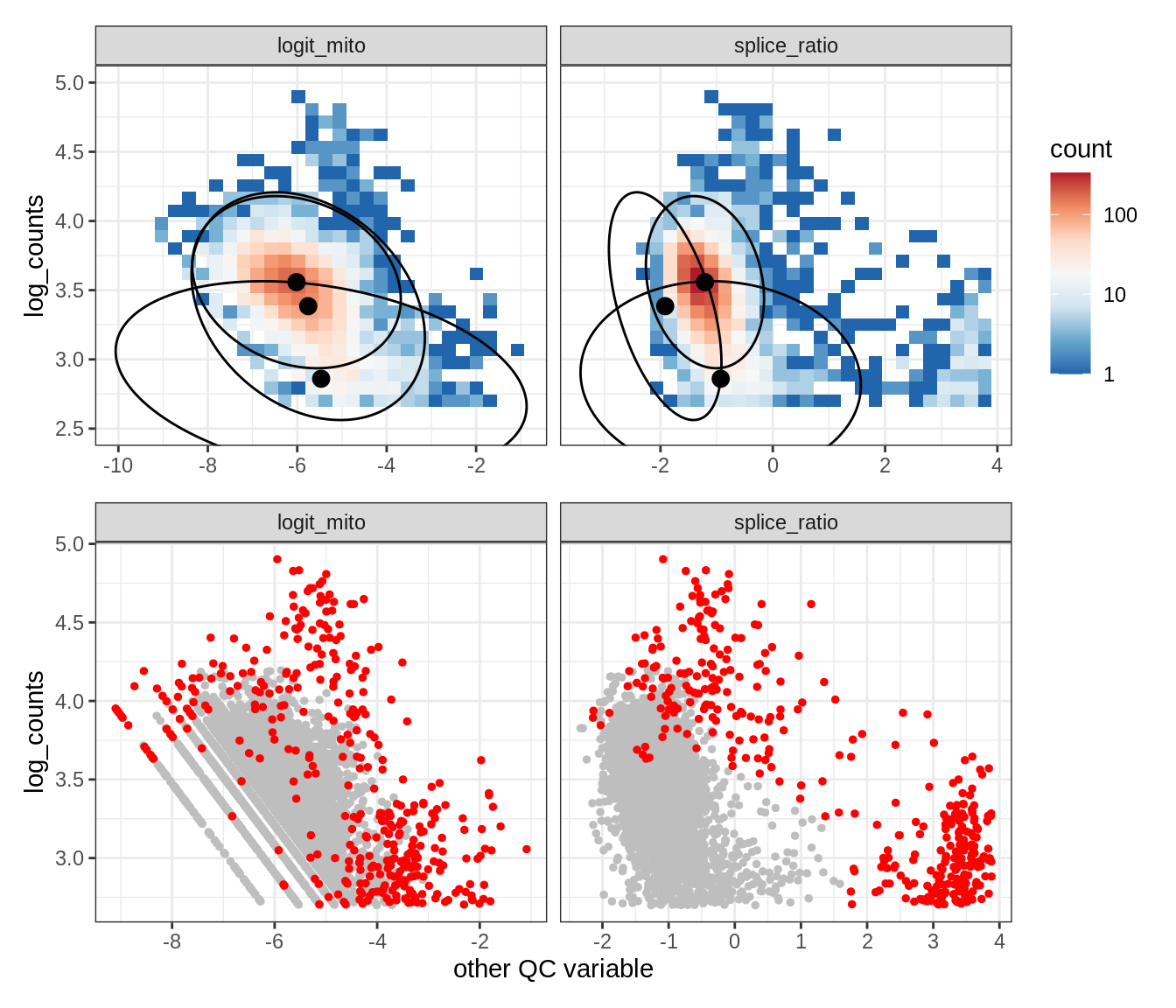
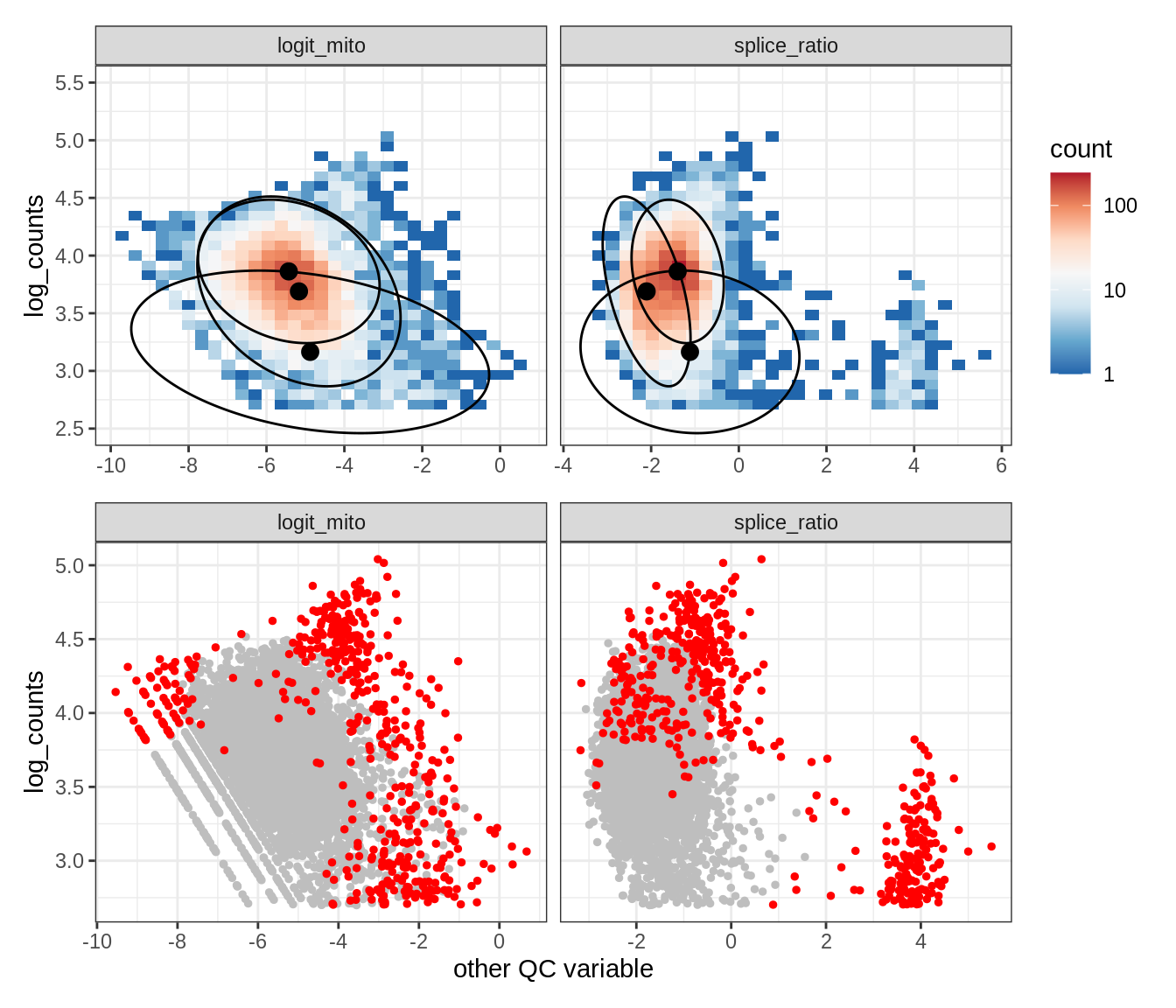
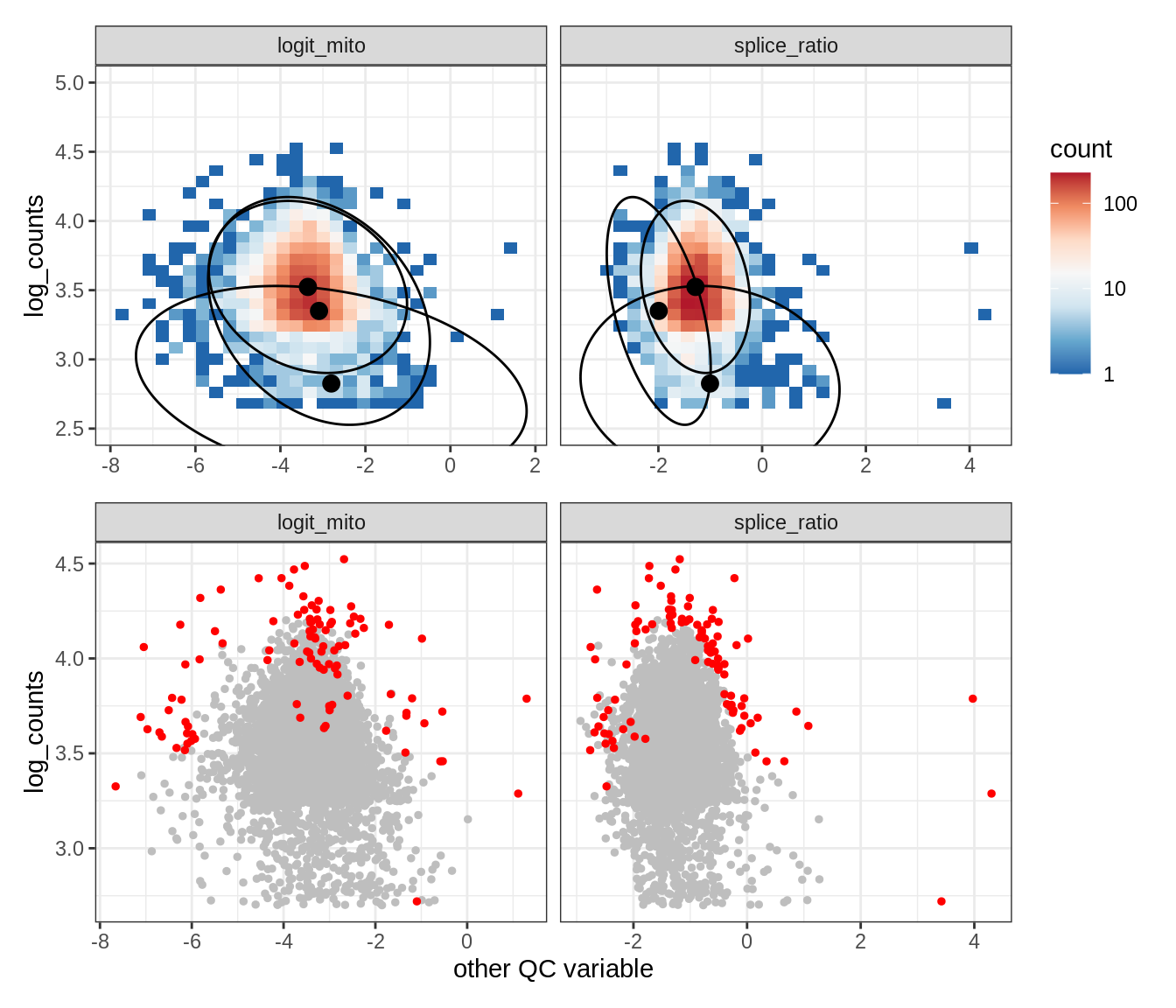
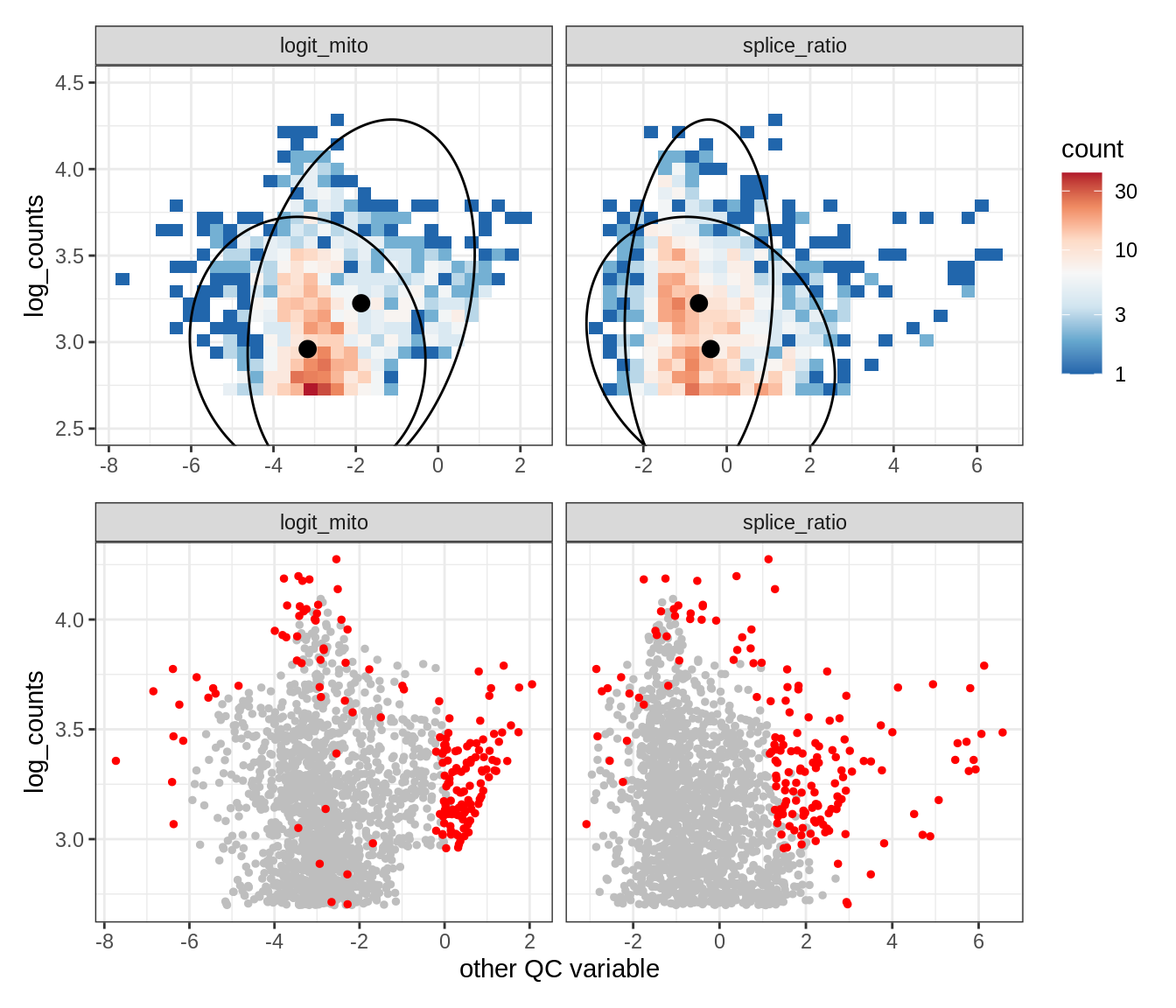
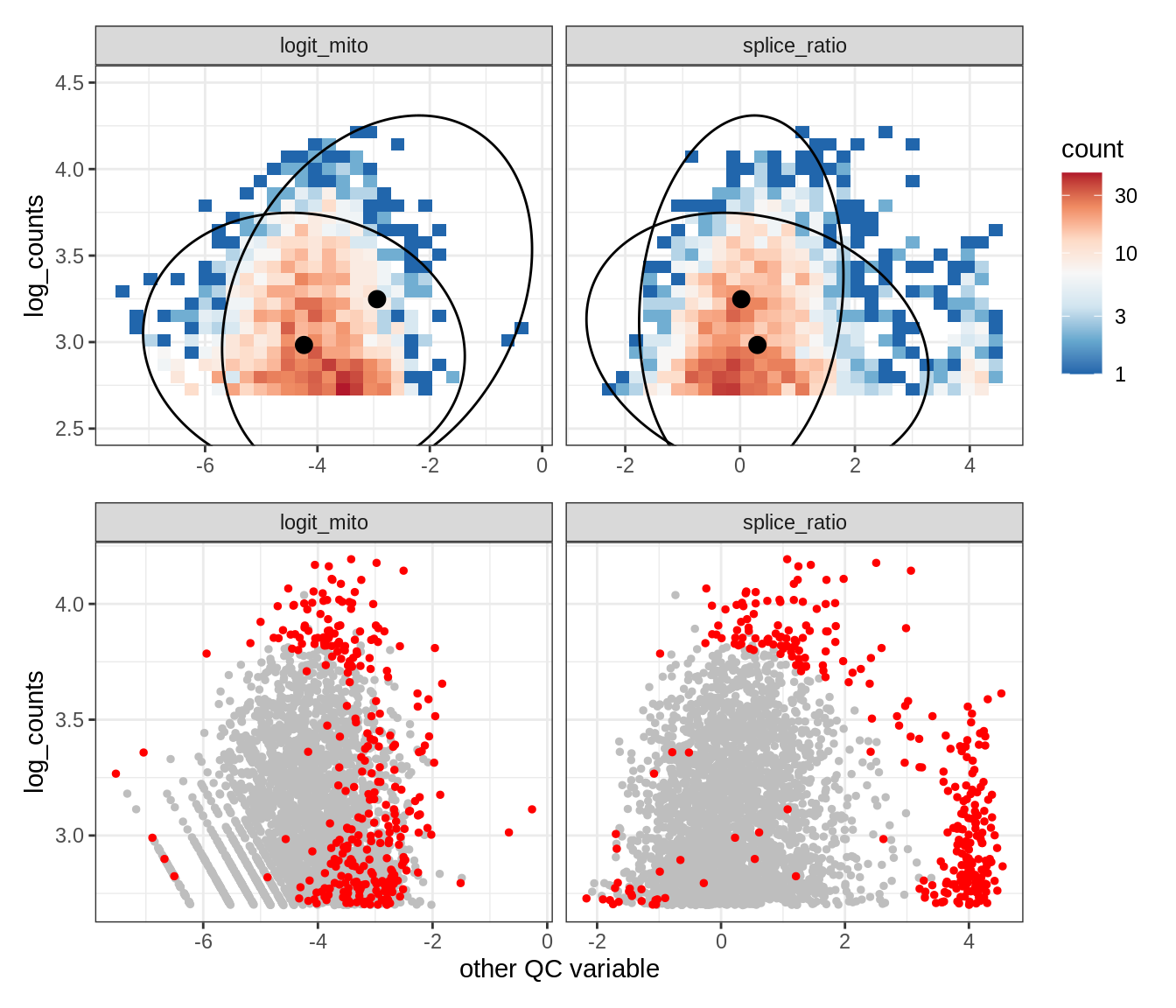
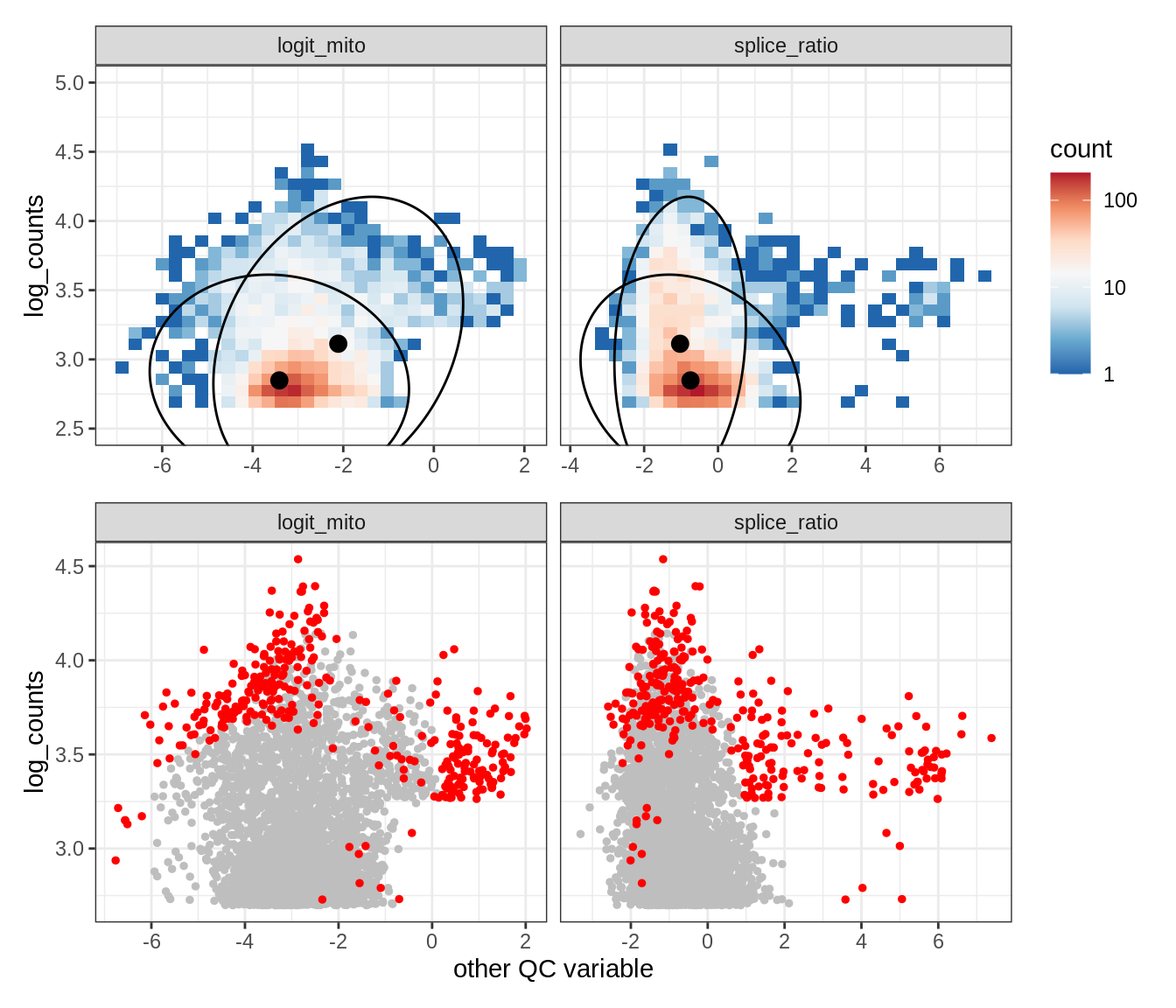
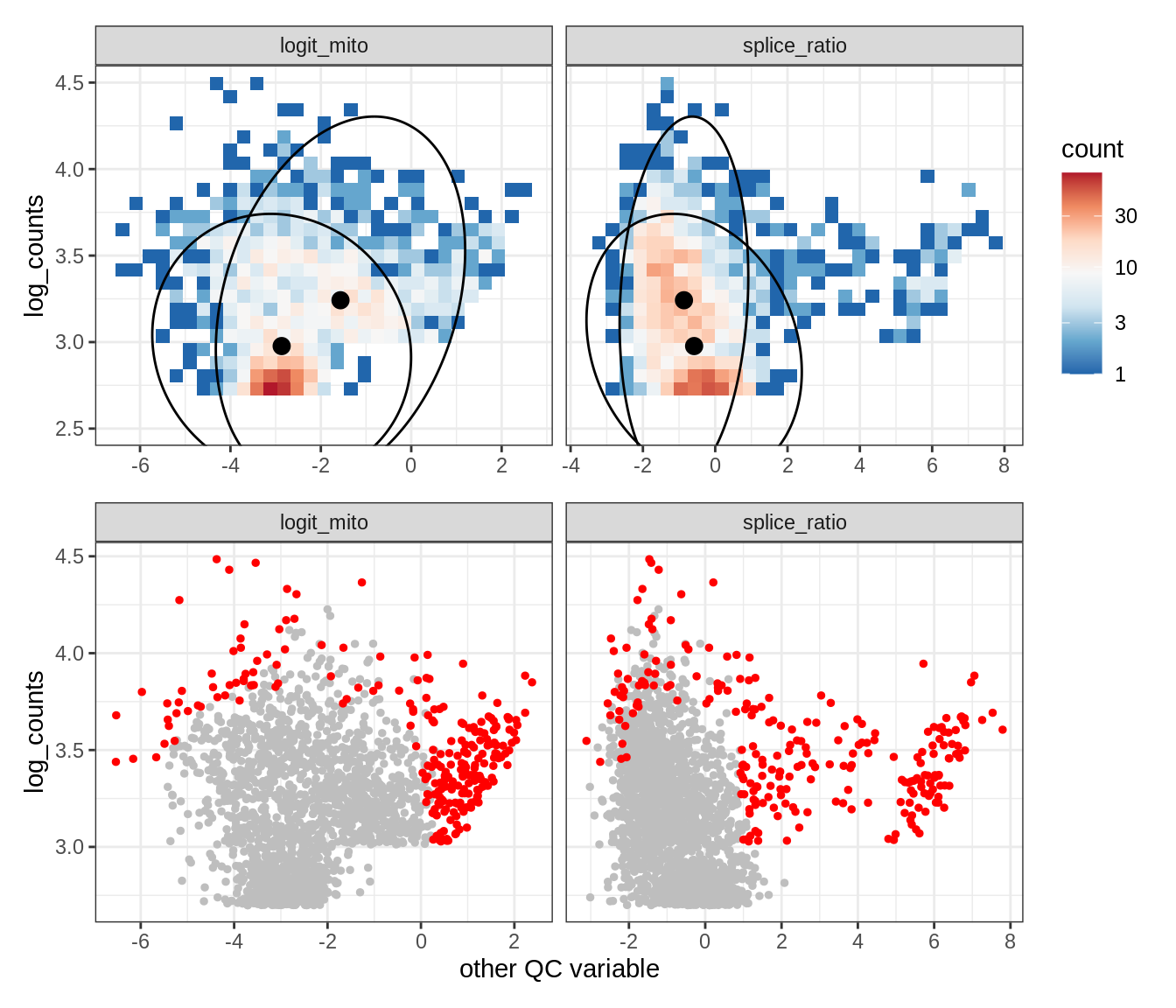
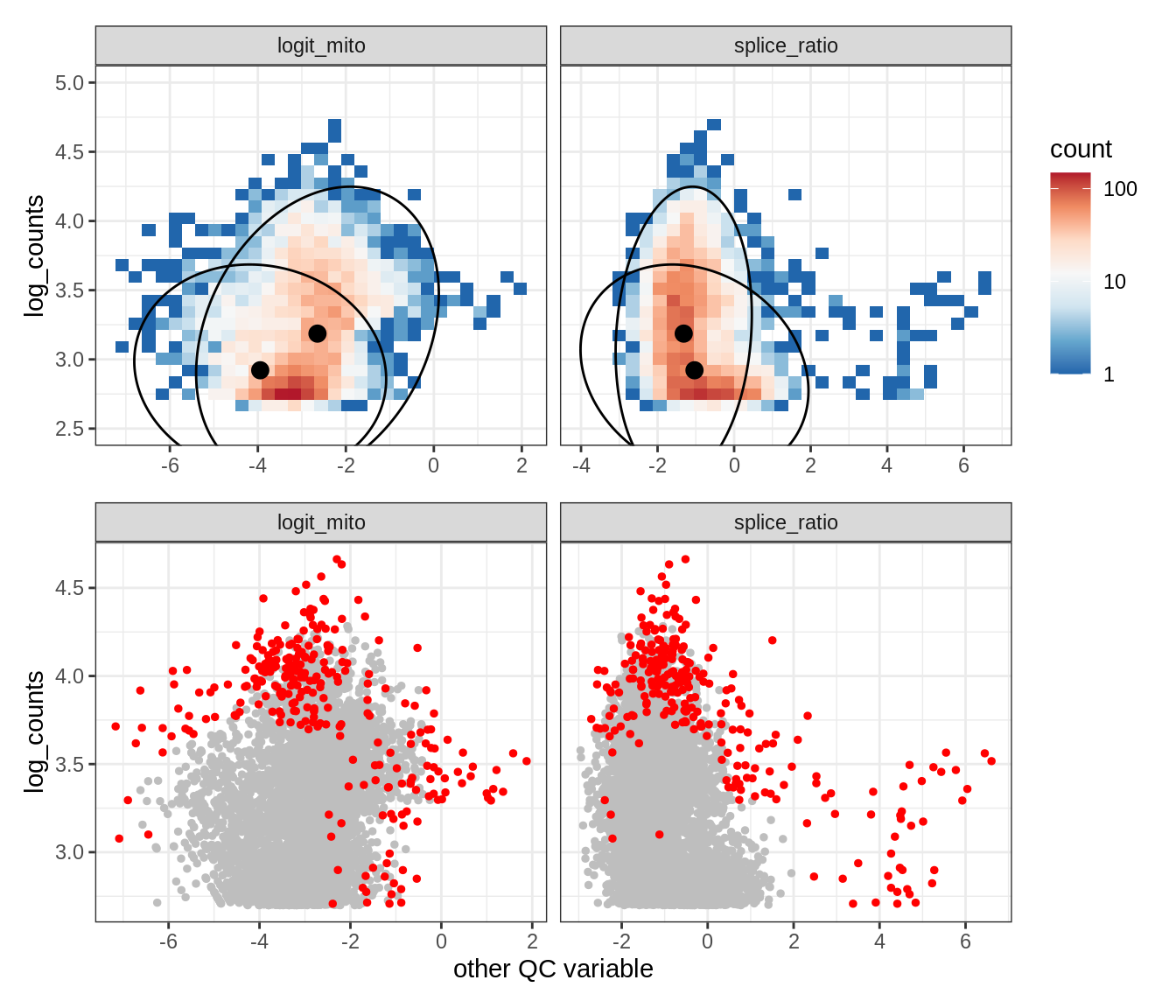
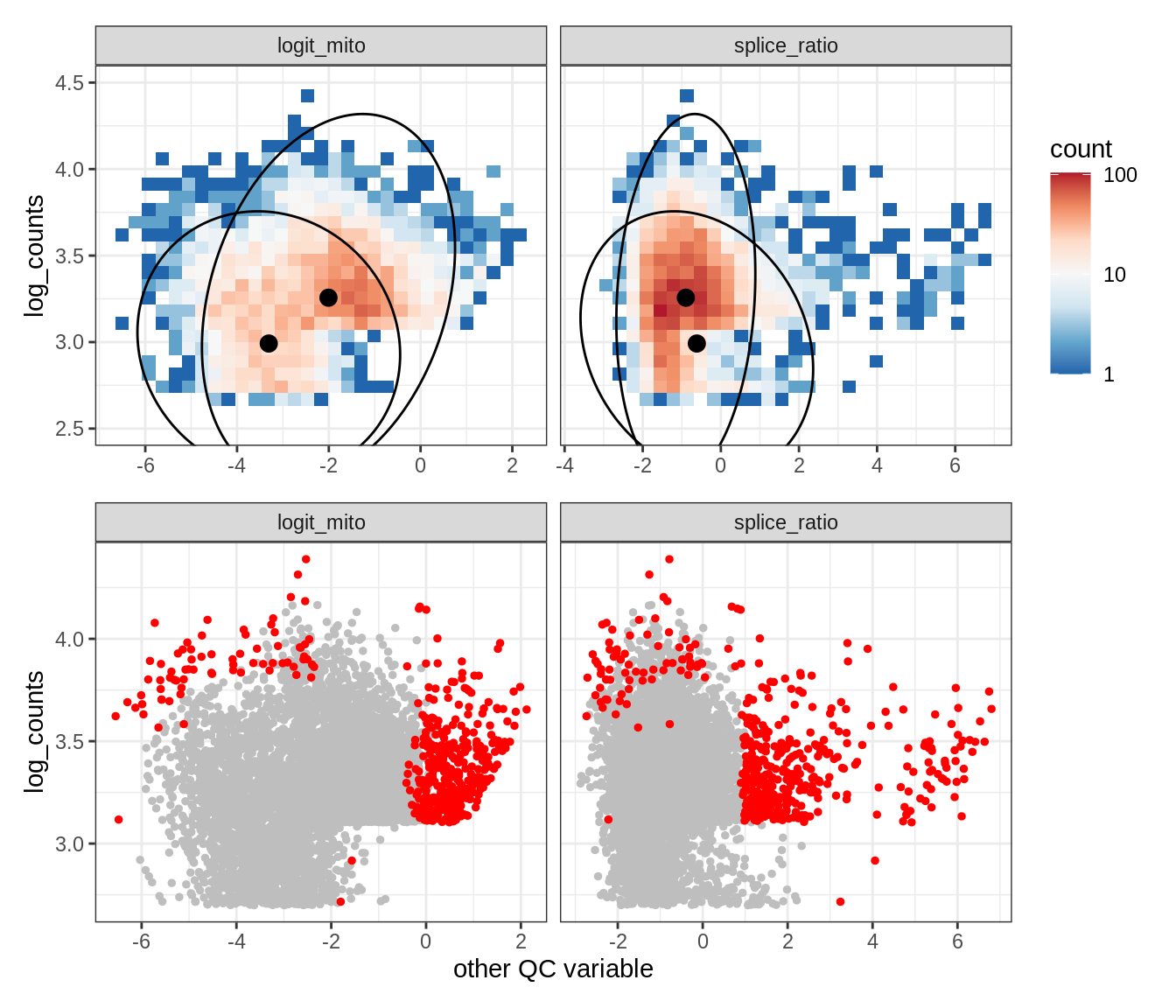
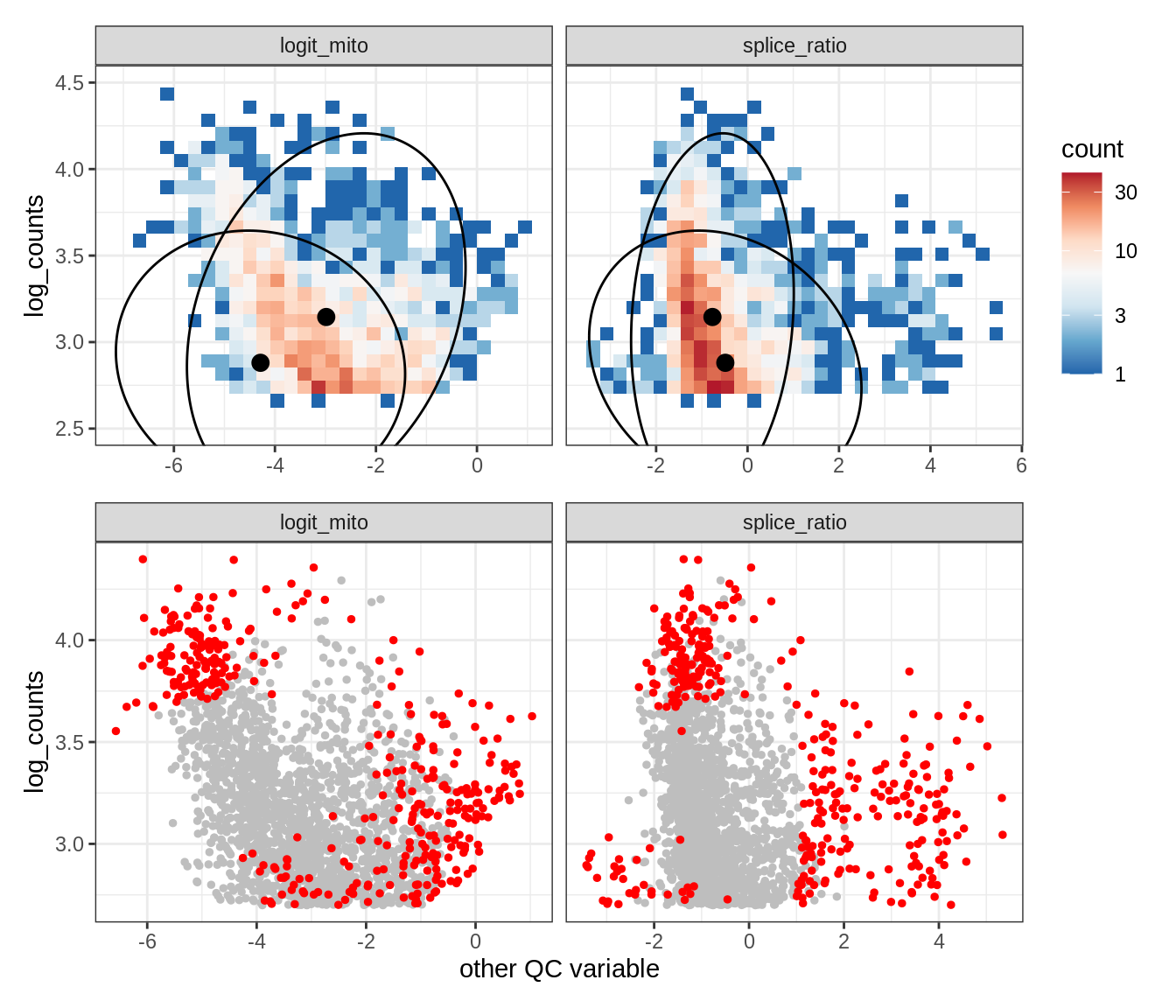
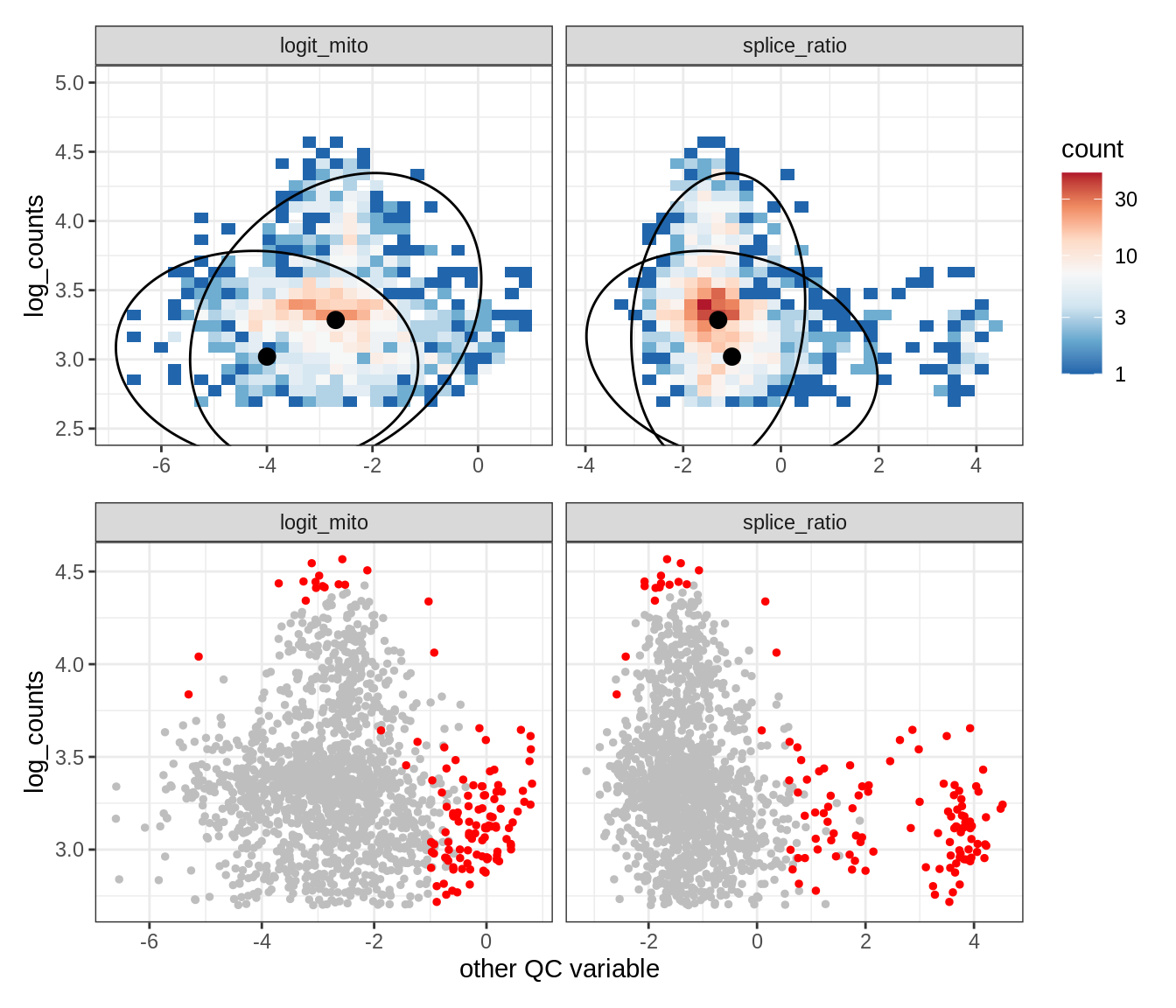
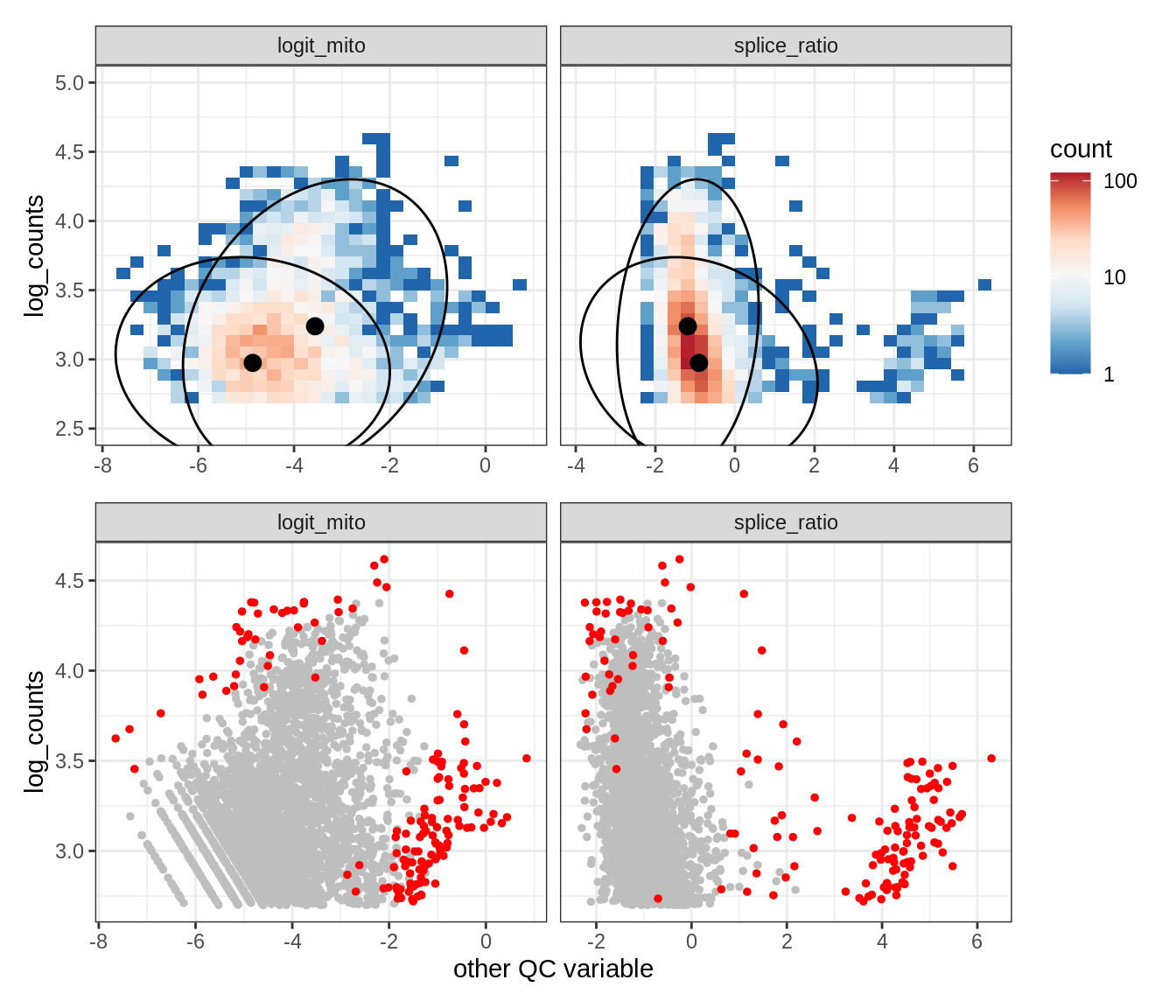
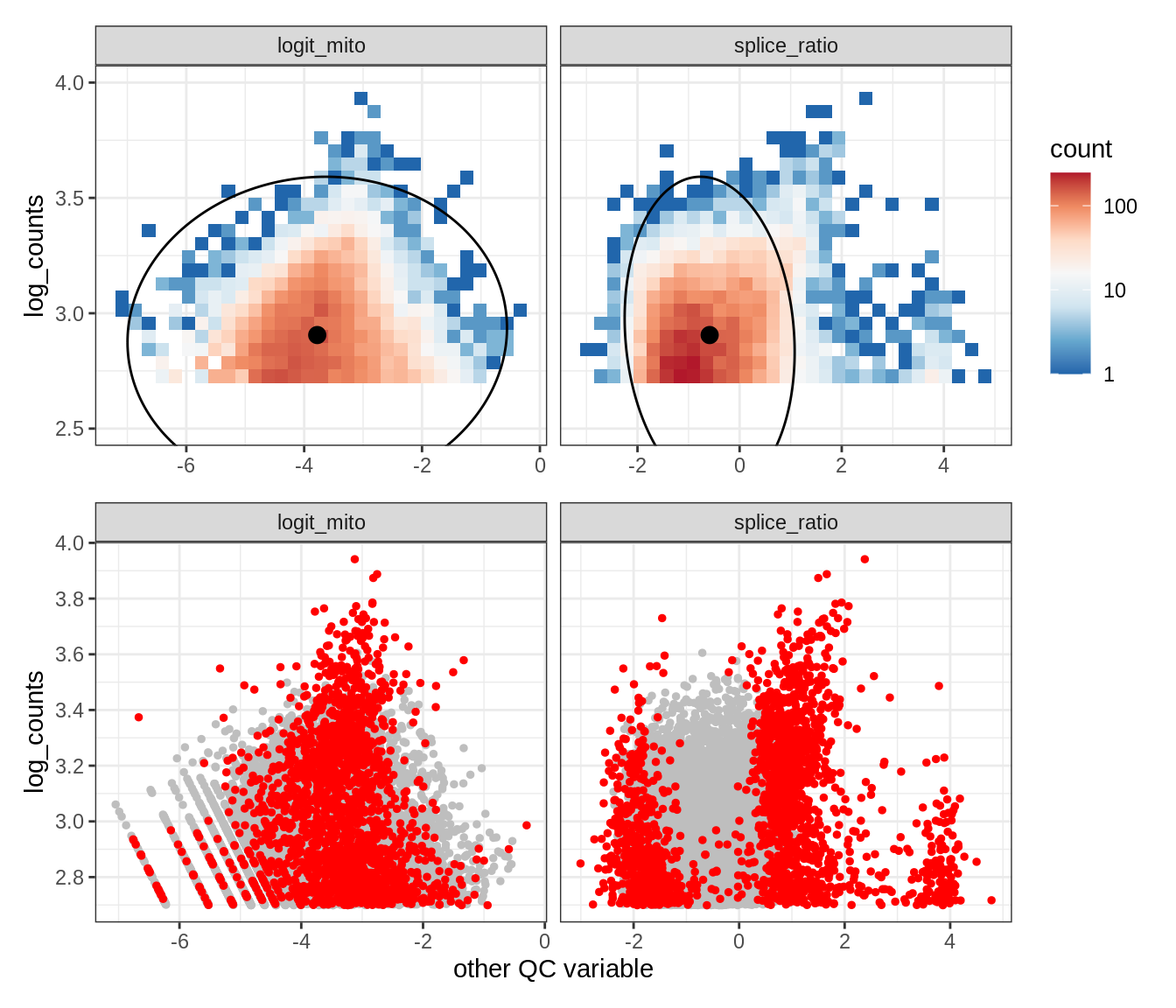
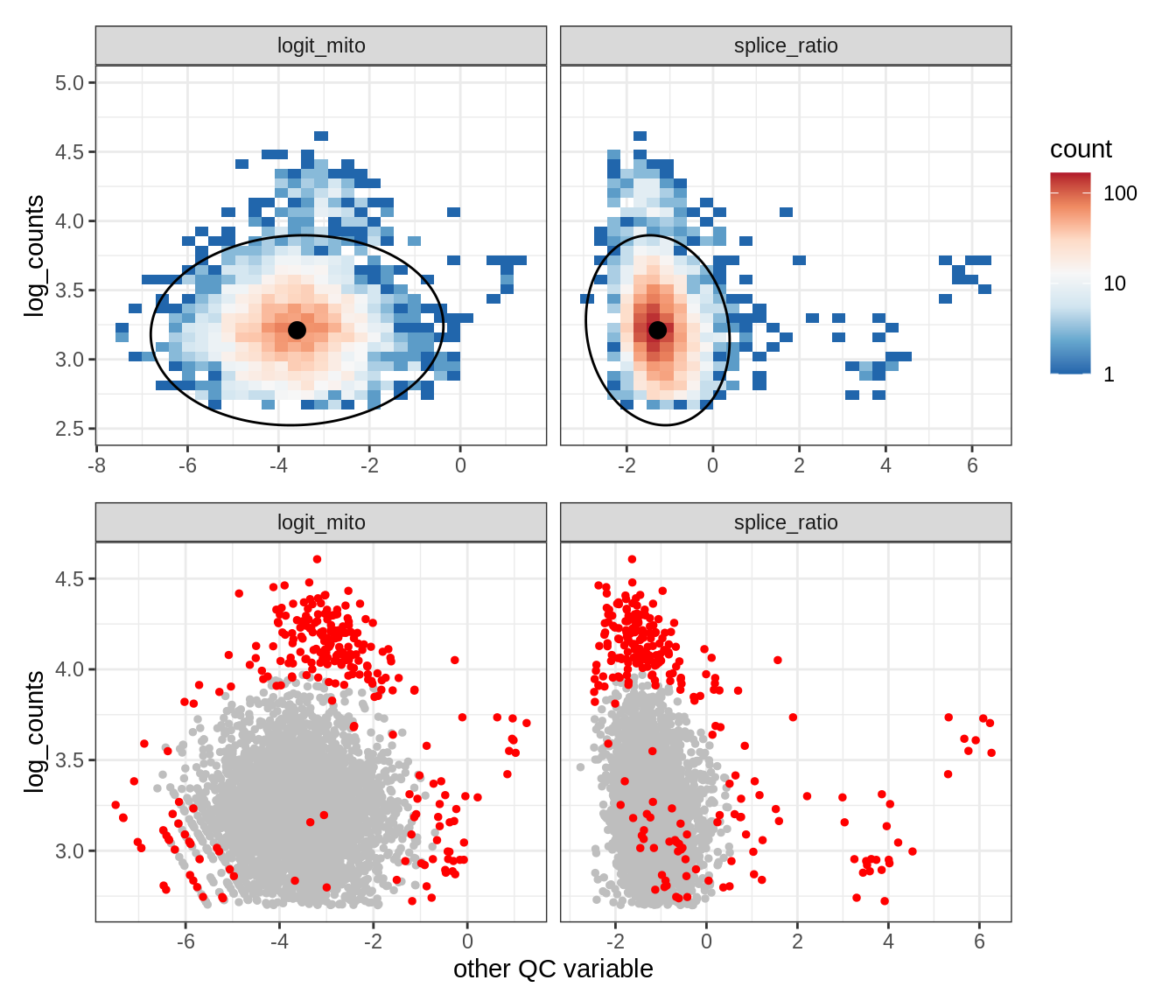
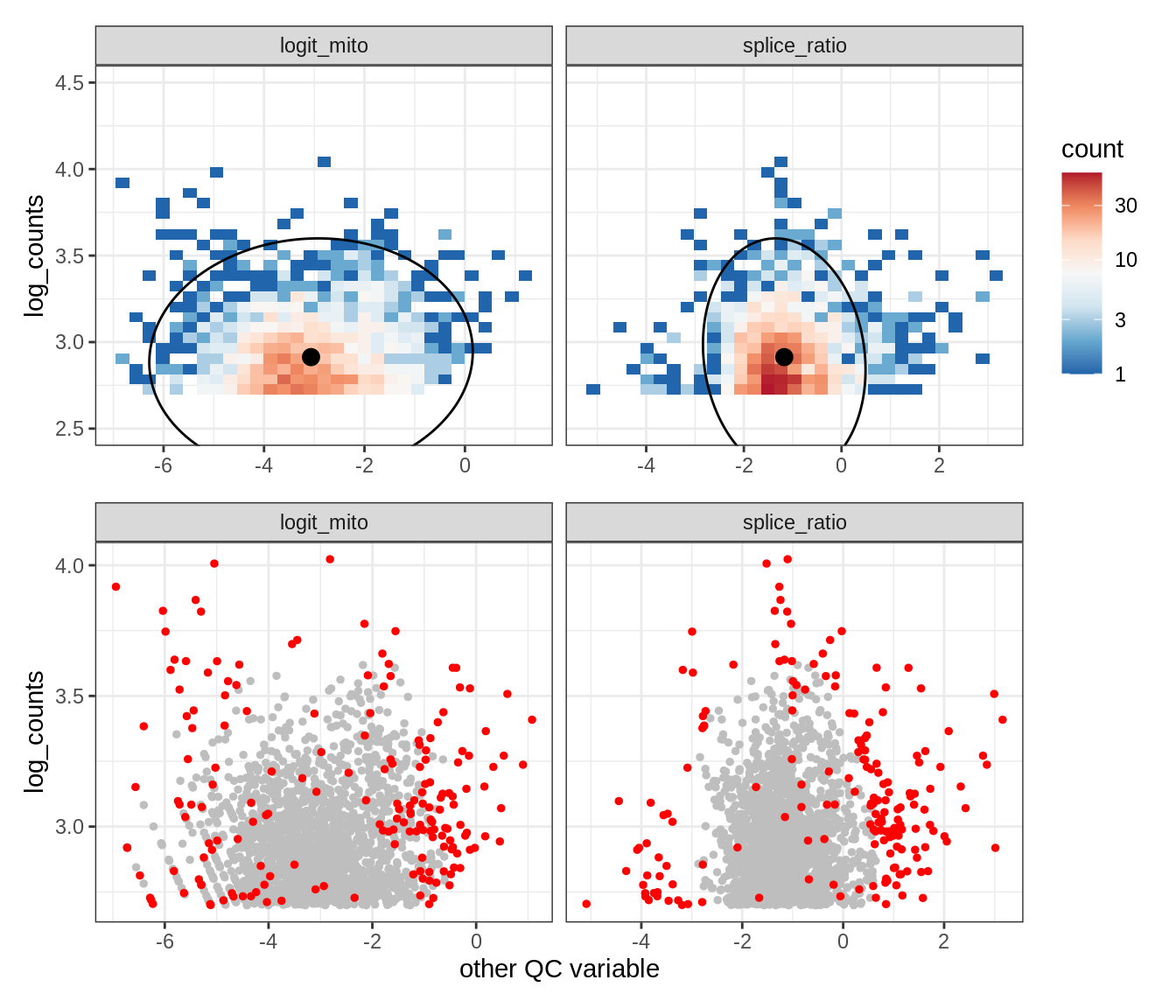
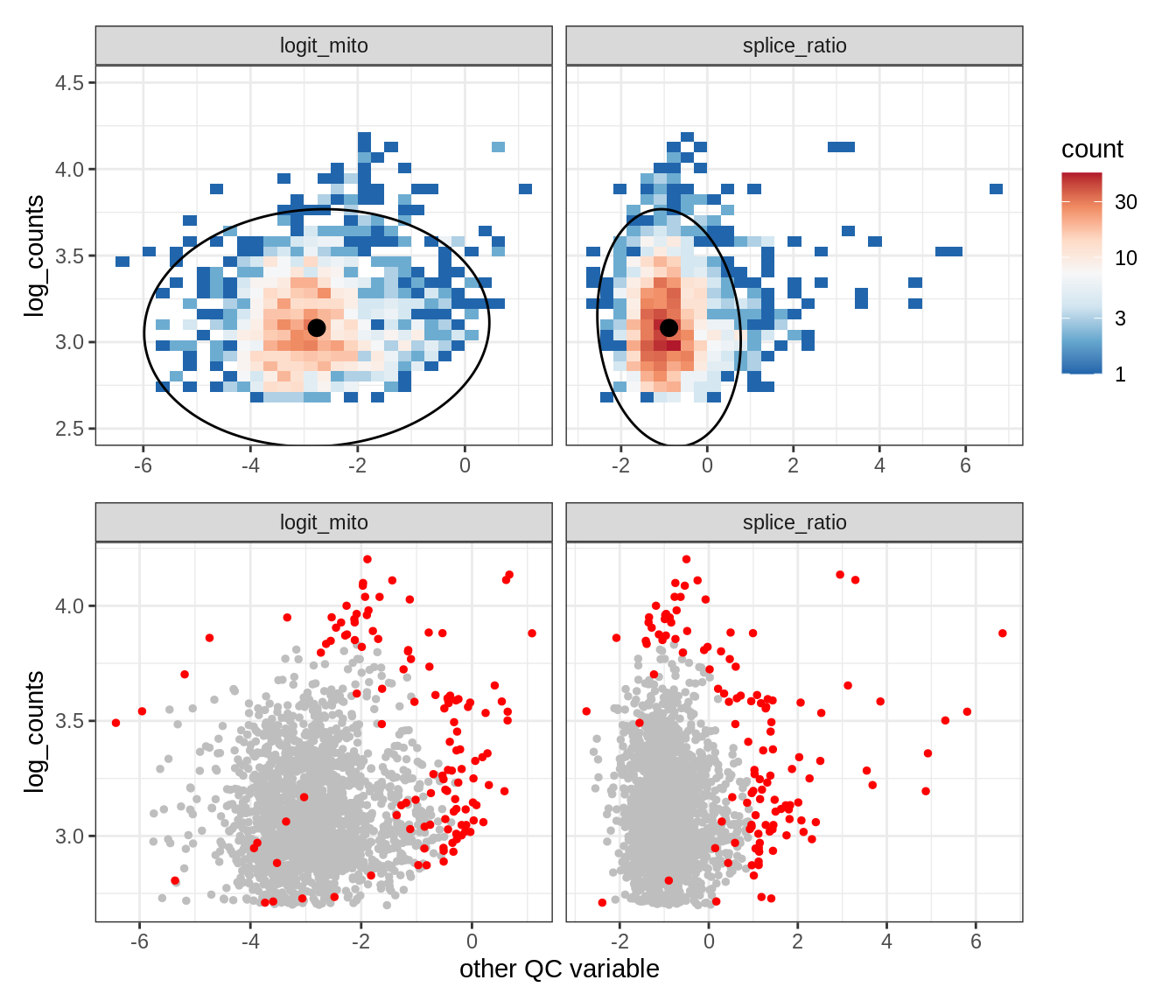
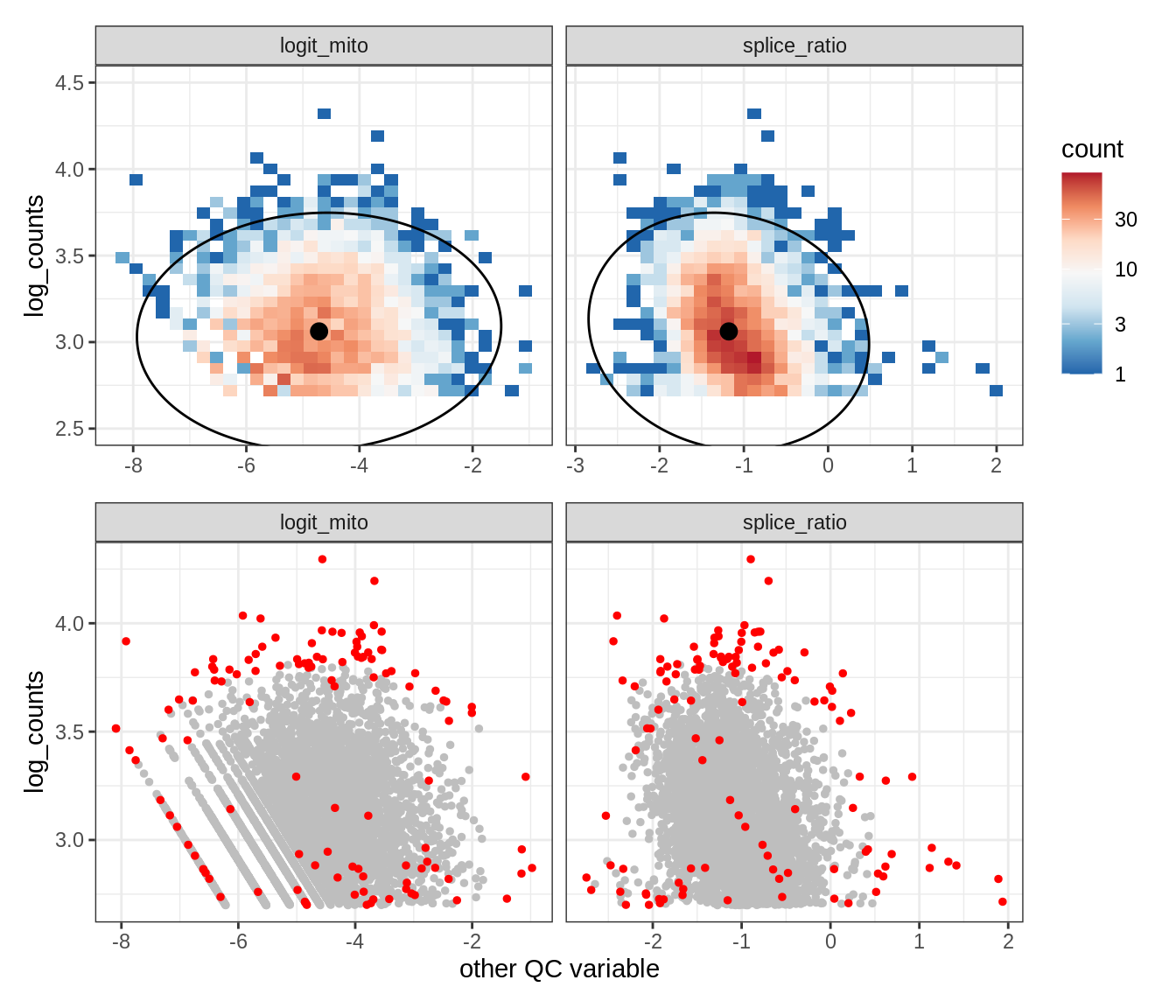
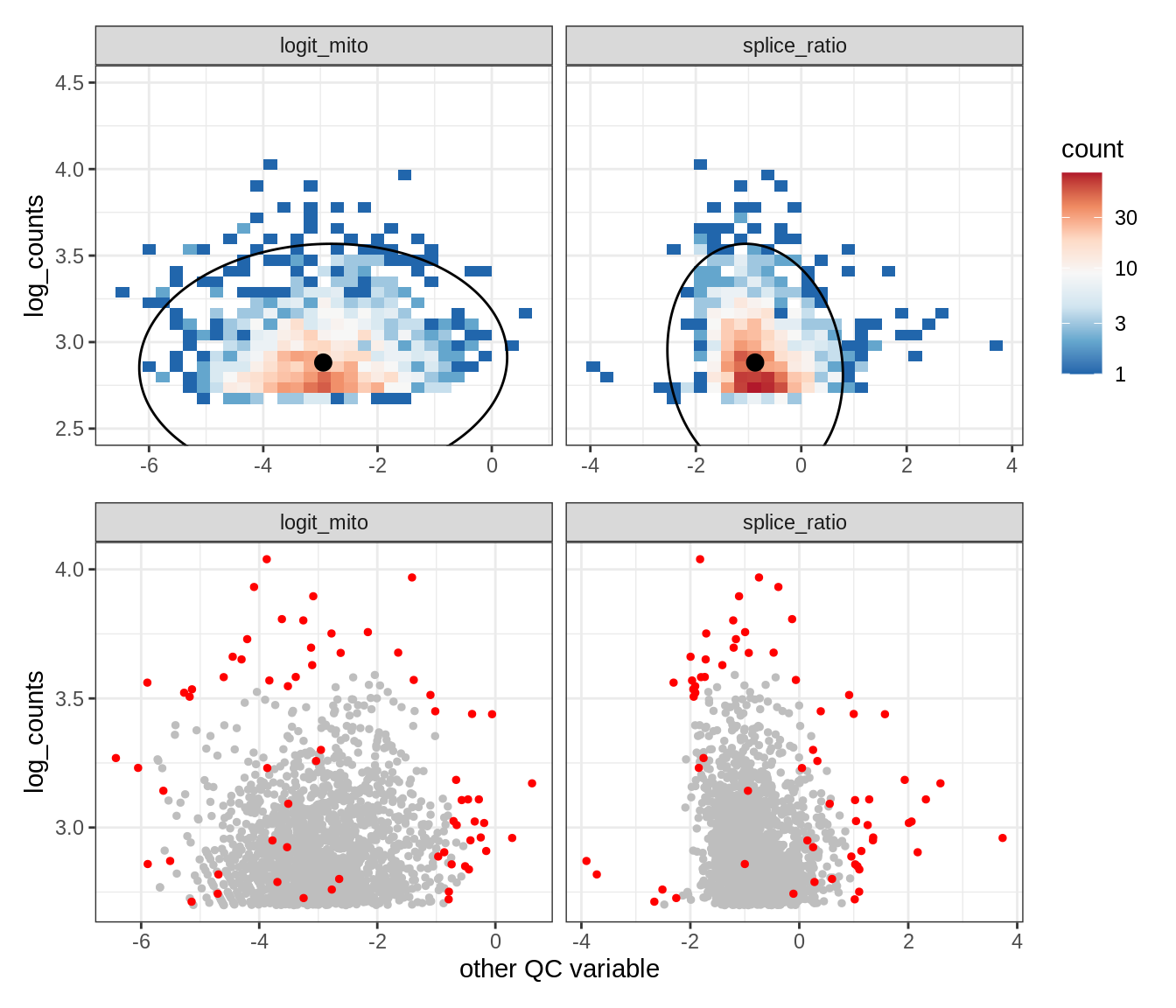
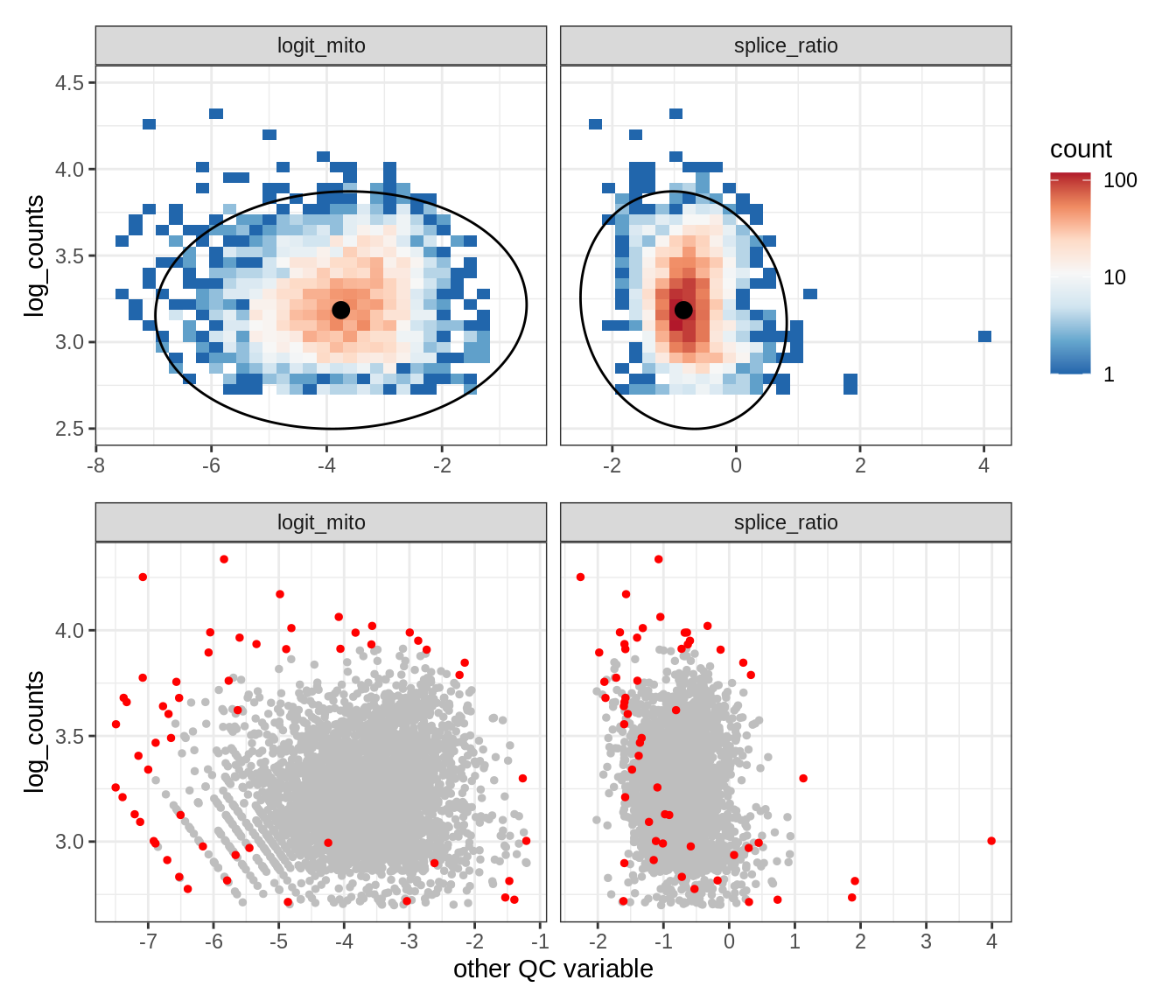
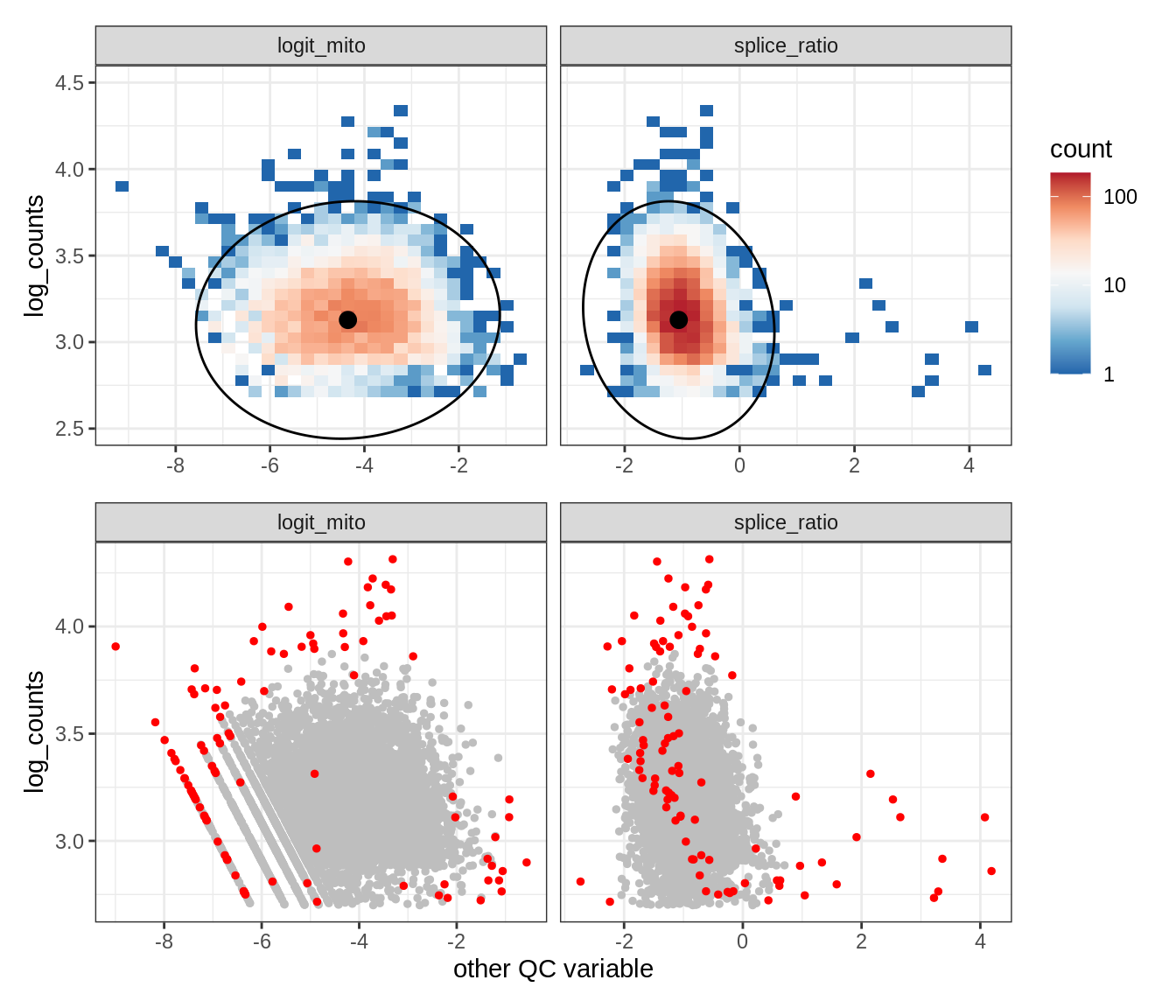
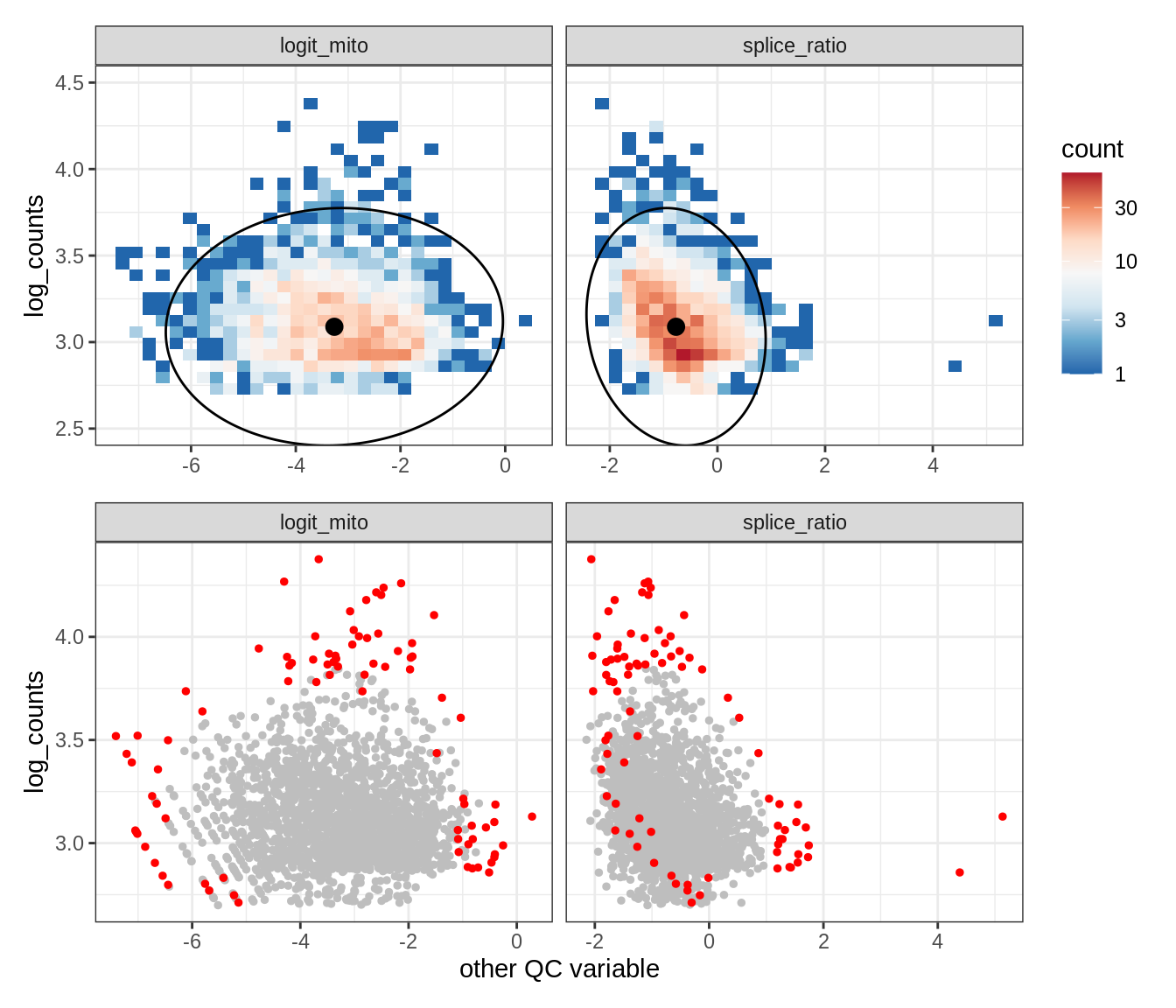
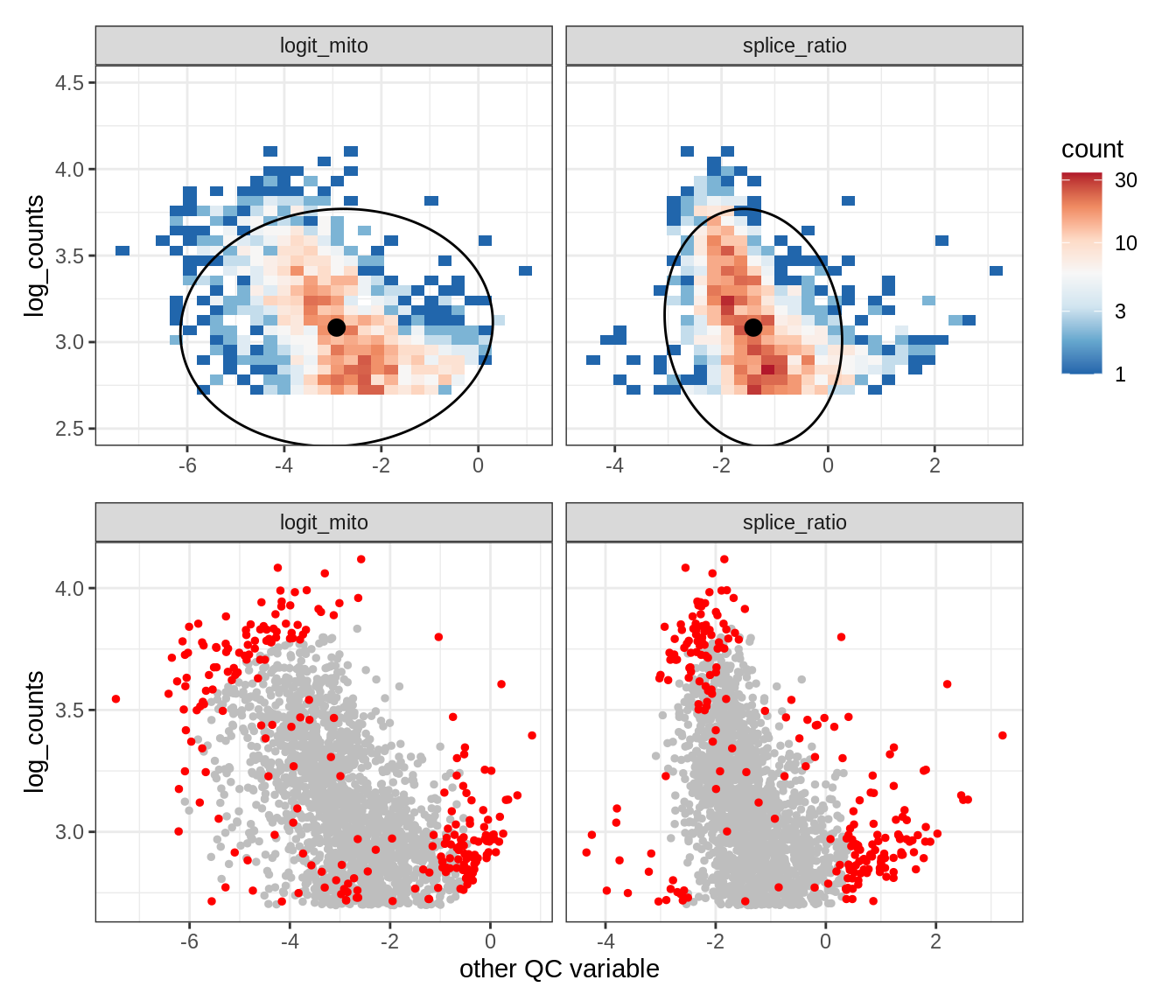
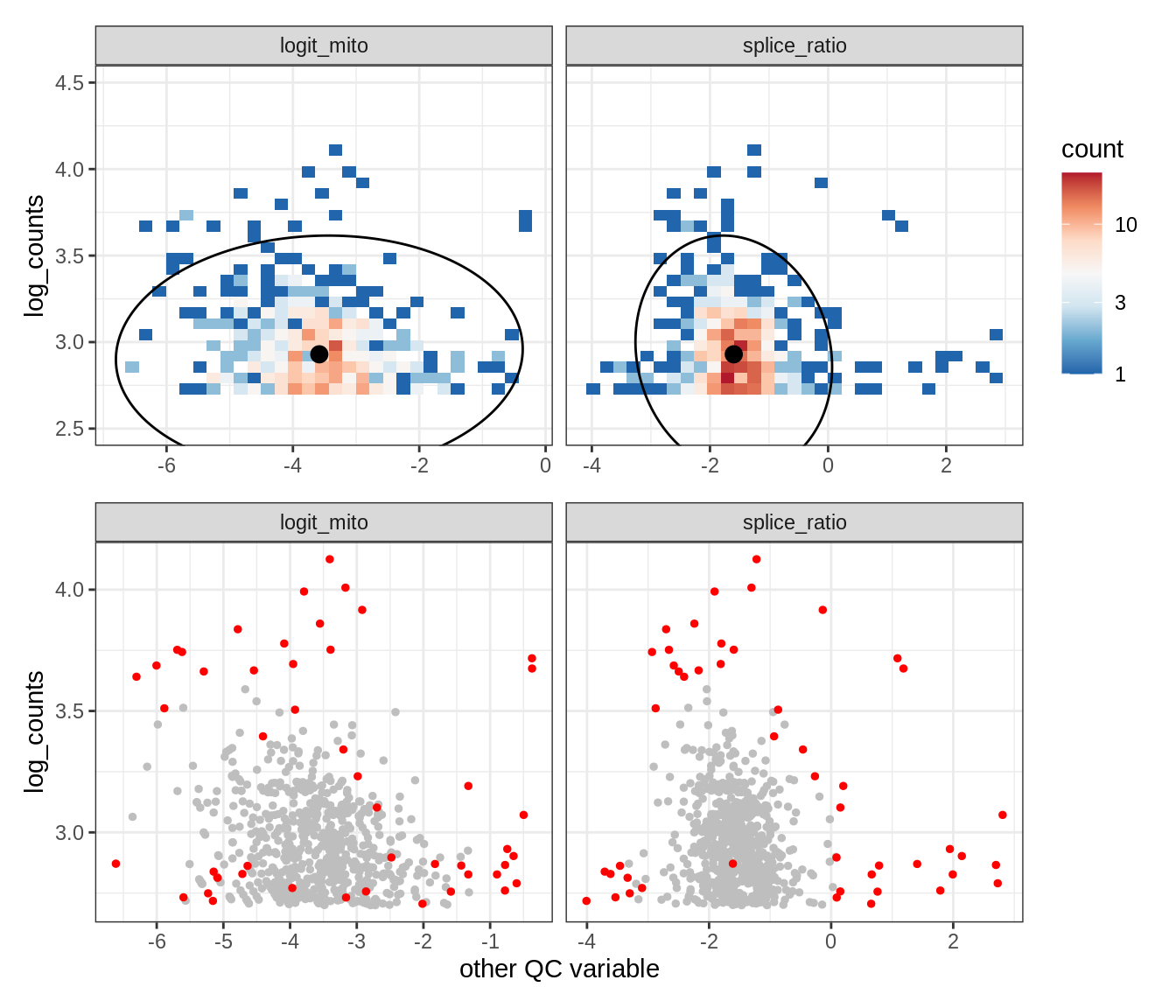
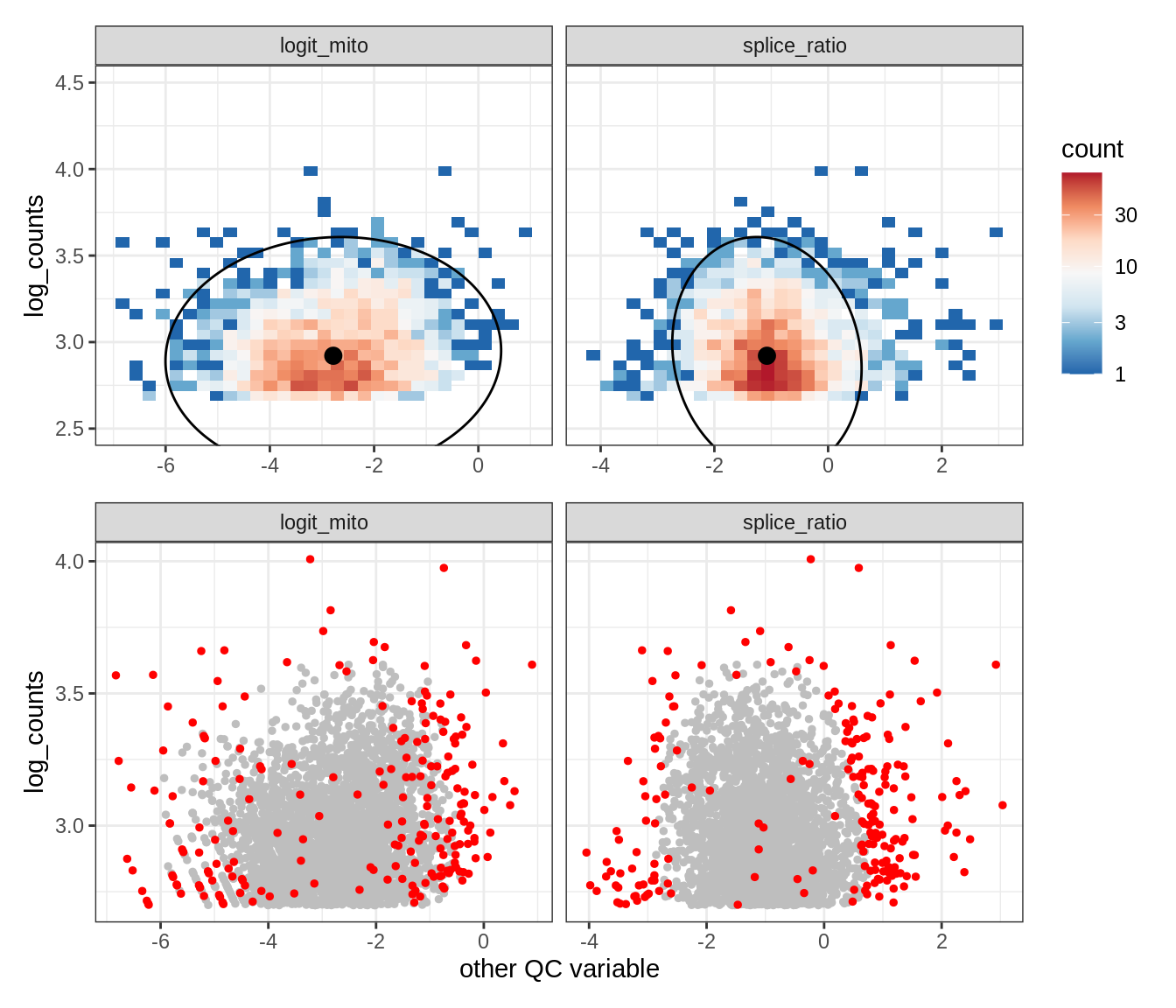
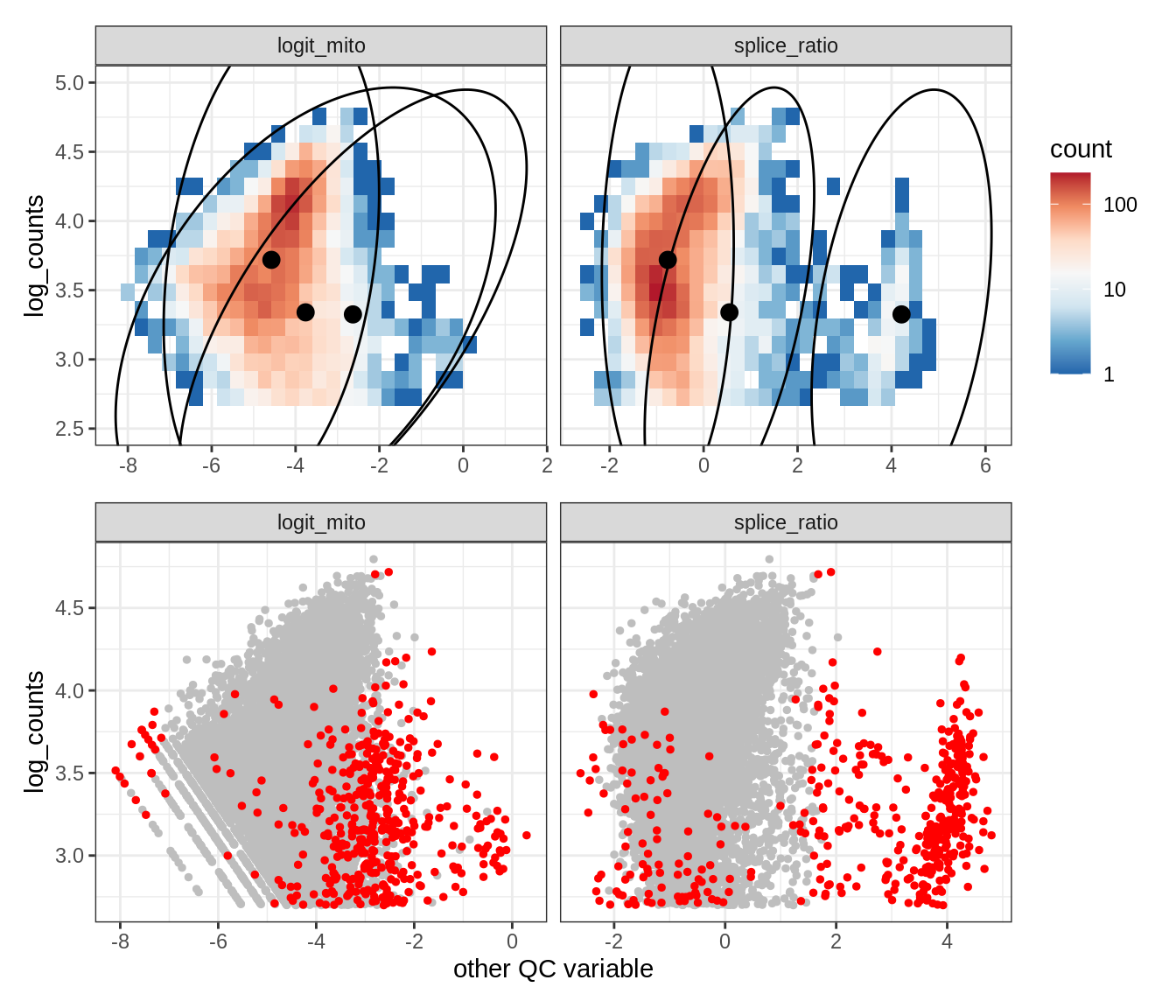
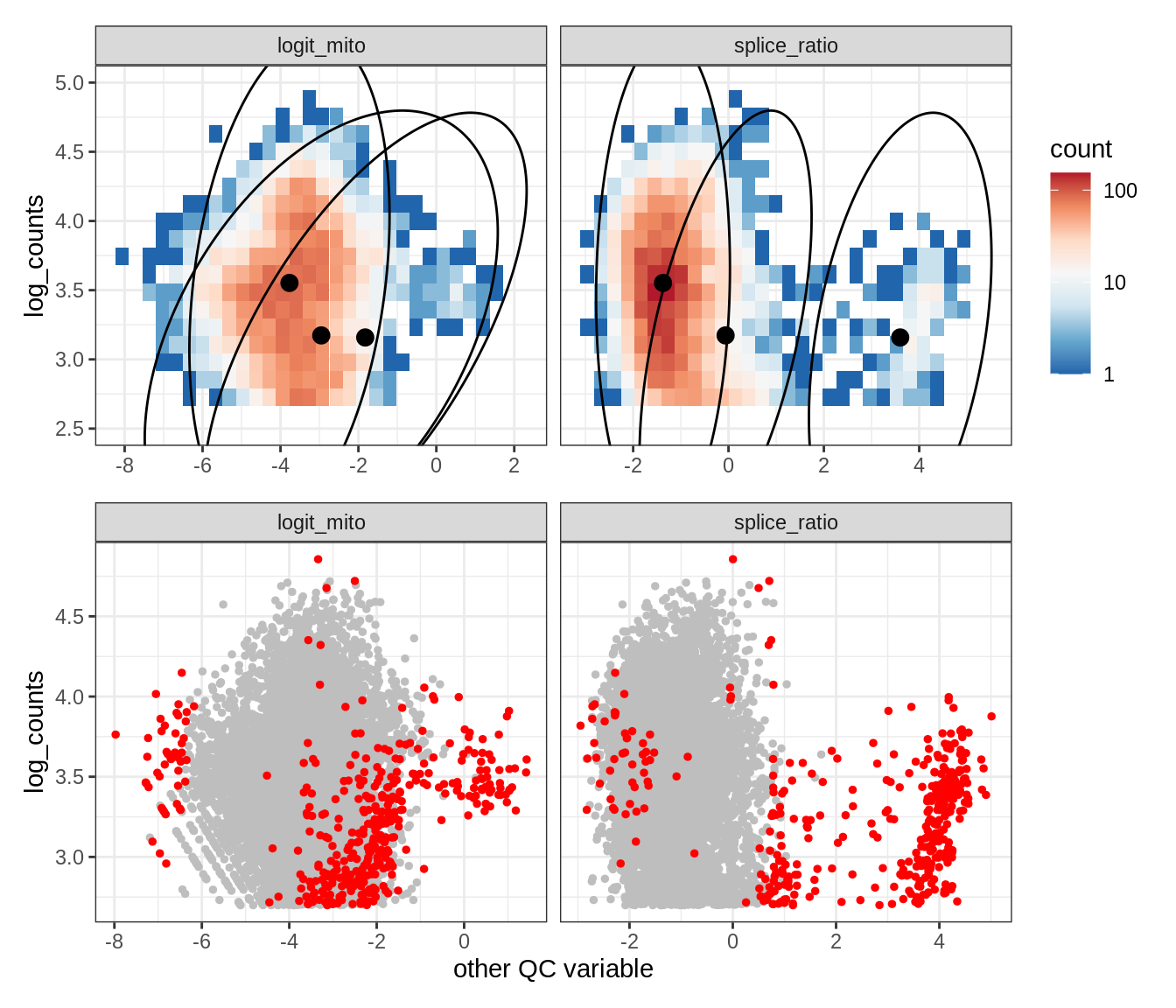
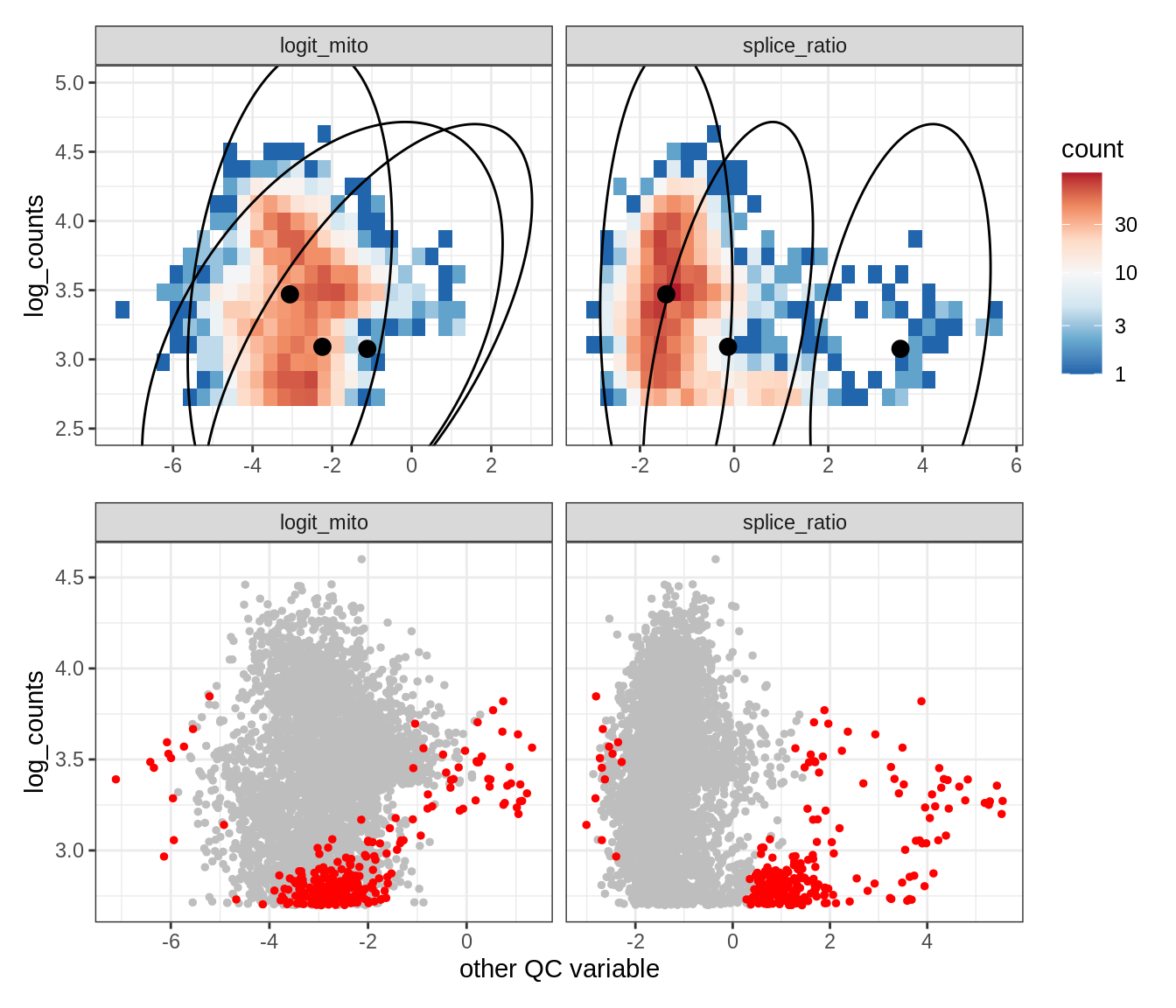
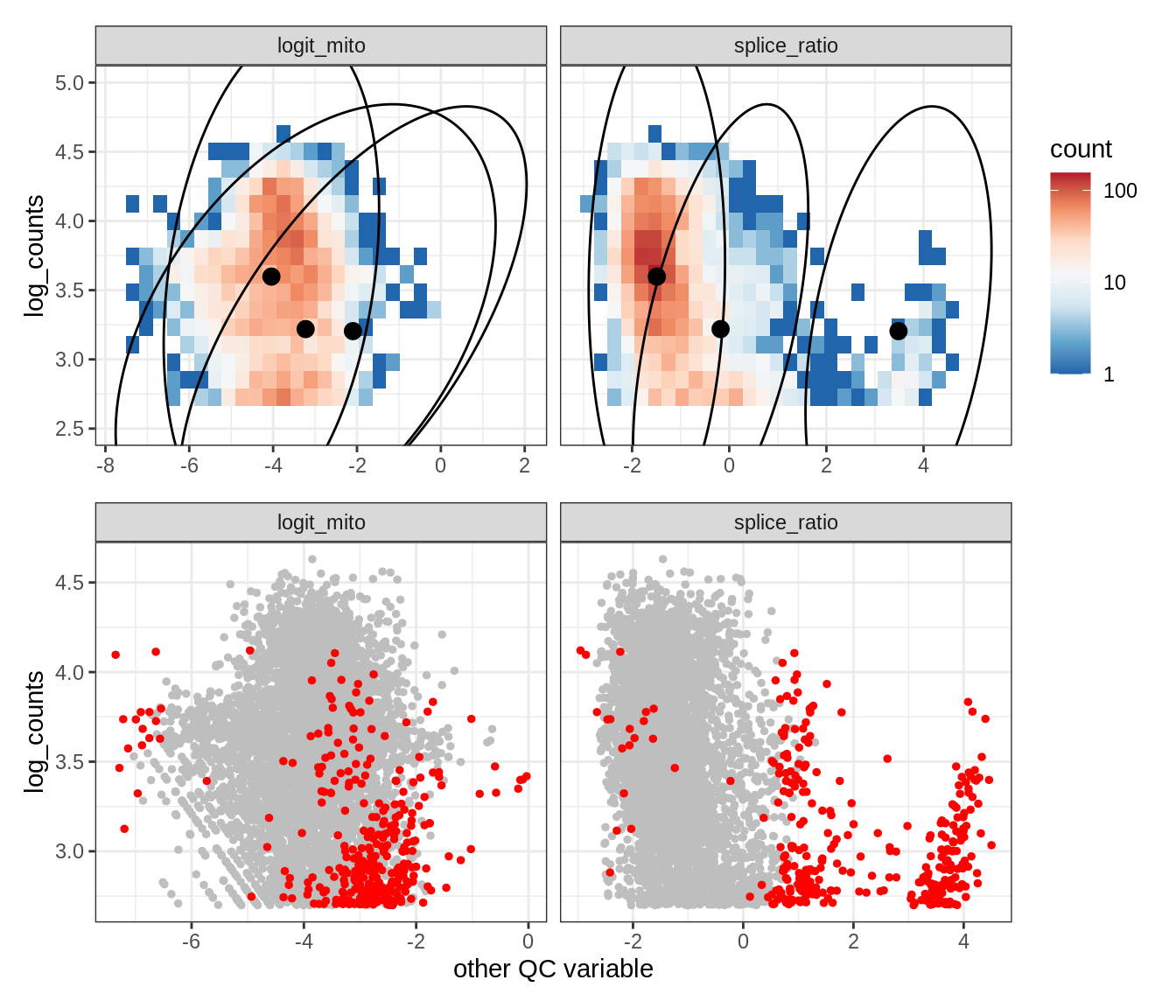
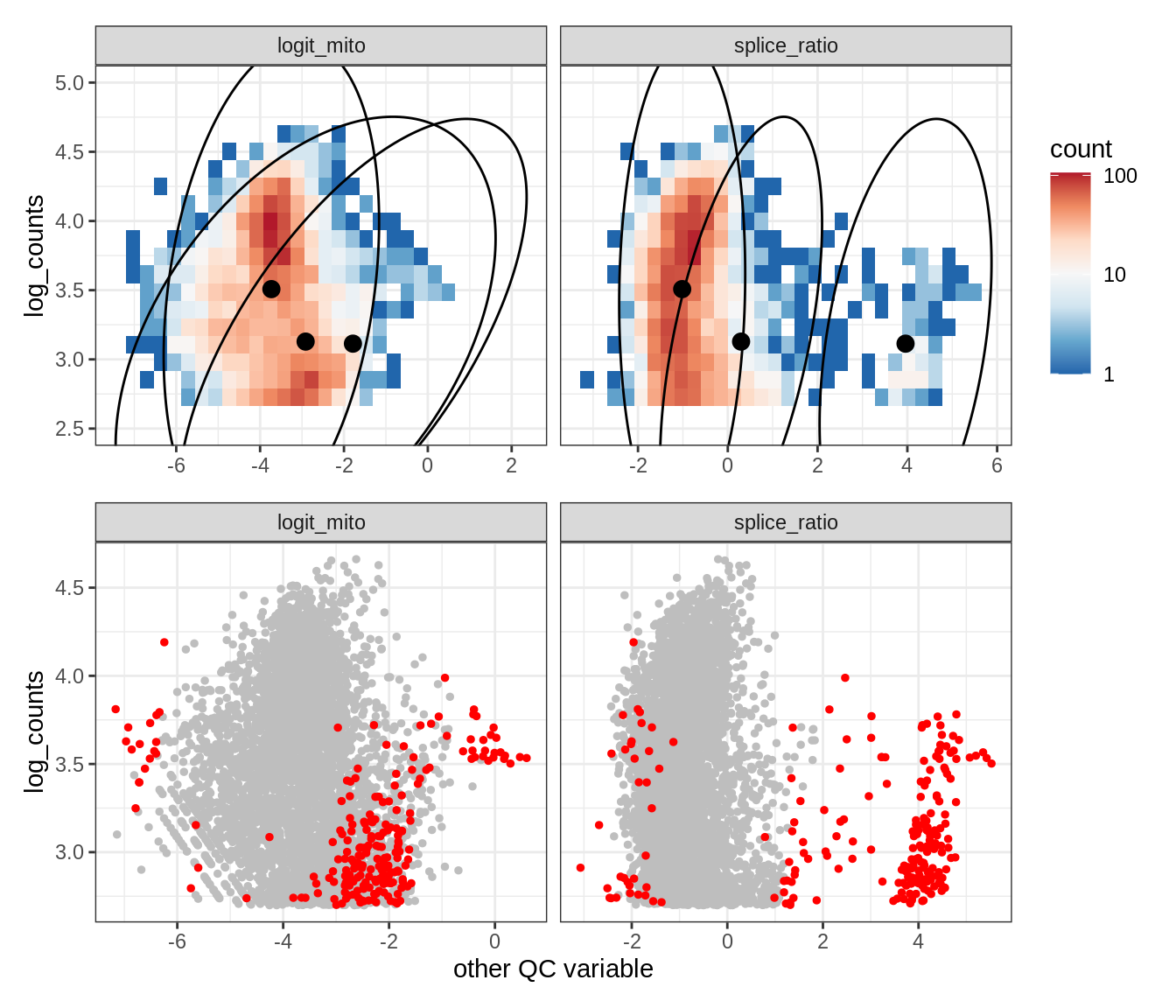
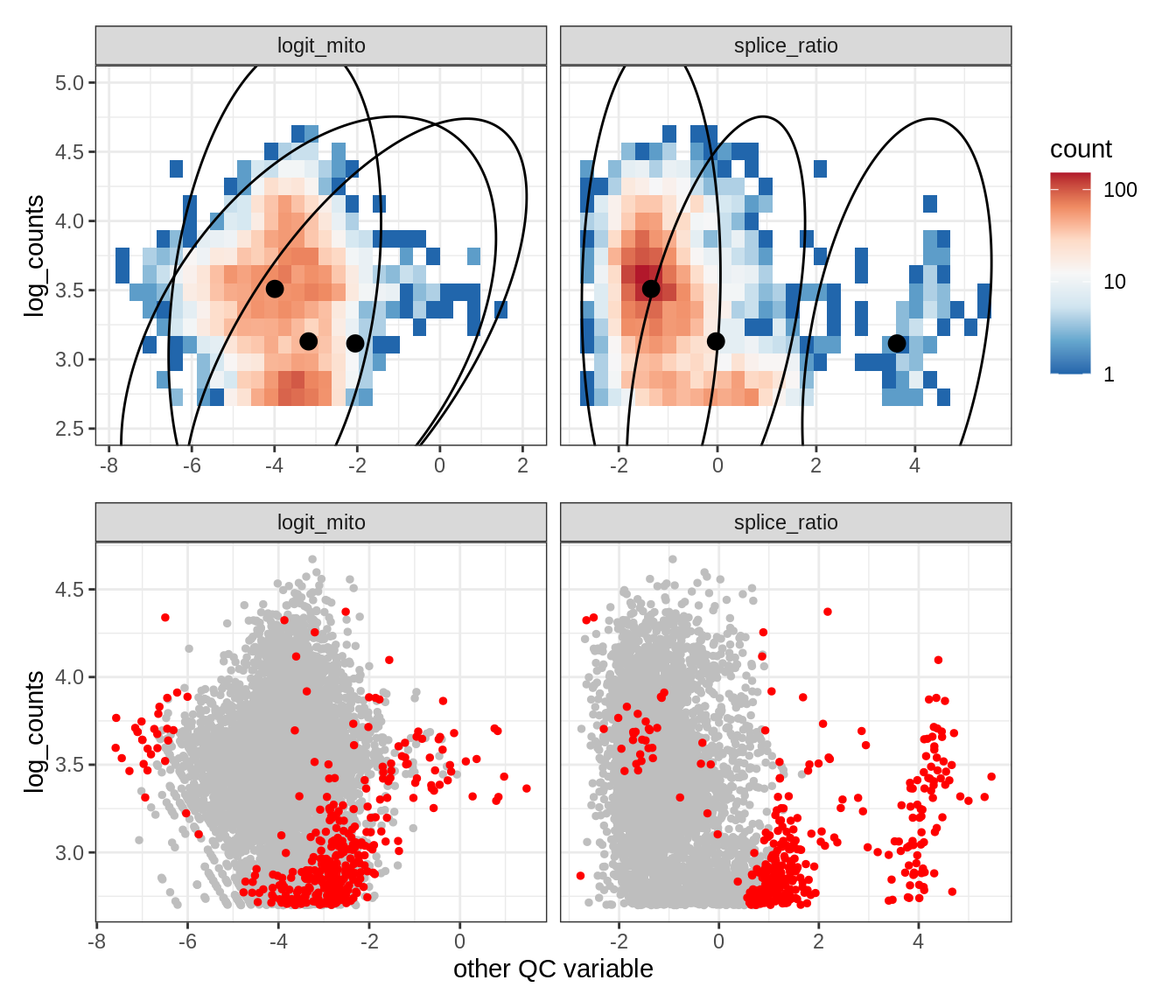
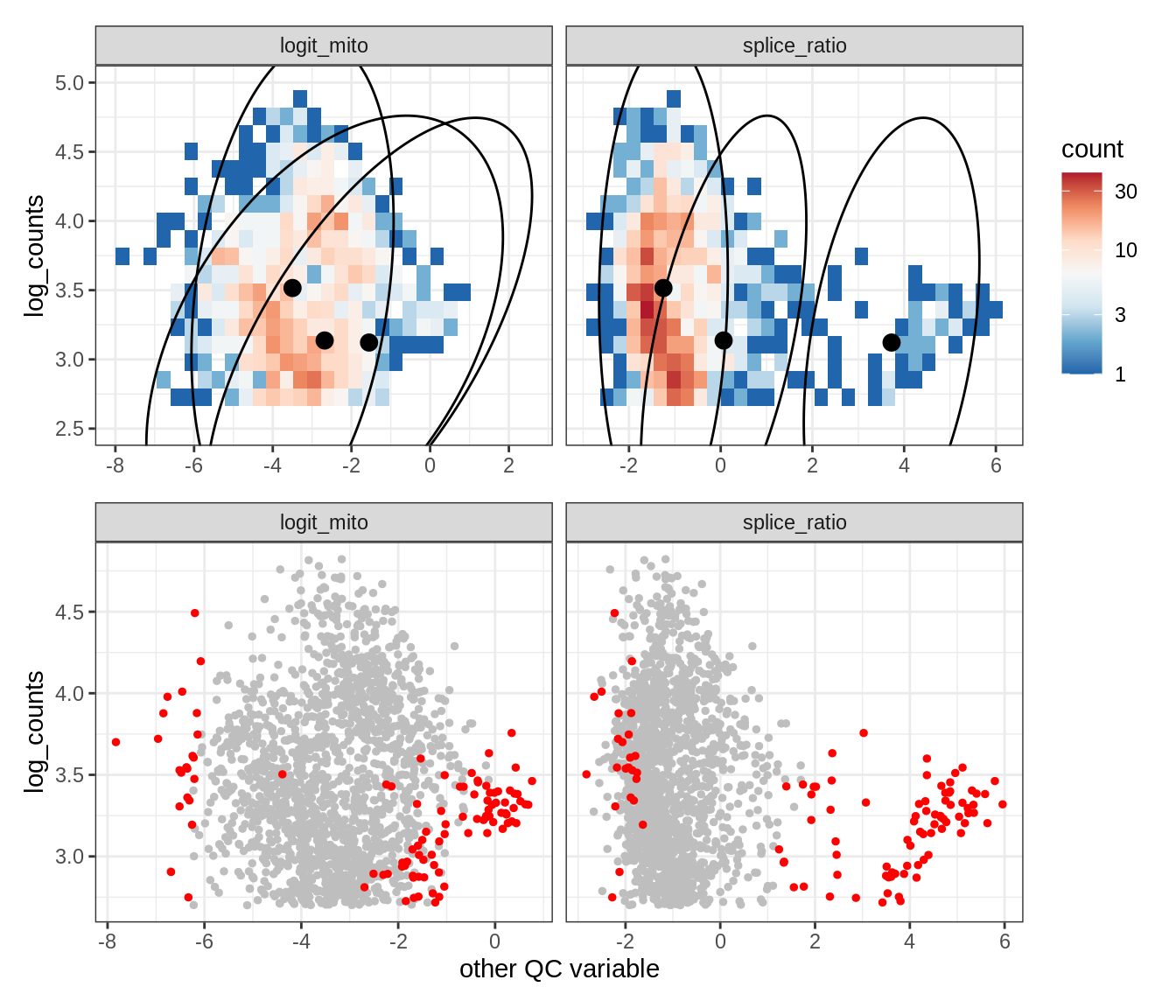
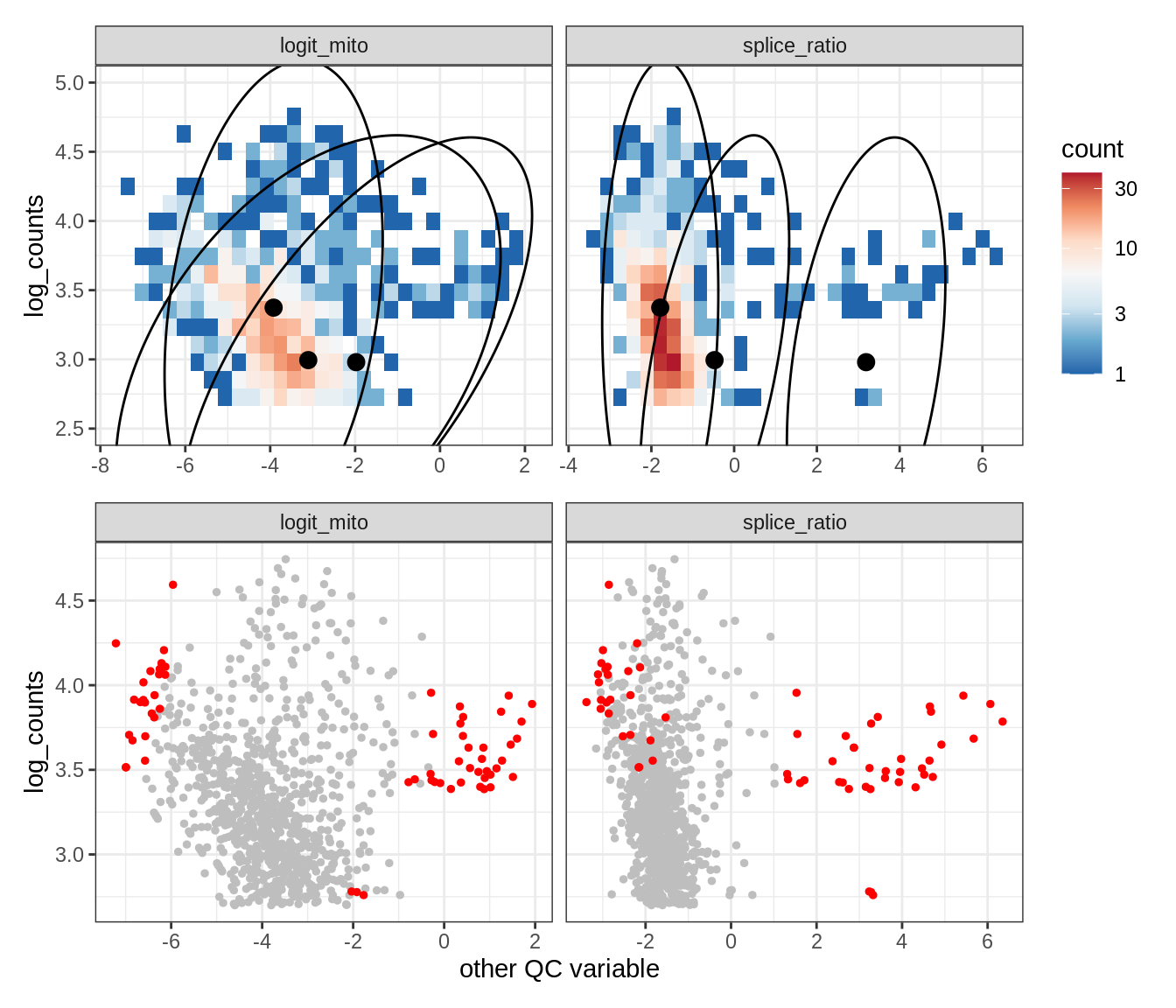
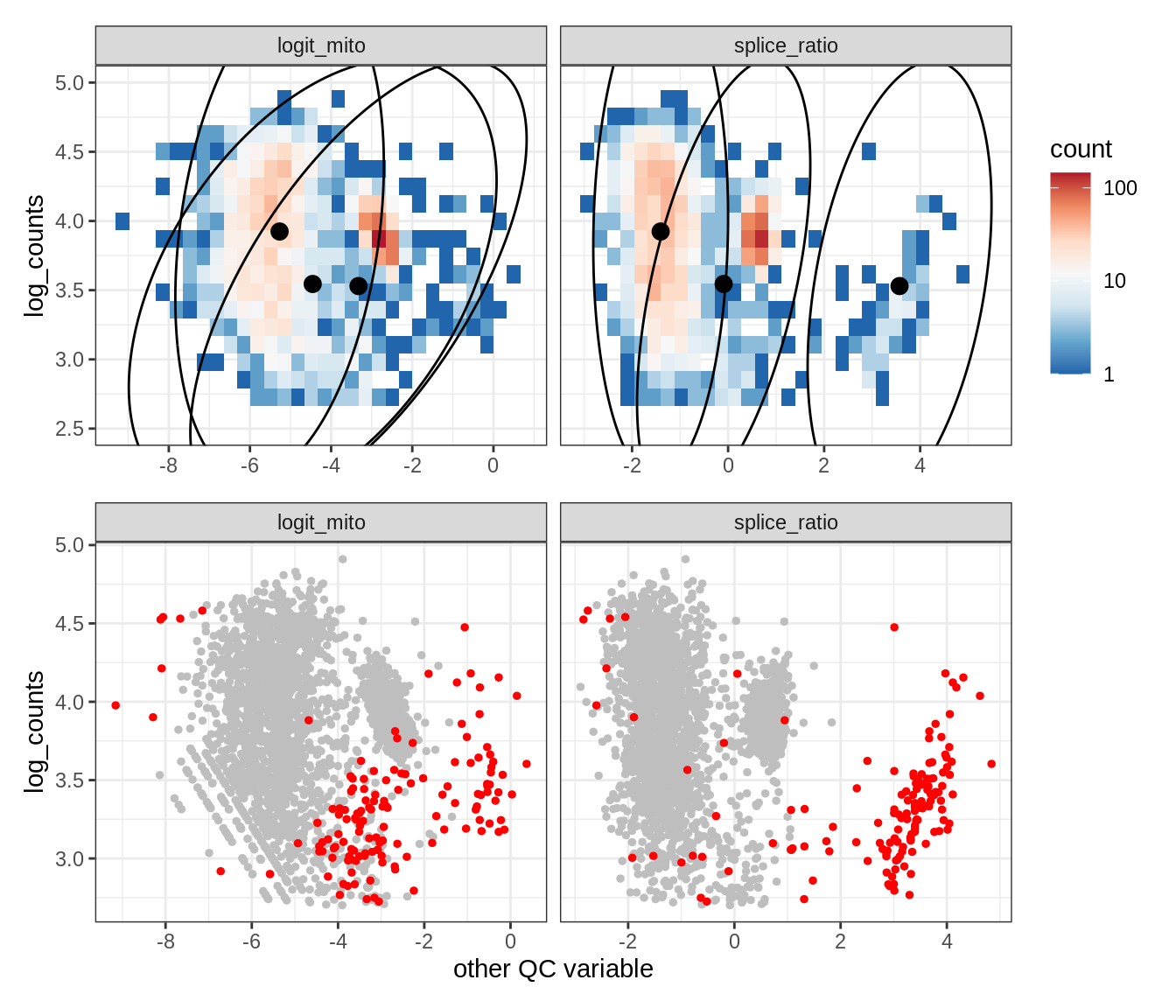
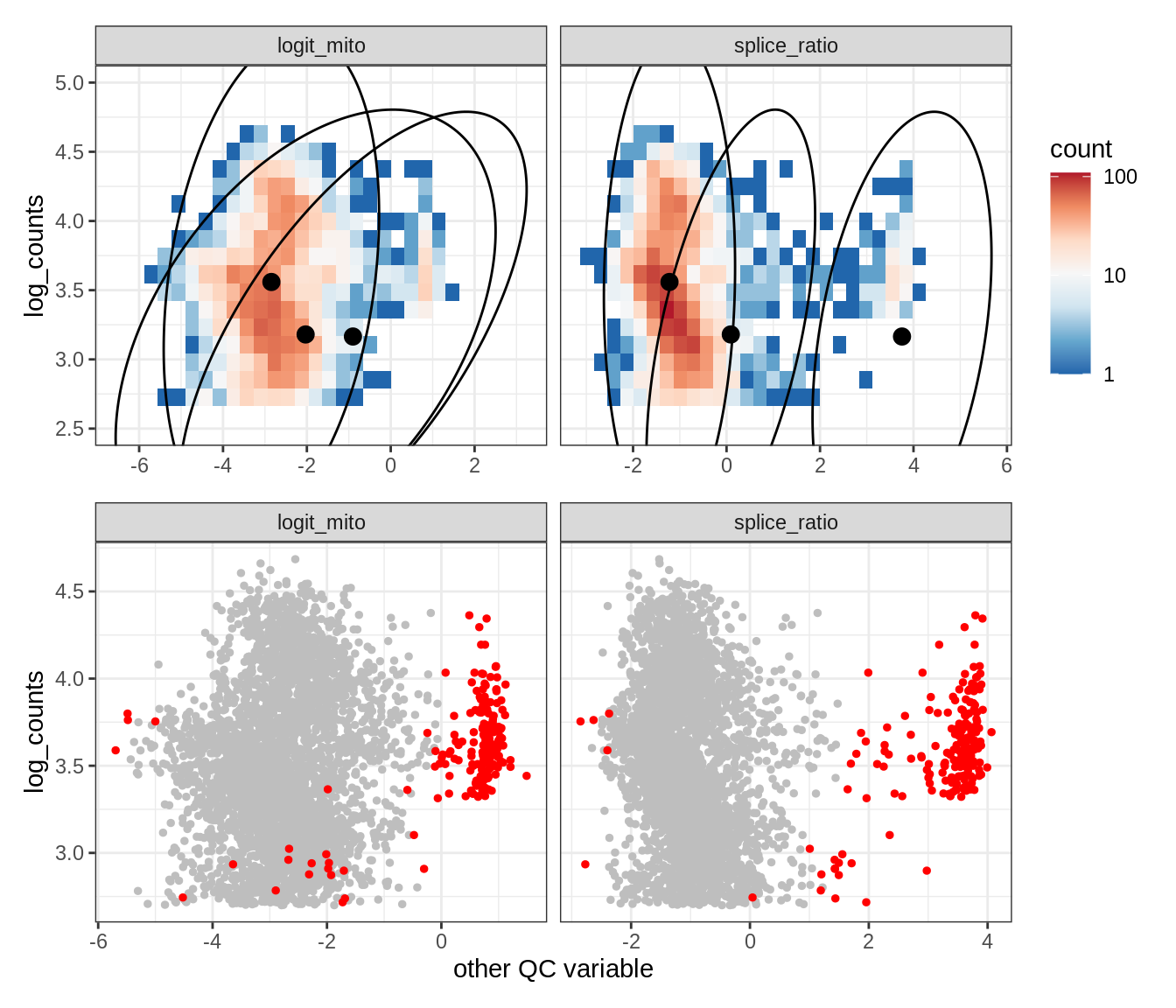
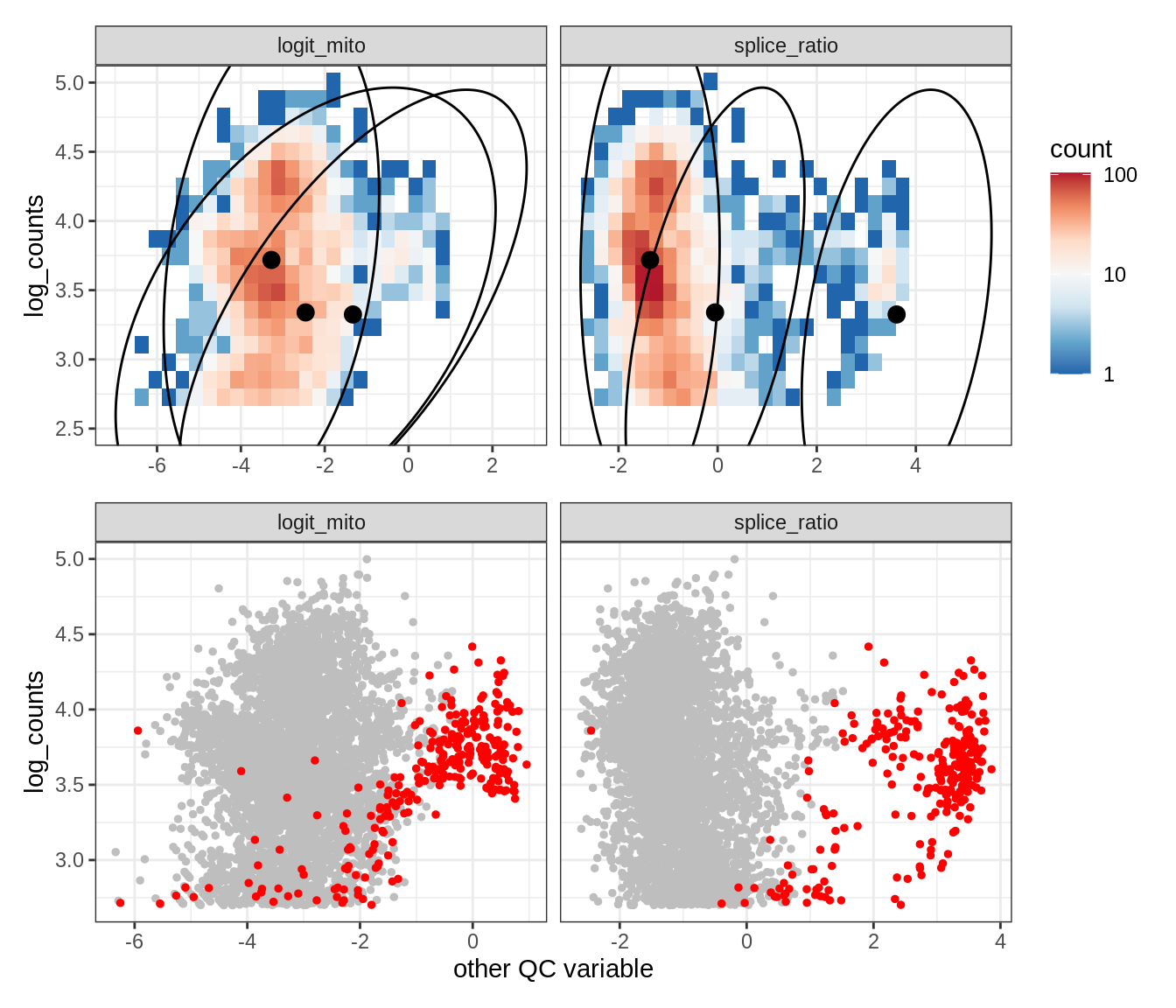
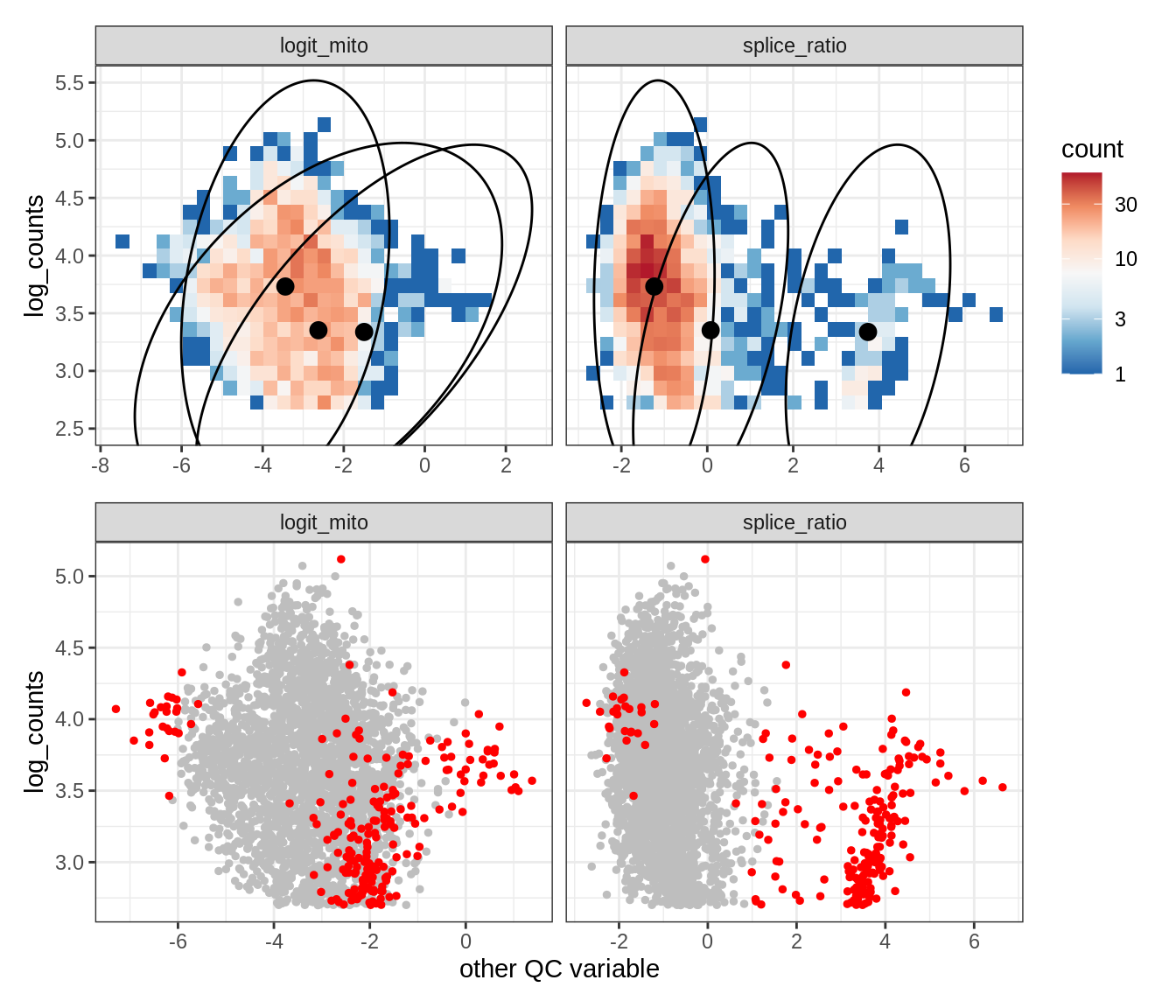
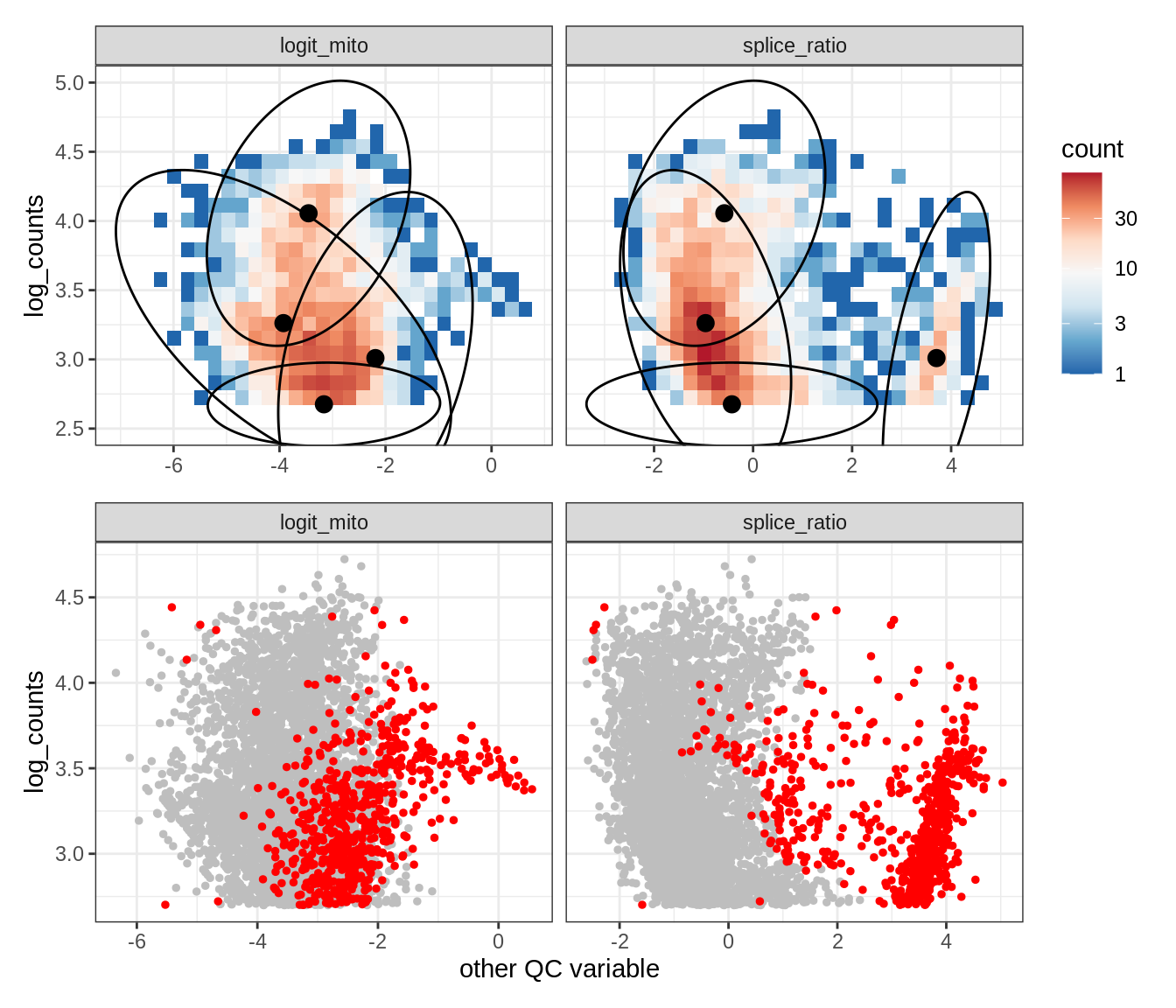
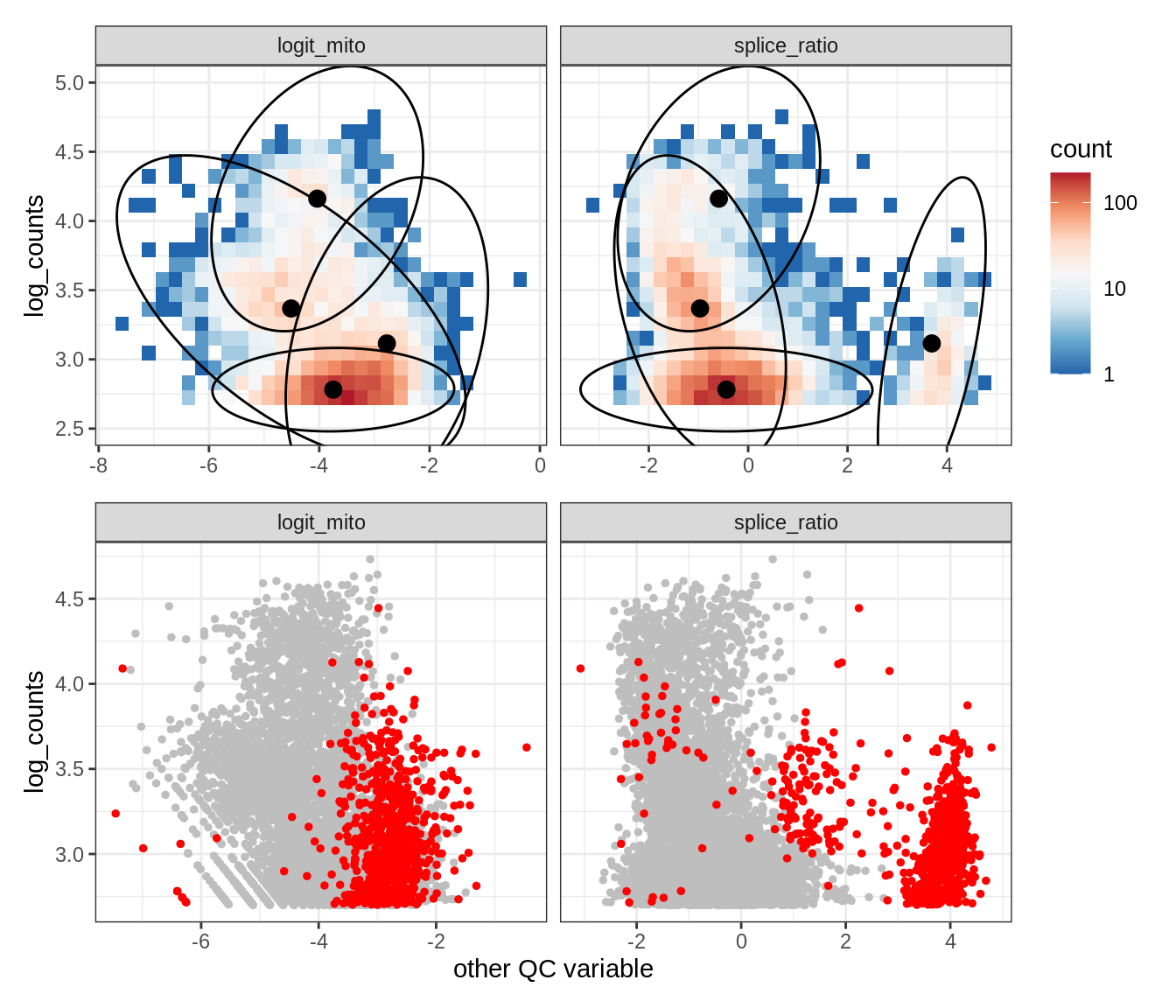
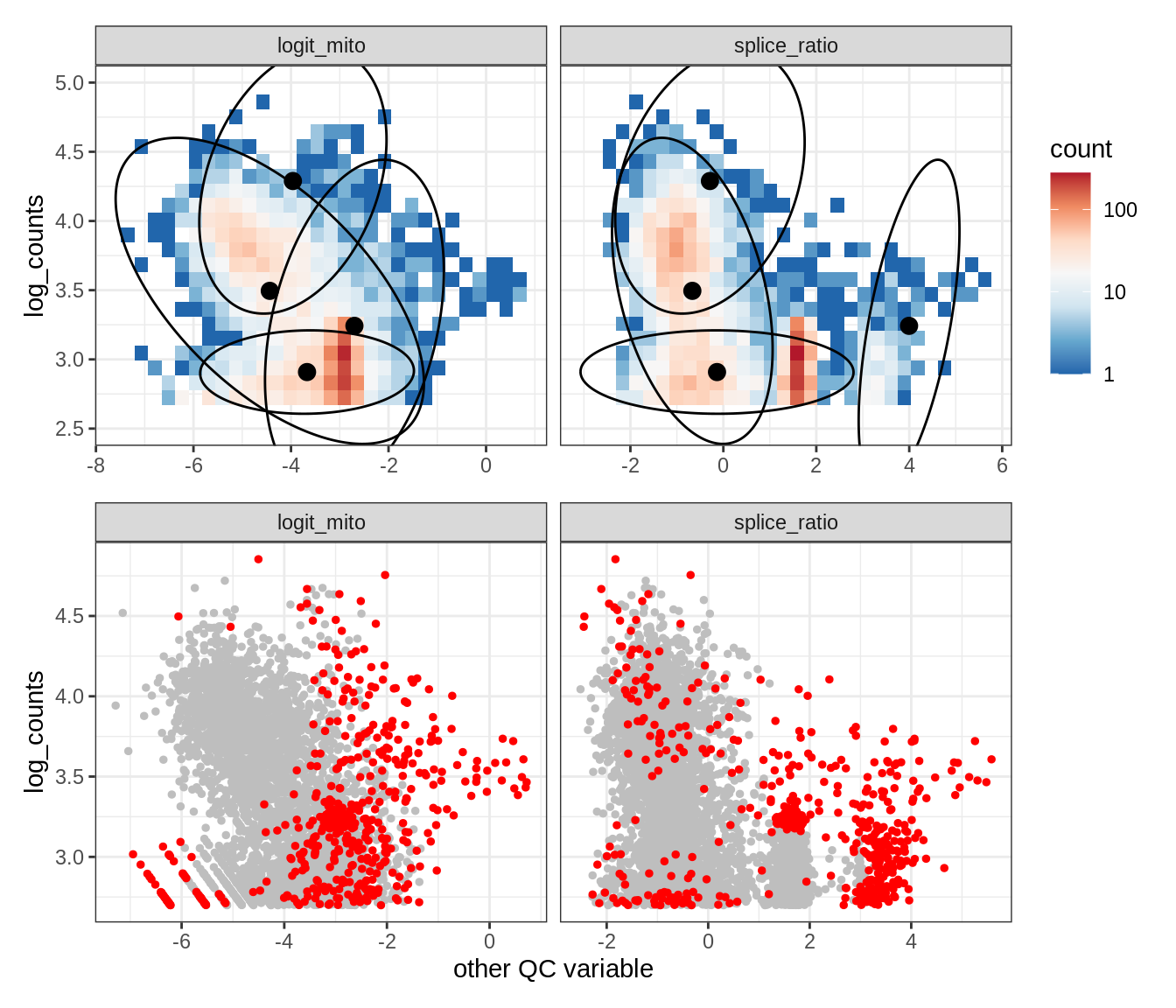
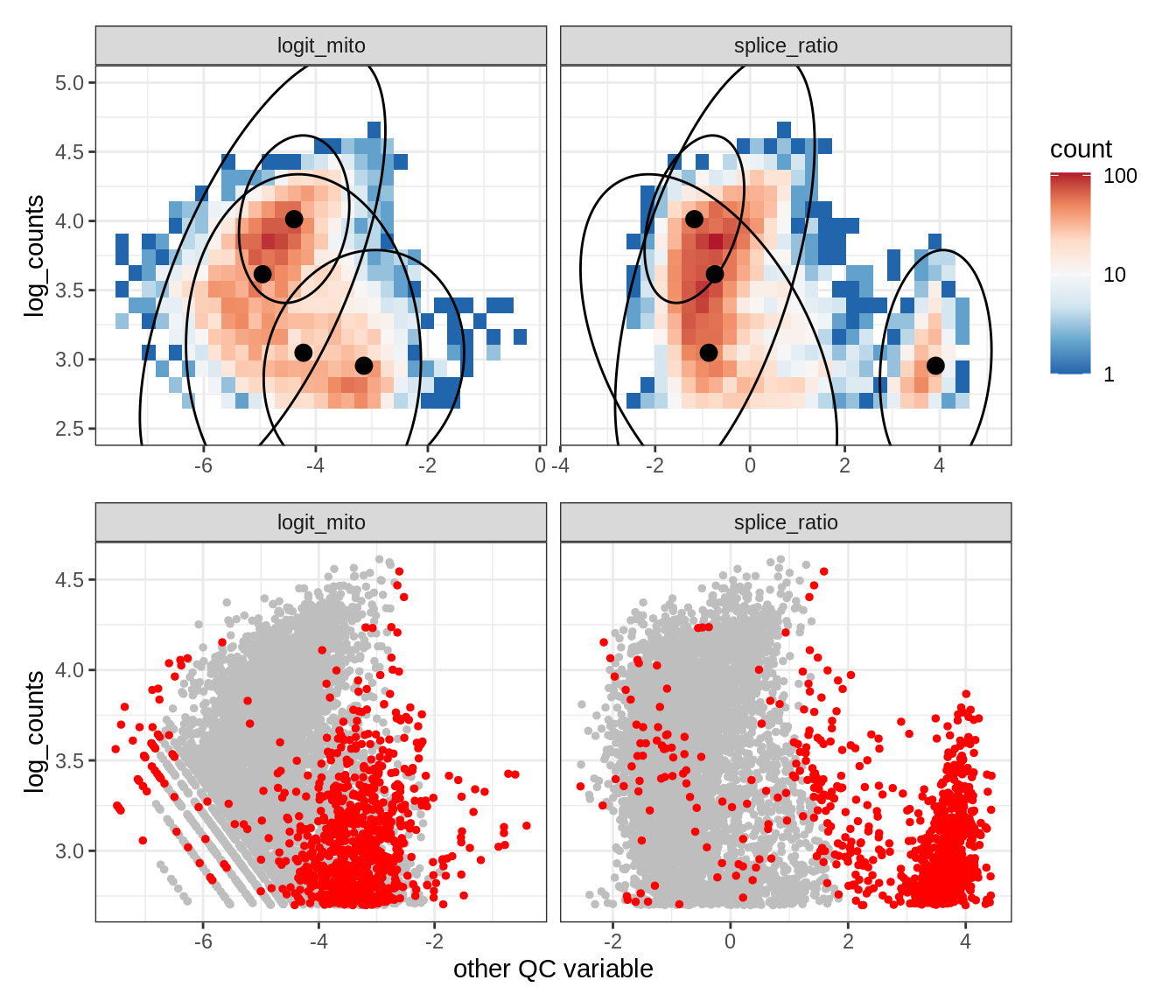
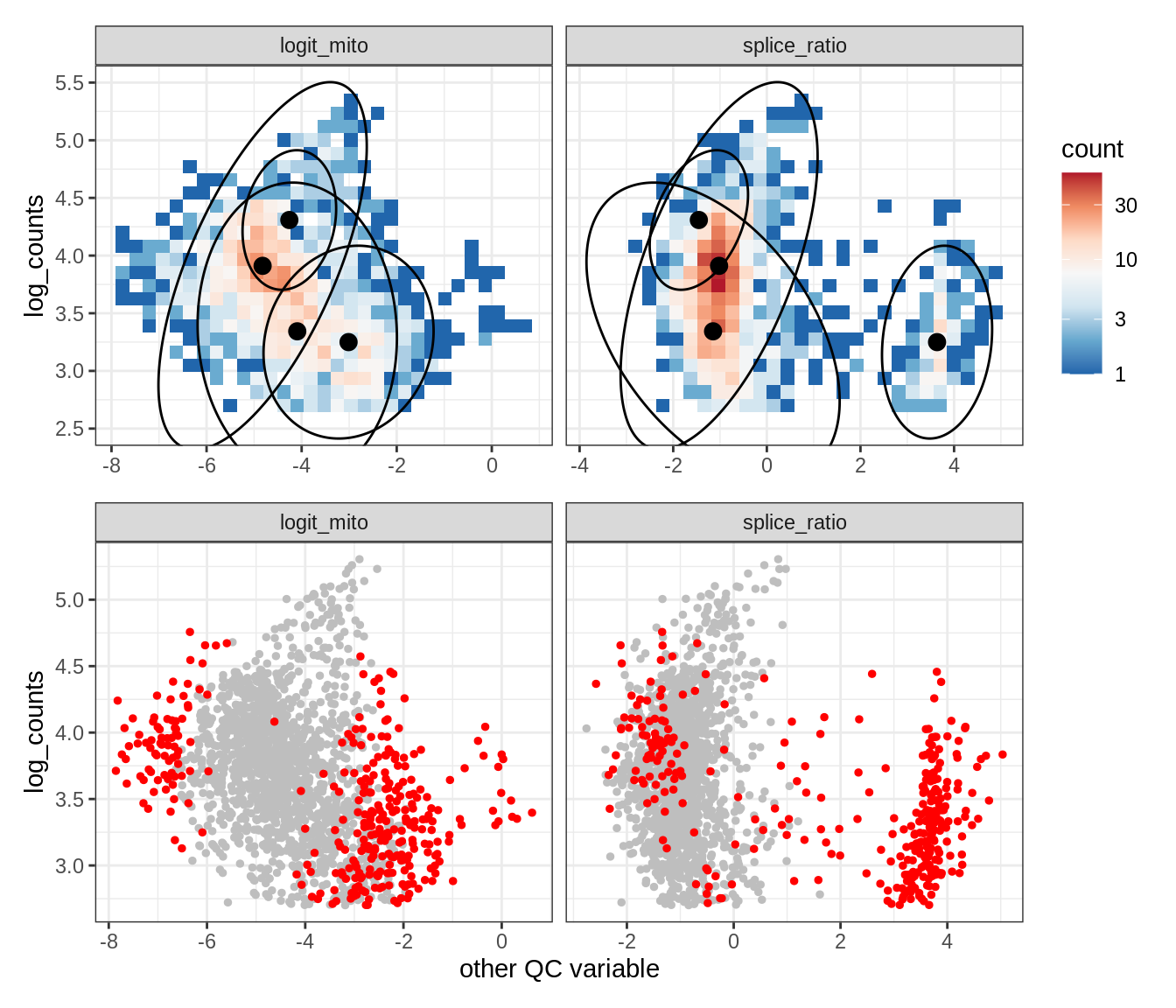
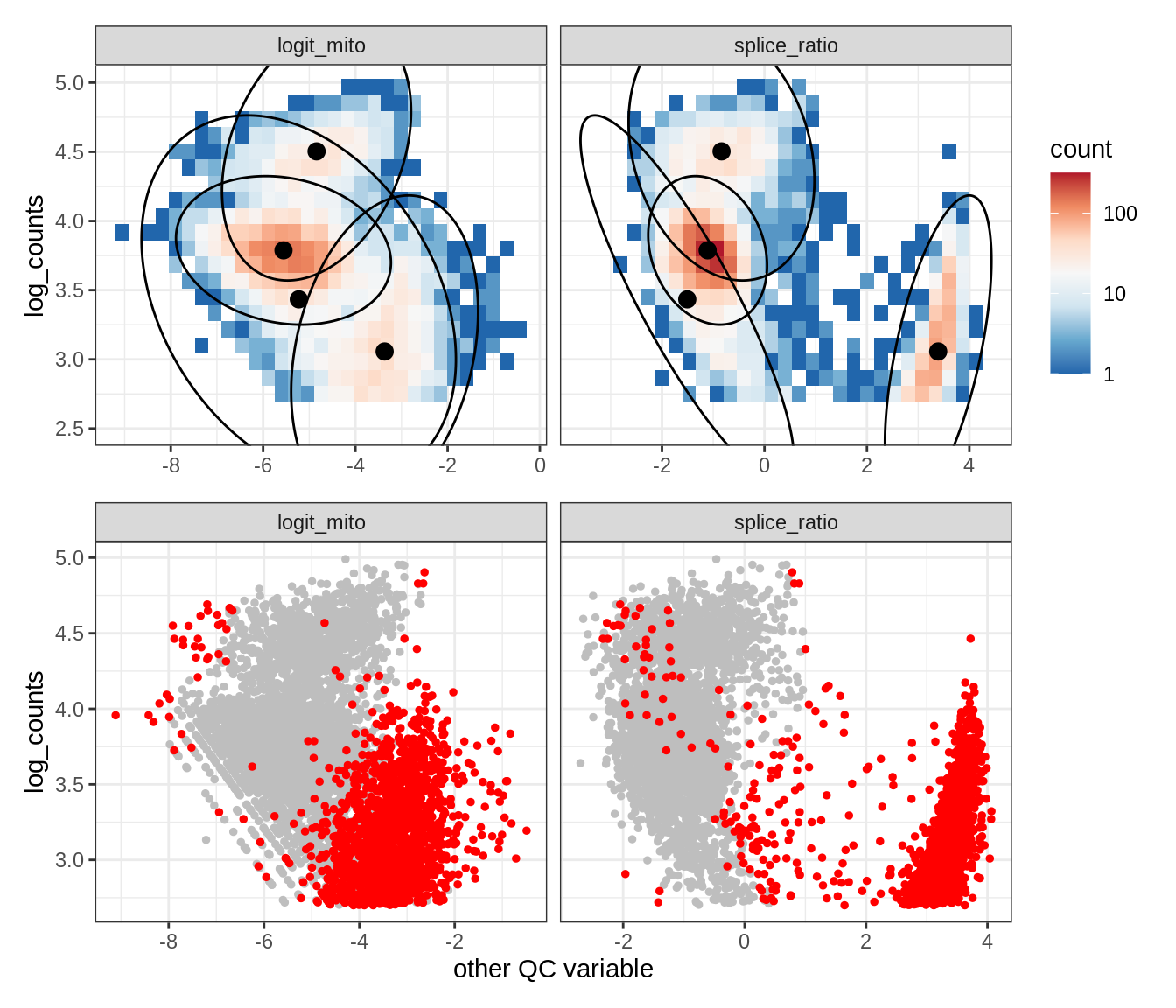
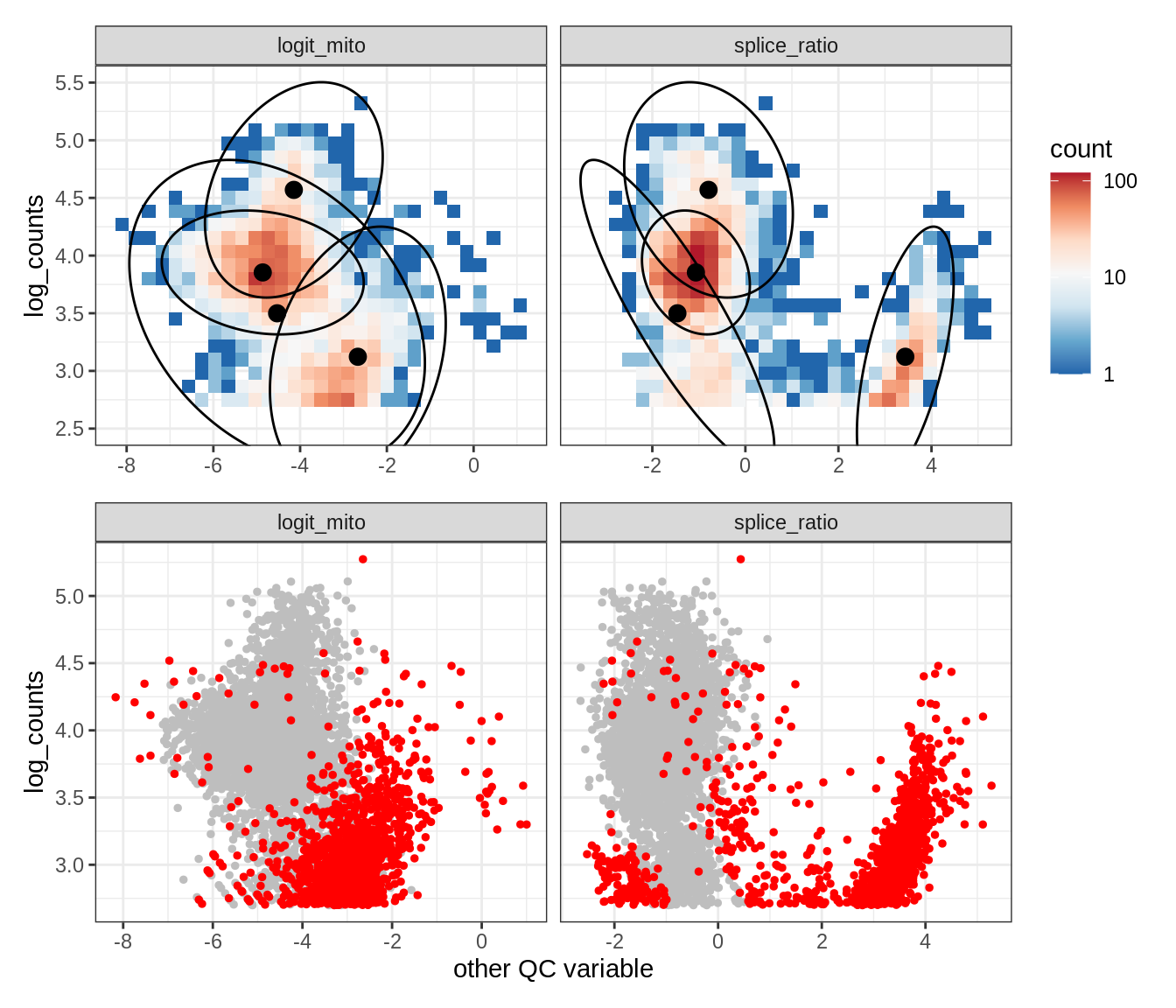
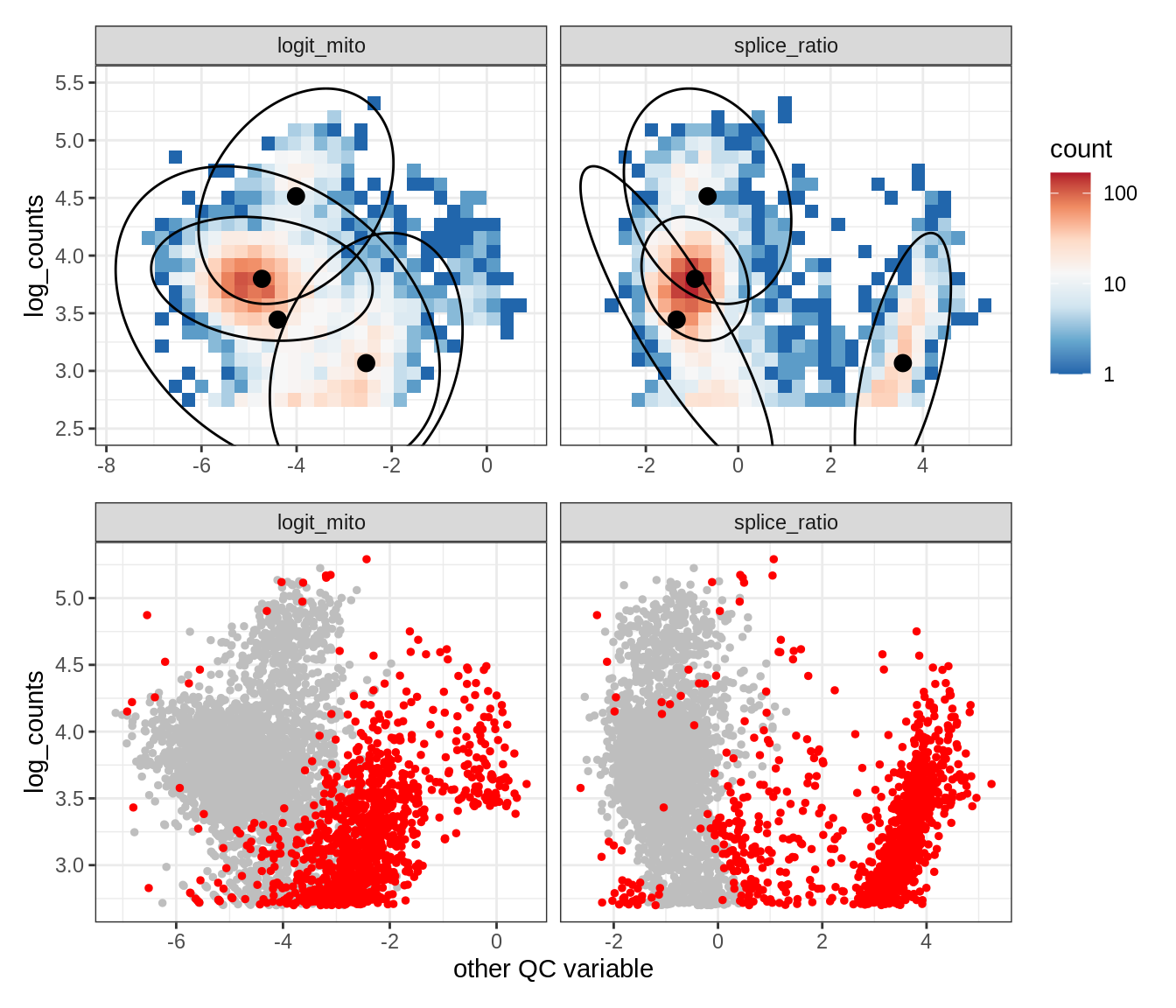
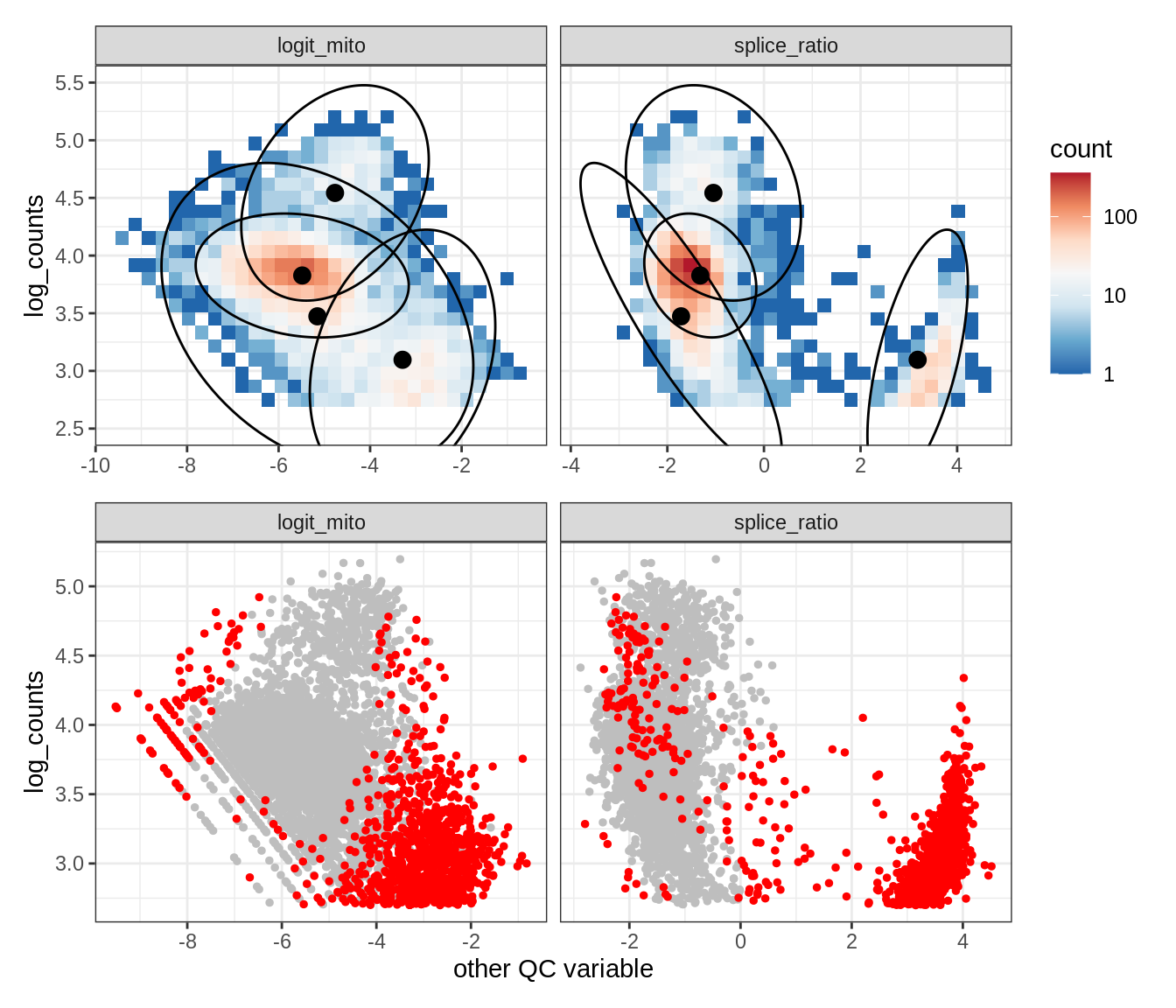
Plot SampleQC model fits and outliers over QC biaxials
group_list = levels(colData(qc_obj)$group_id)
for (g in group_list) {
cat('### ', g, '{.tabset}\n')
# which samples?
samples_g = sort(colData(qc_obj)$sample_id[ colData(qc_obj)$group_id == g ])
for (s in samples_g) {
cat('#### ', s, ' \n')
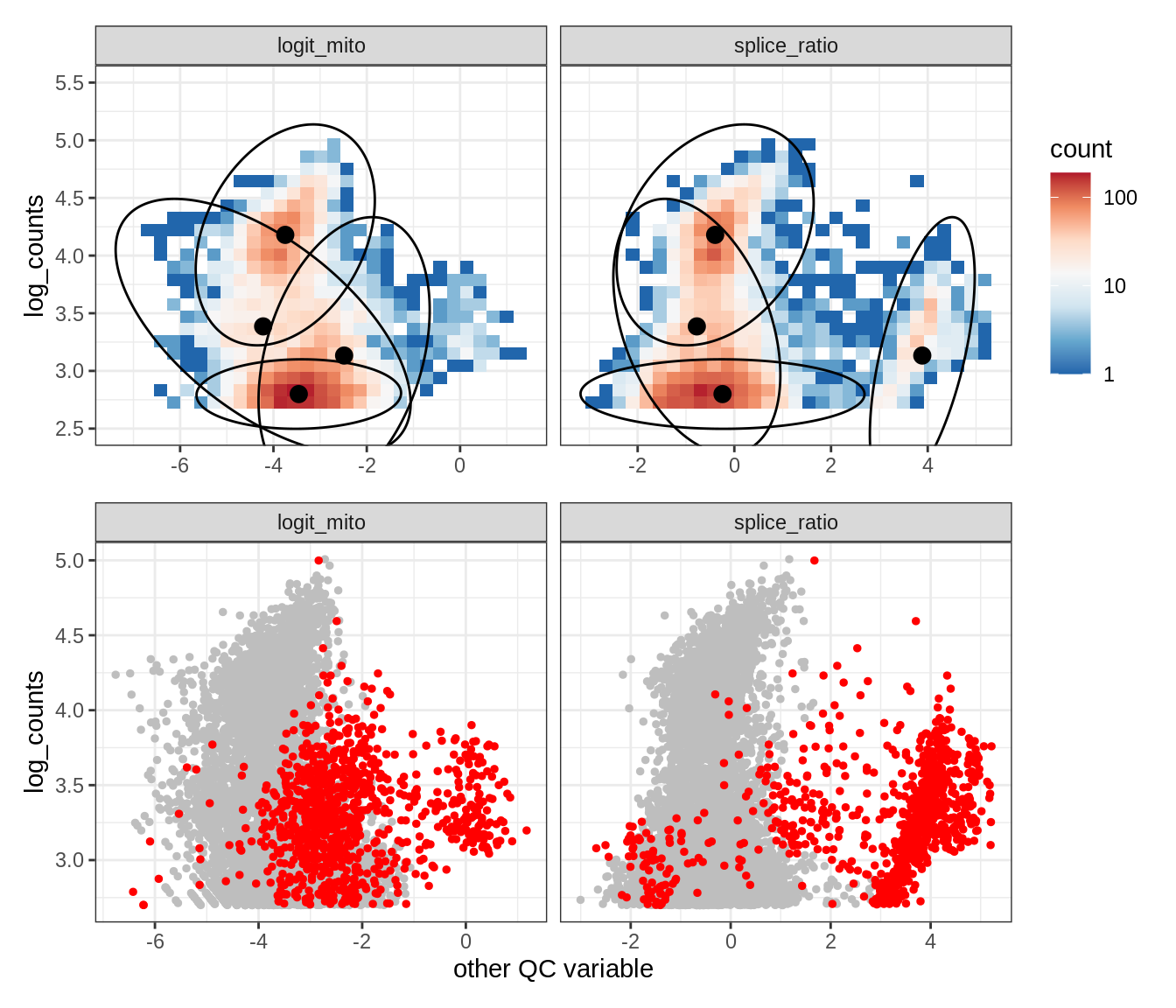
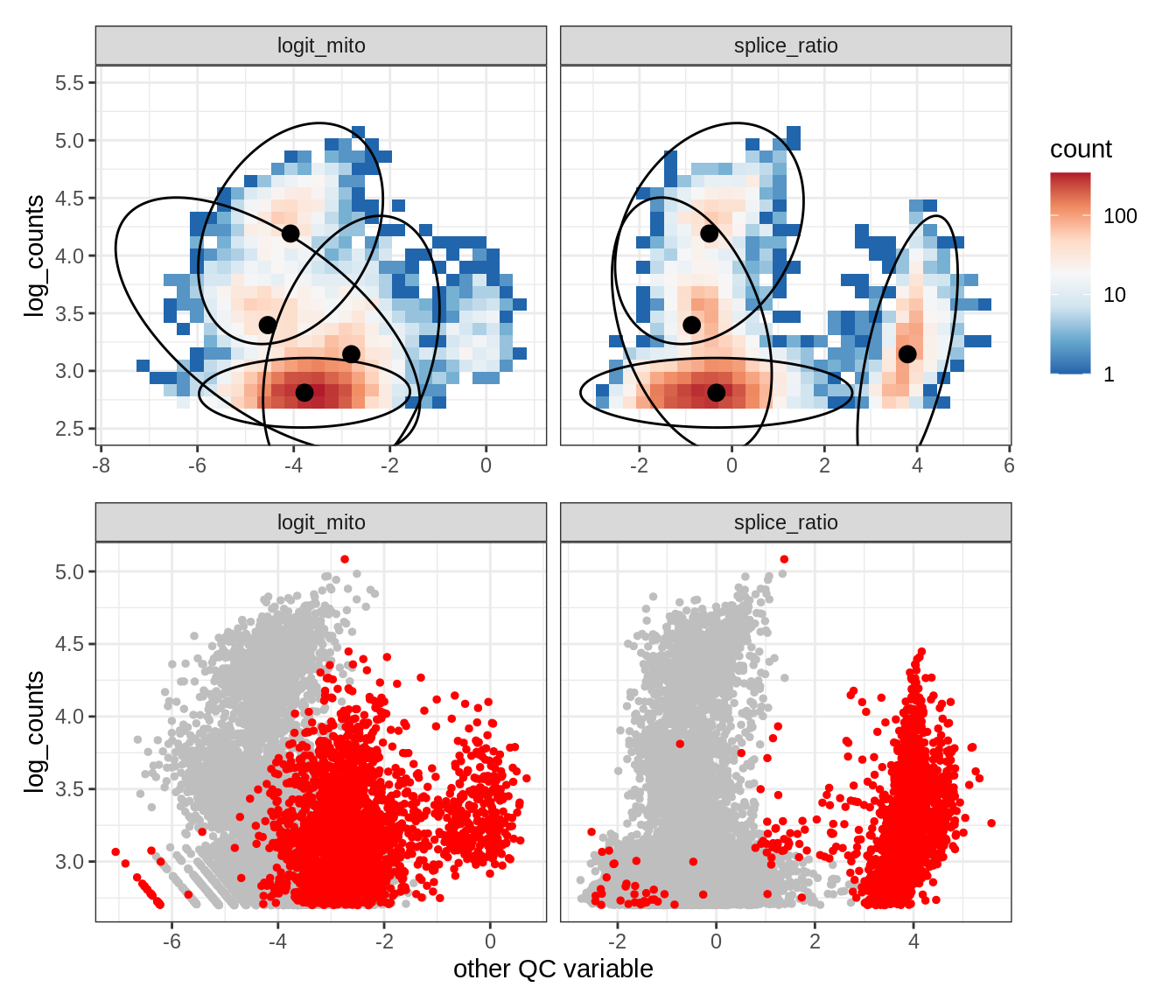
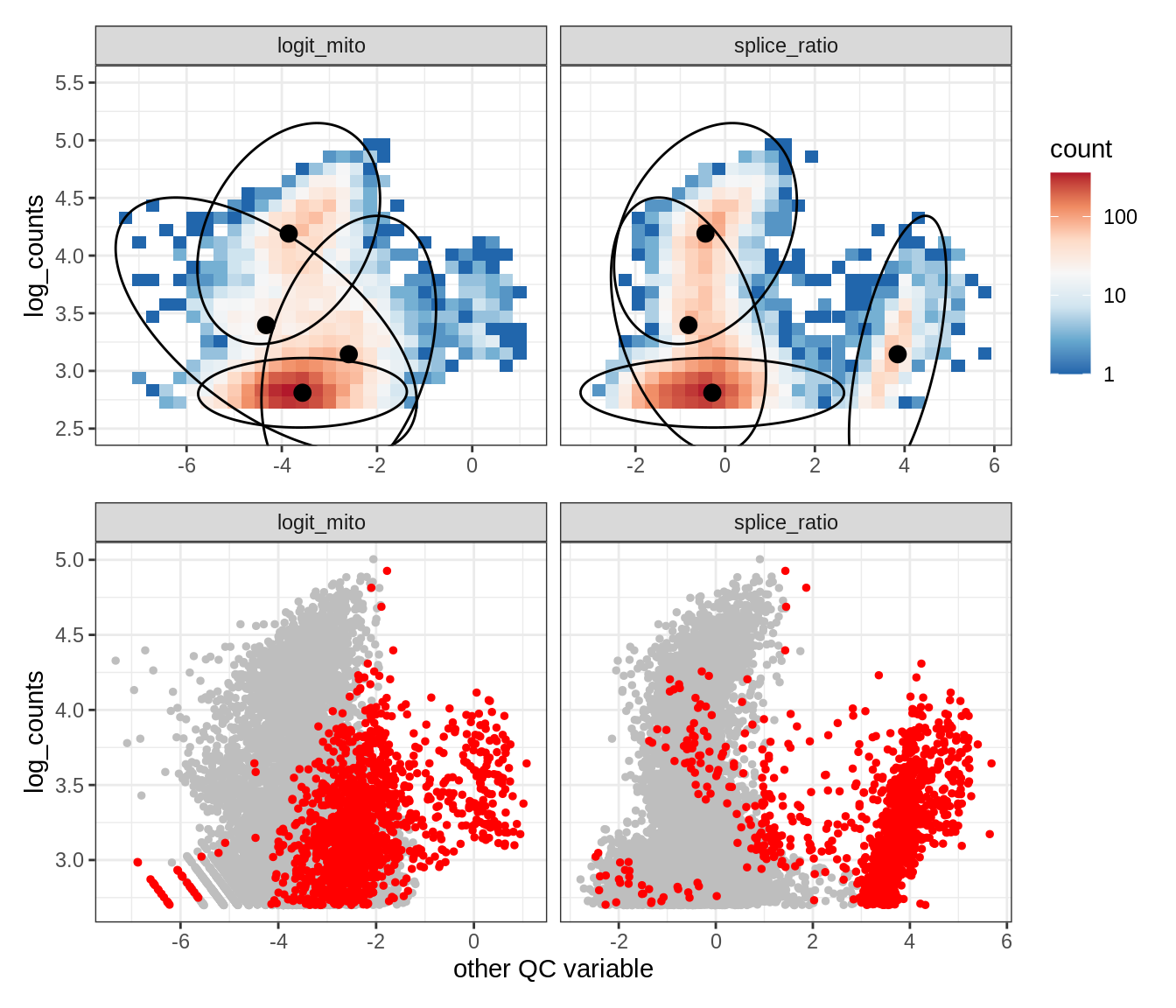
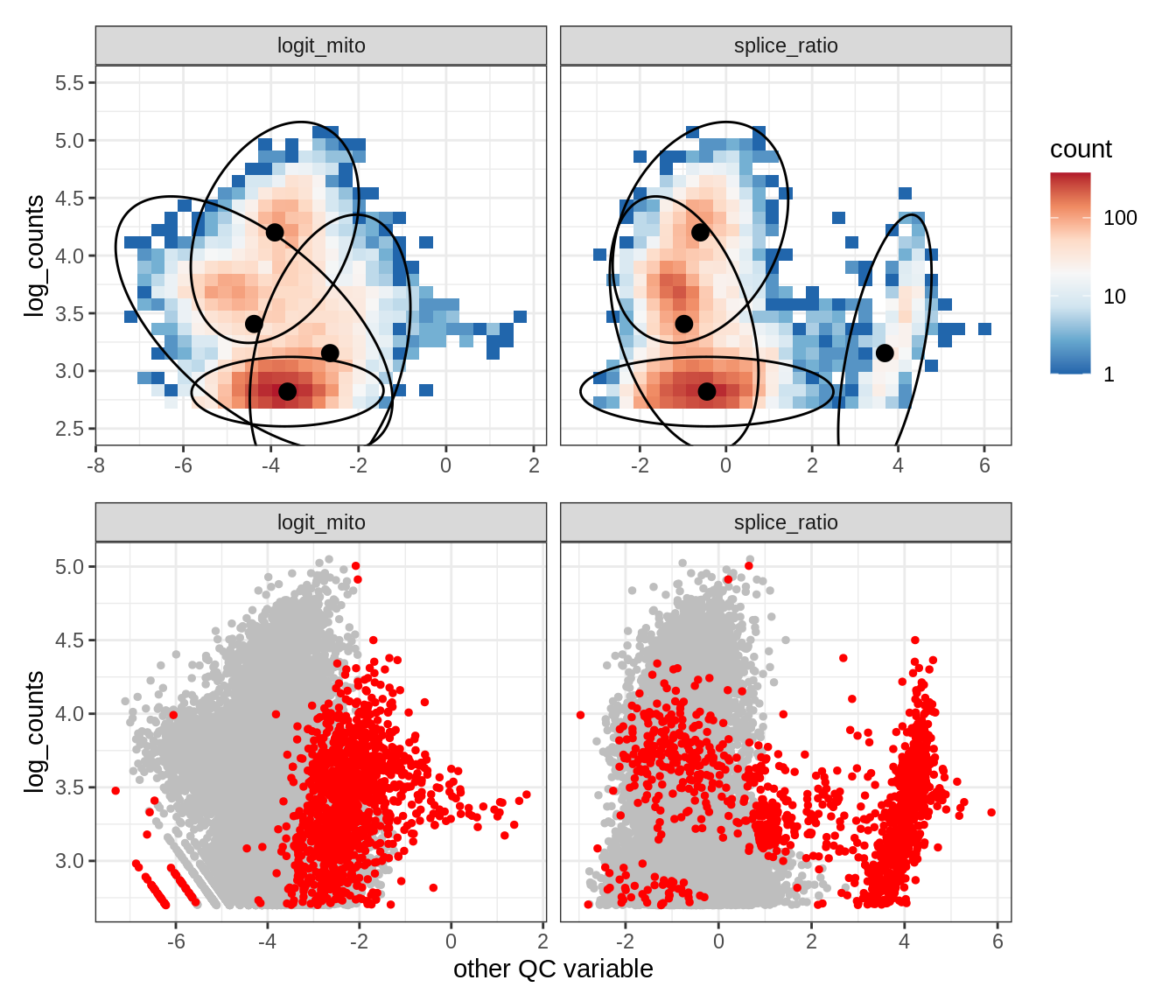
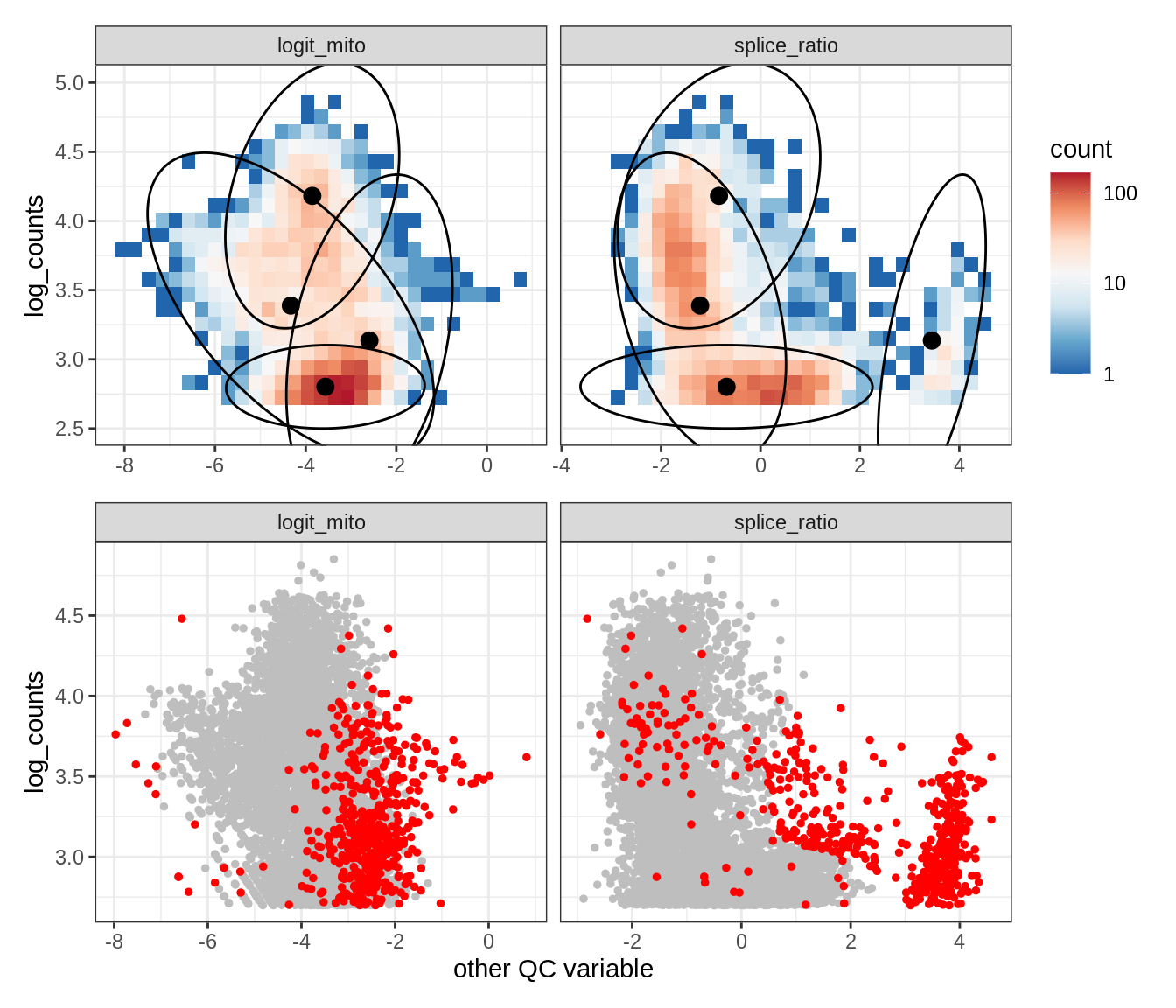
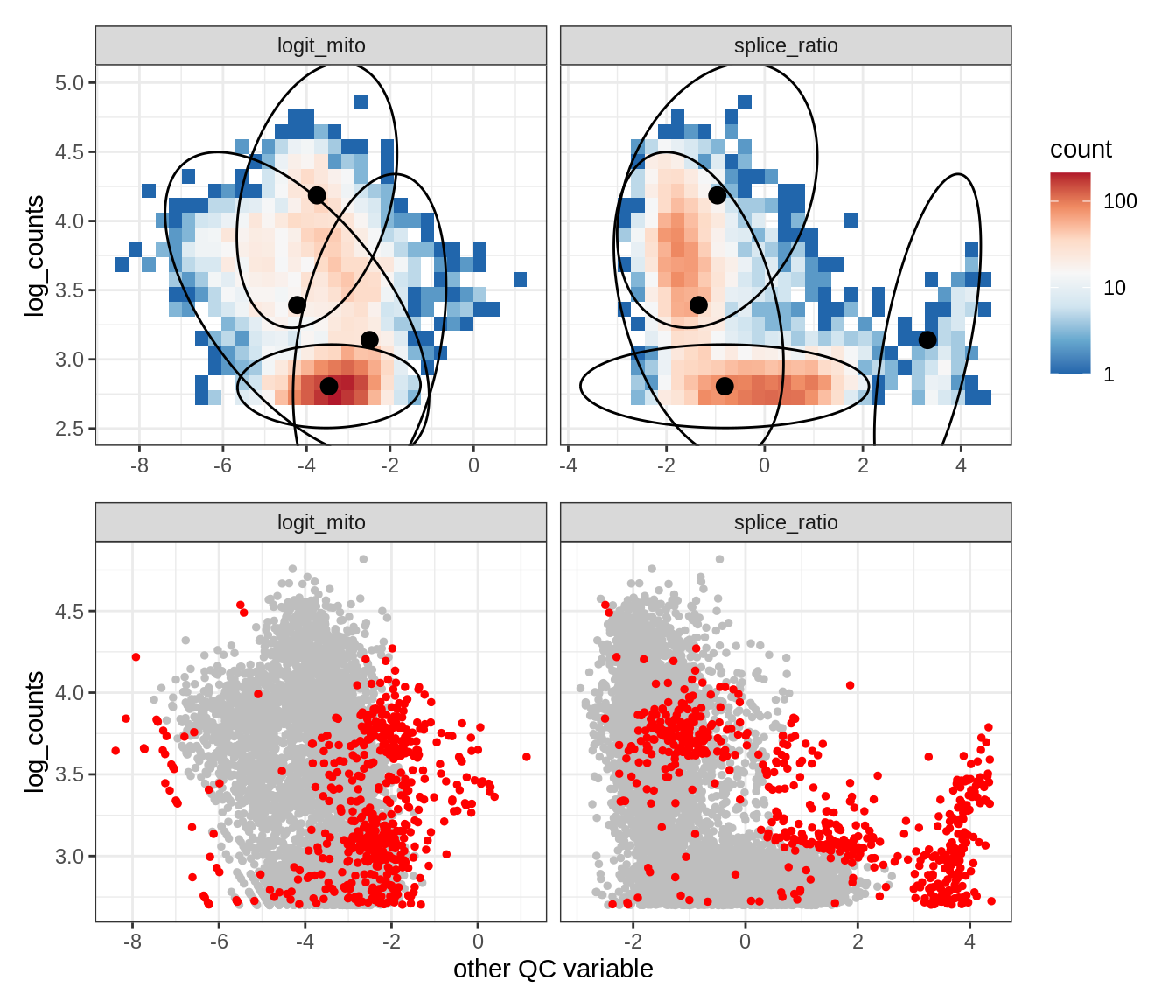
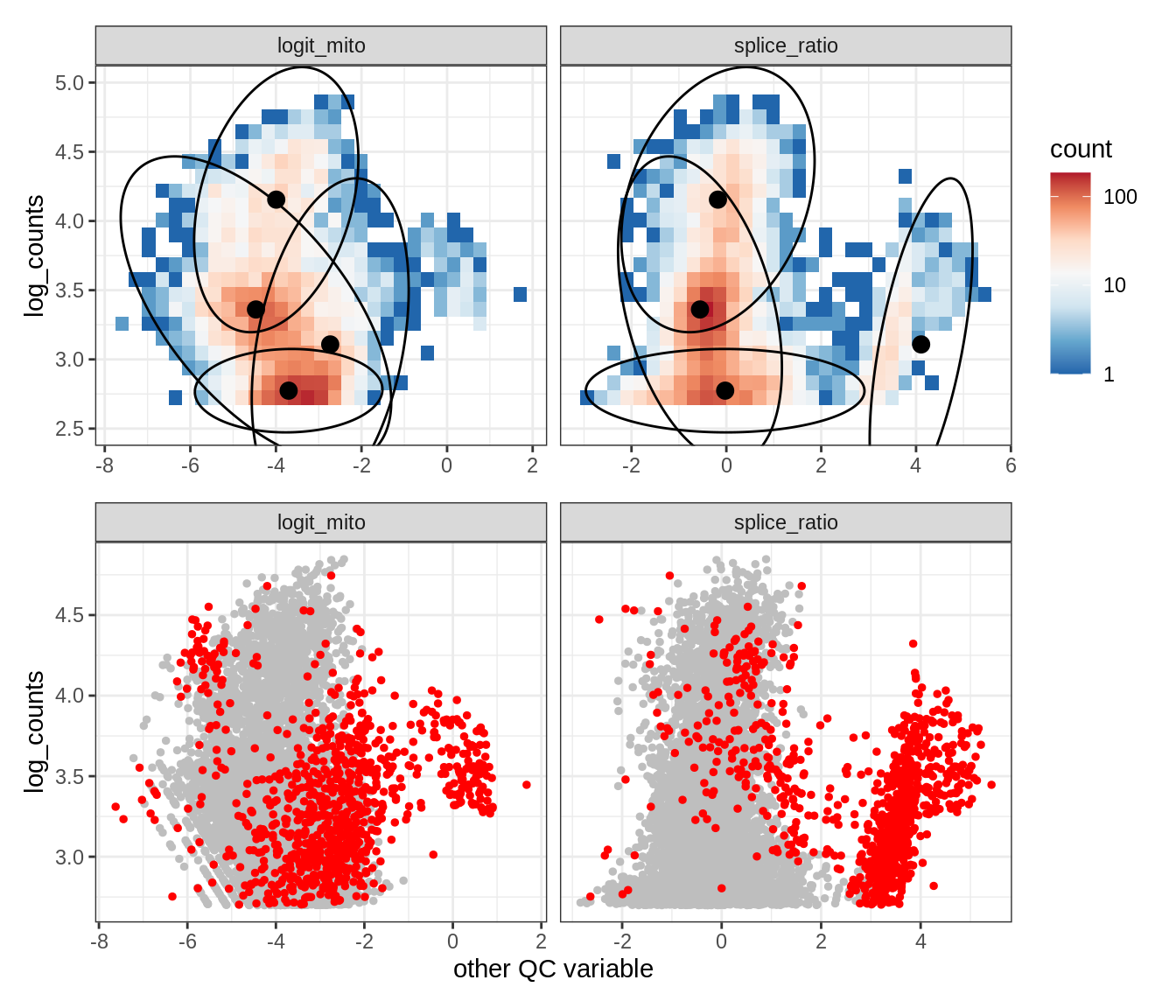
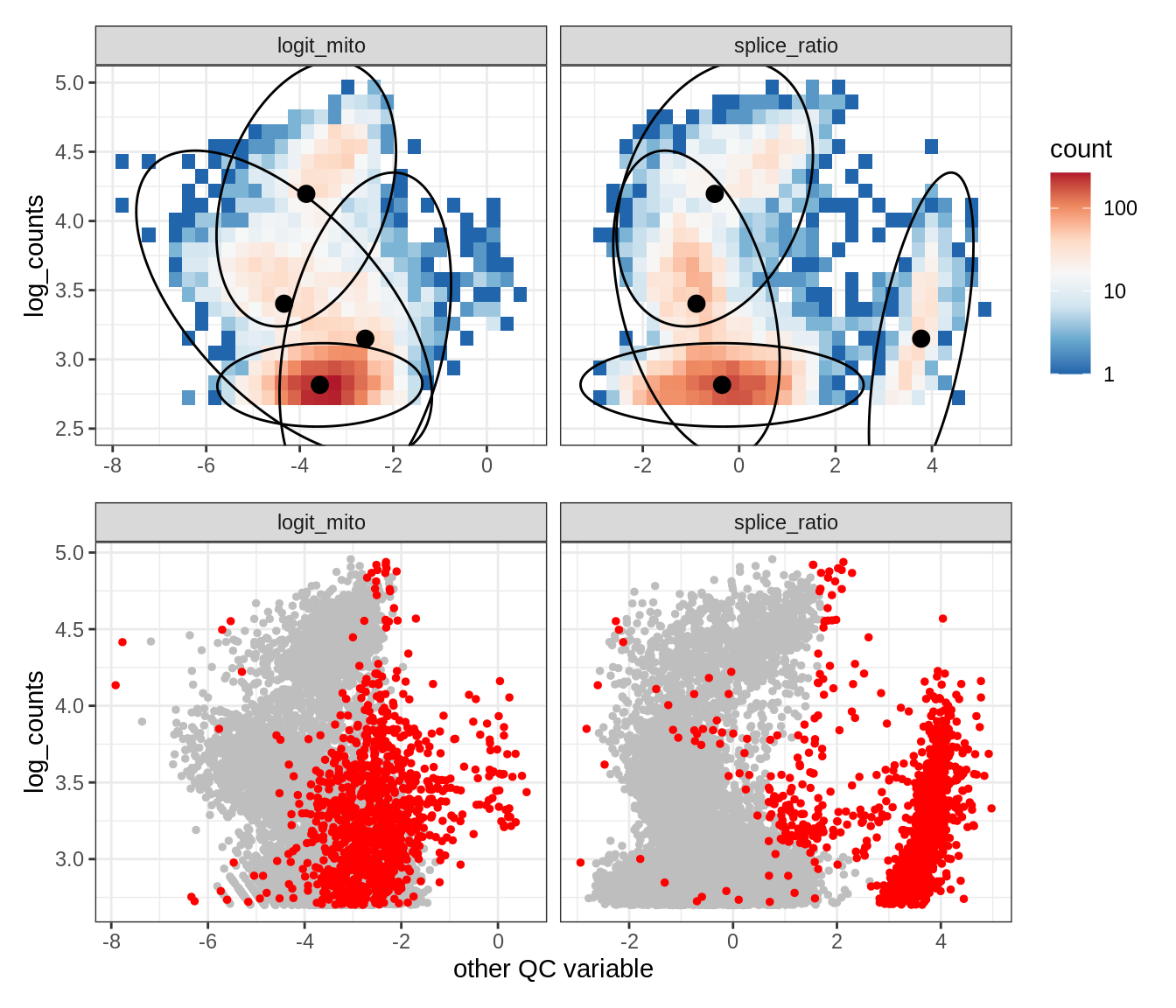
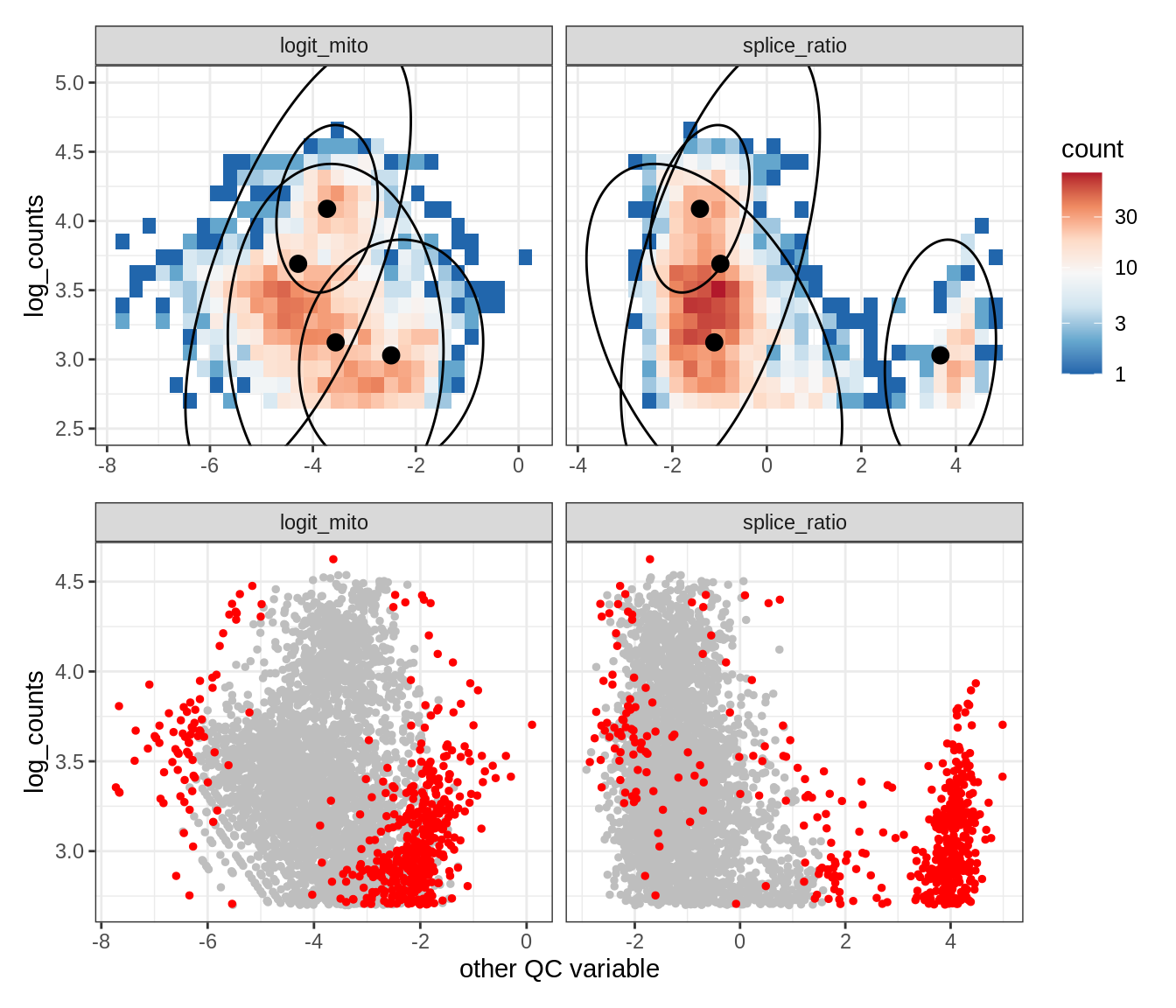
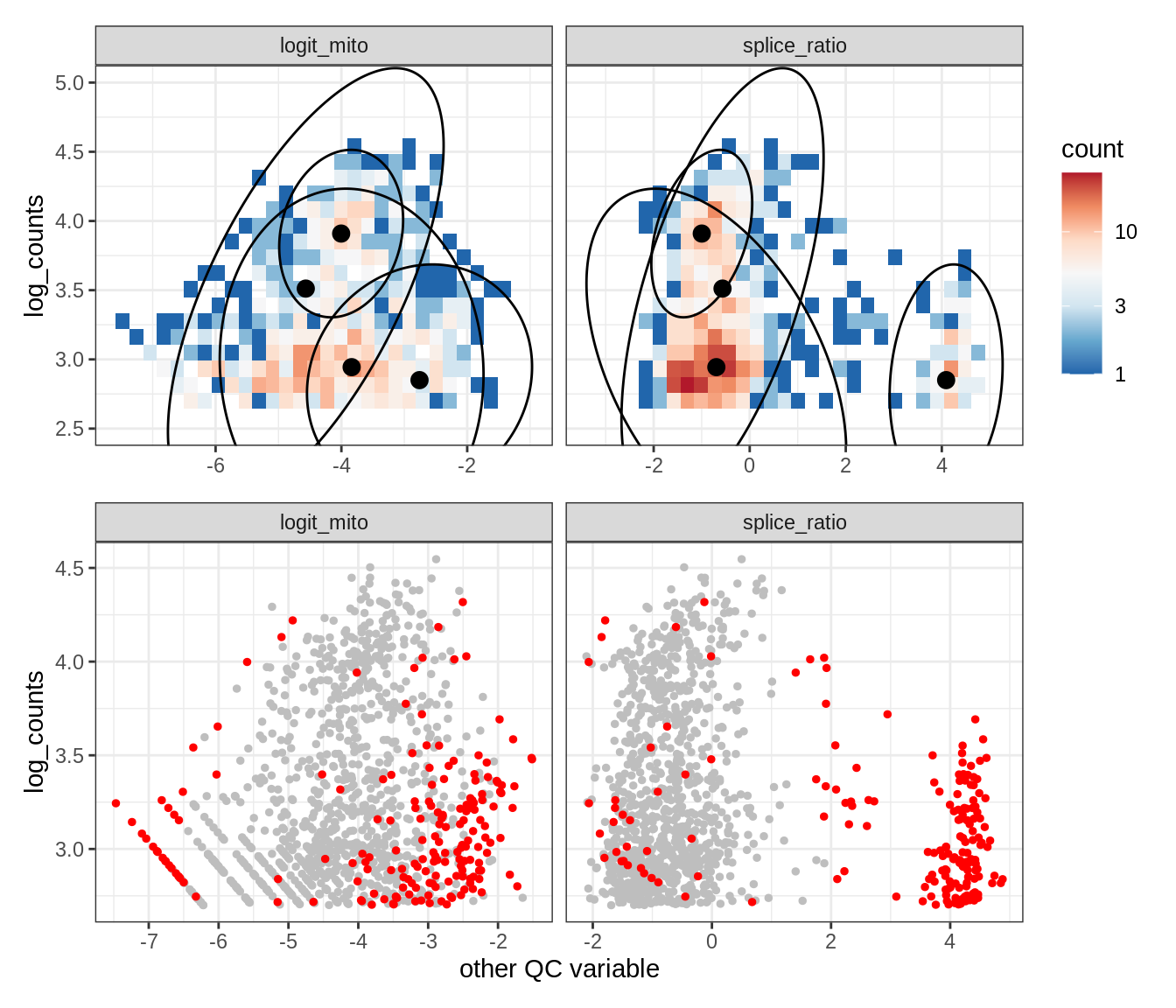
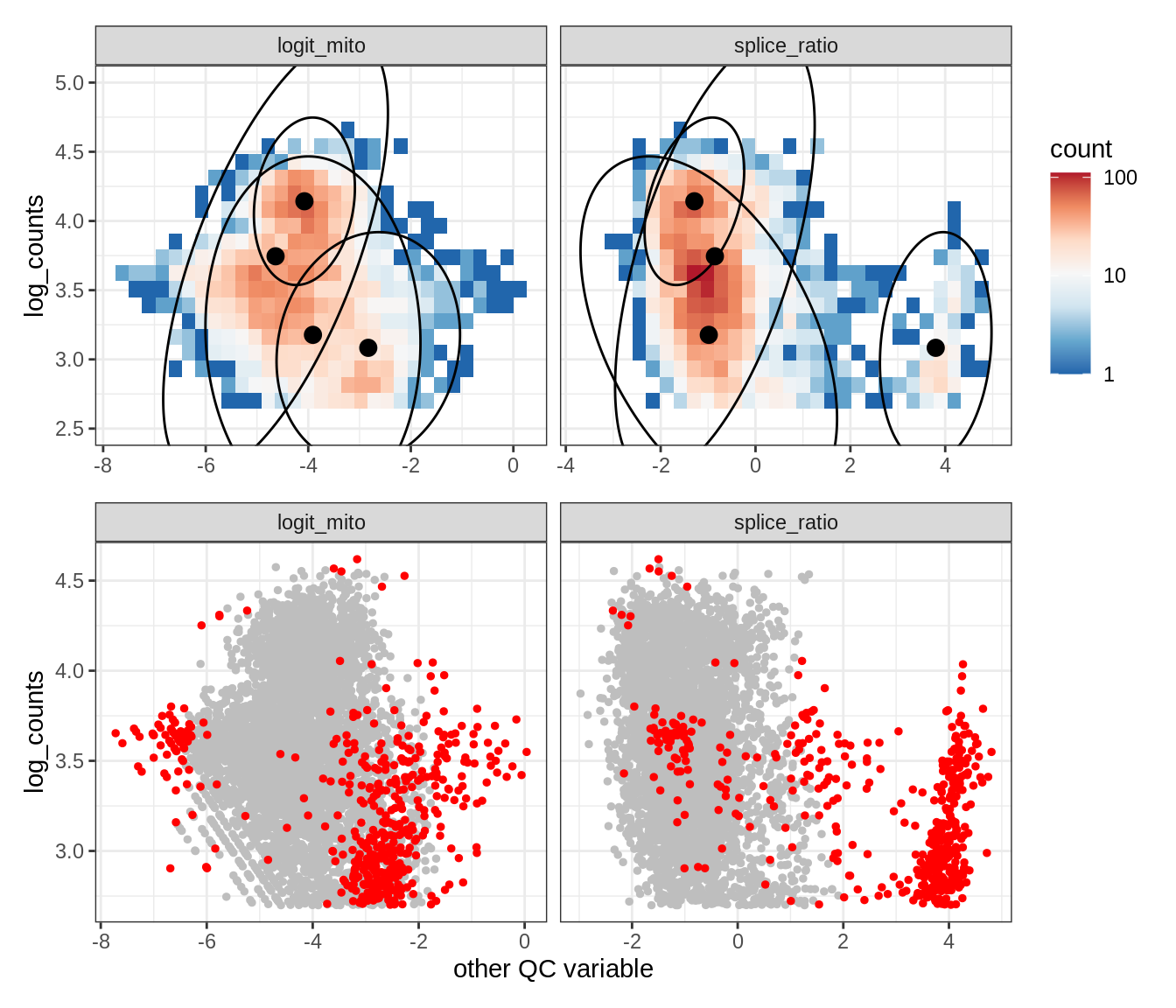
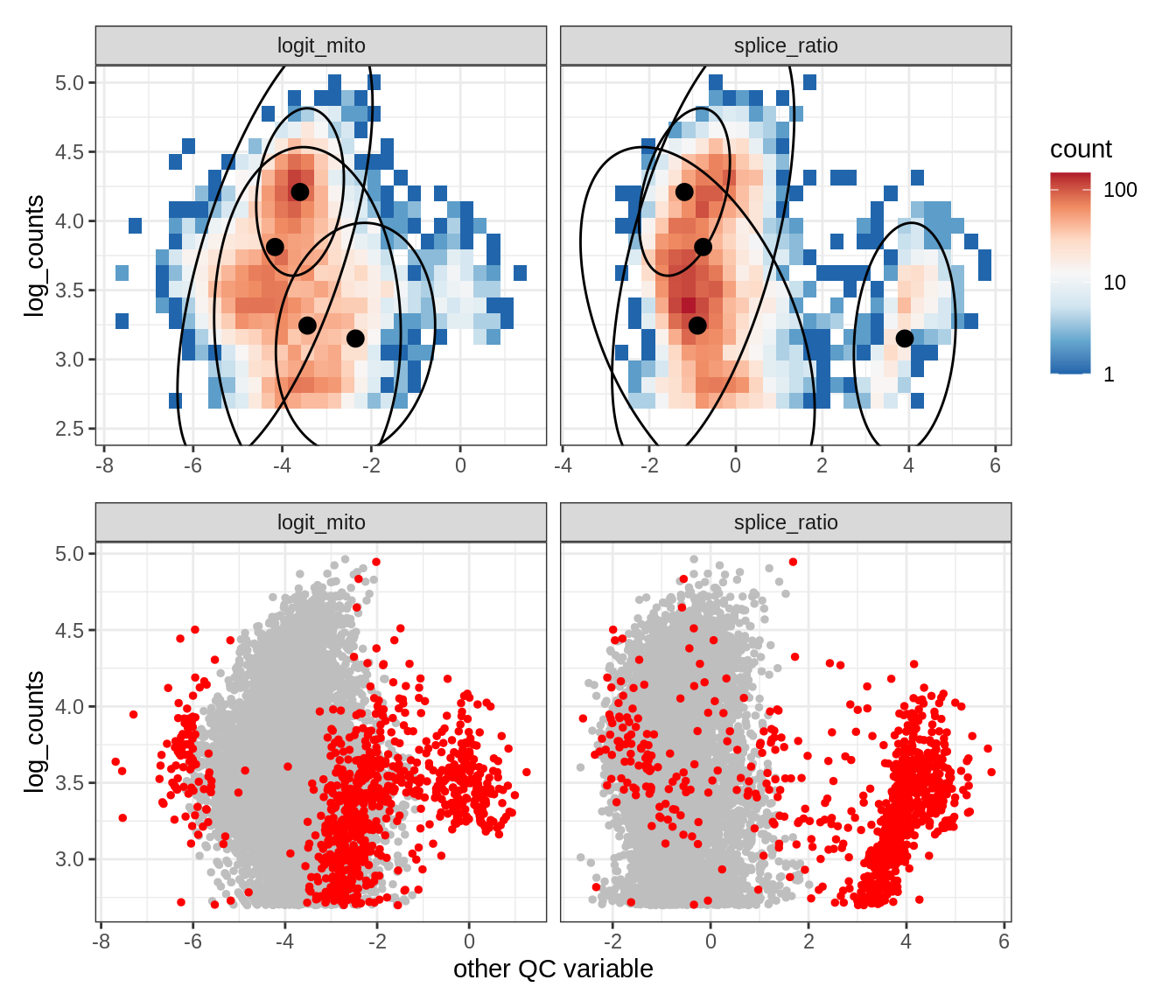
g_fit = plot_fit_over_biaxials_one_sample(qc_obj, s)
g_out = plot_outliers_one_sample(qc_obj, s, outliers_dt=outliers_dt)
g = g_fit / g_out
print(g)
cat('\n\n')
}
}SG1
EU006
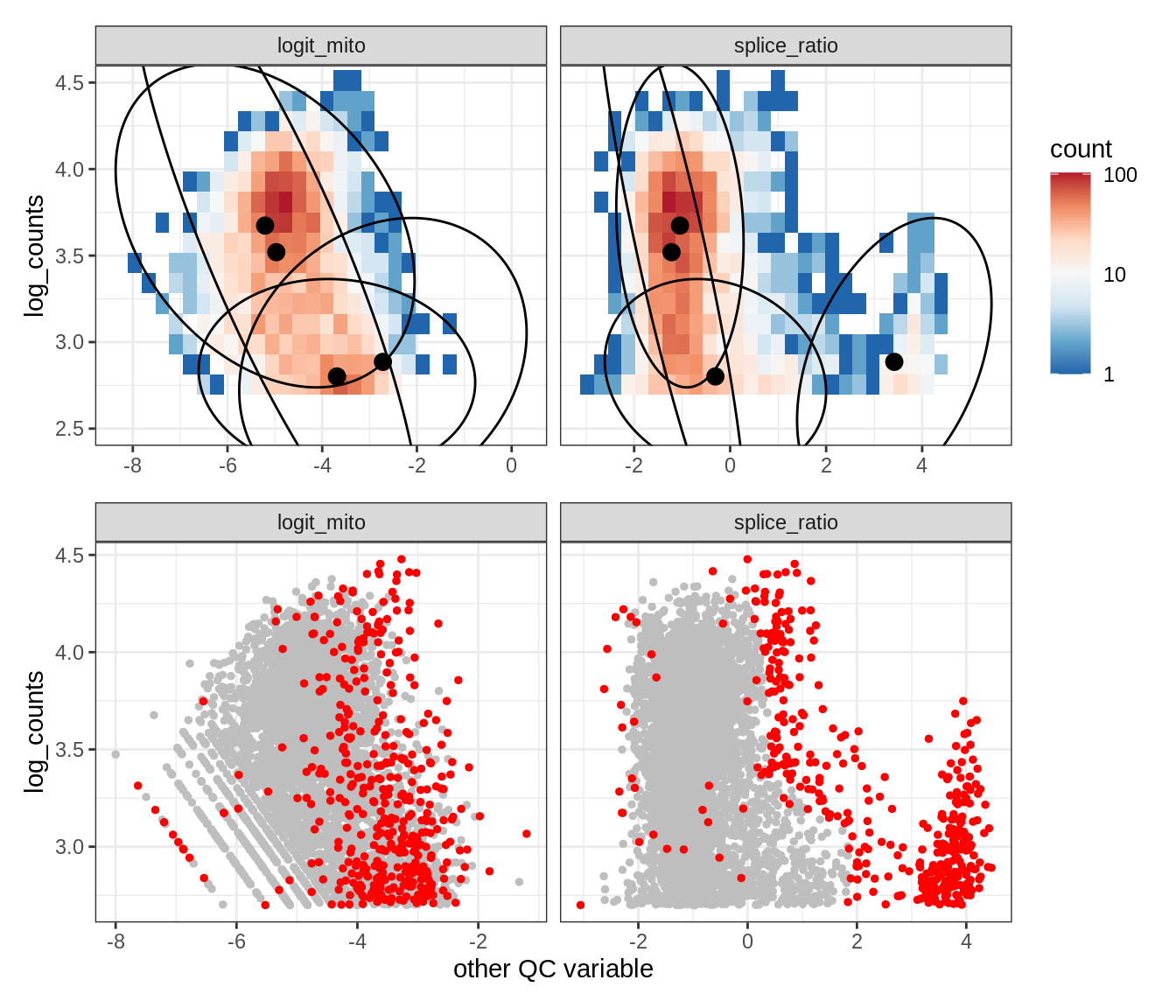
EU013
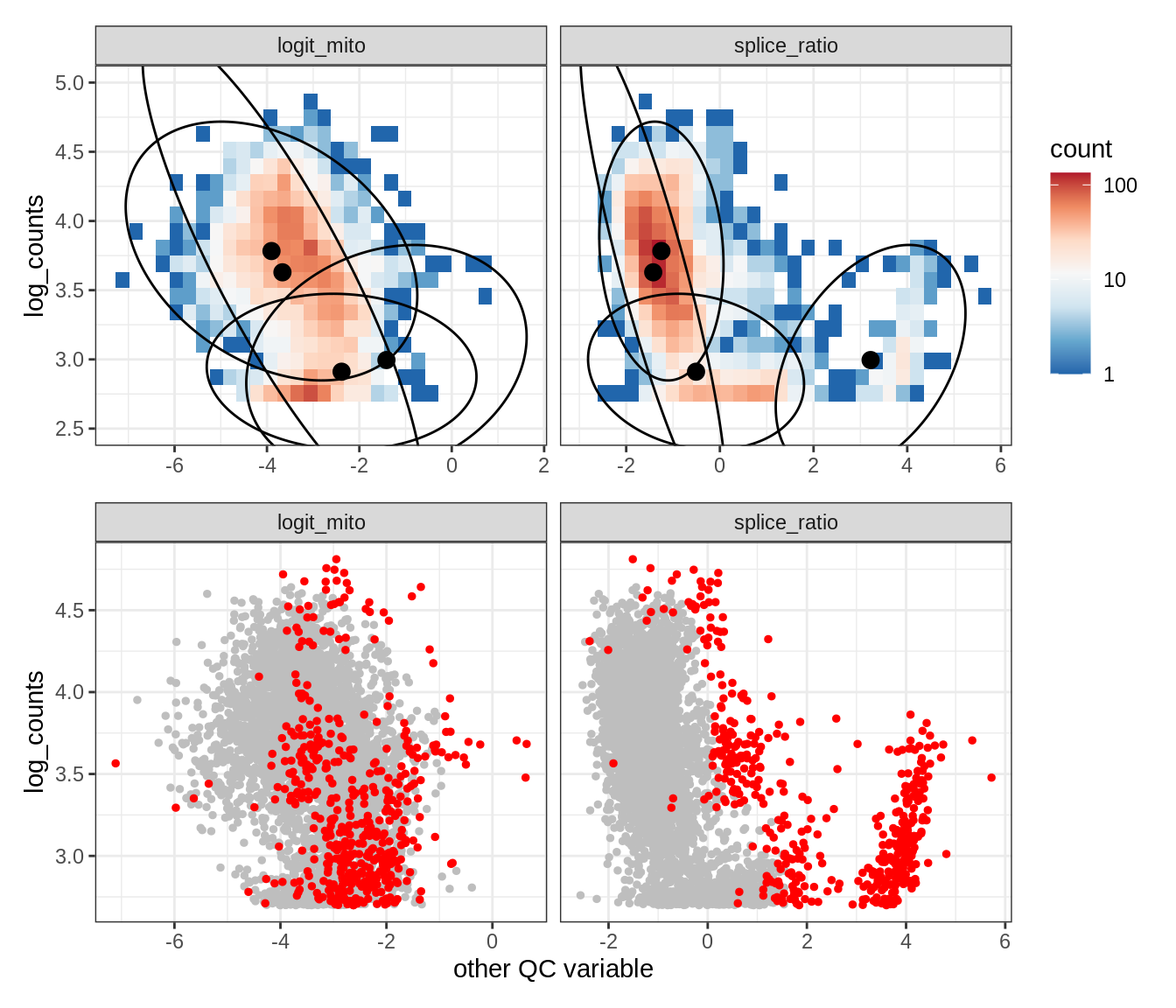
WM102

WM103
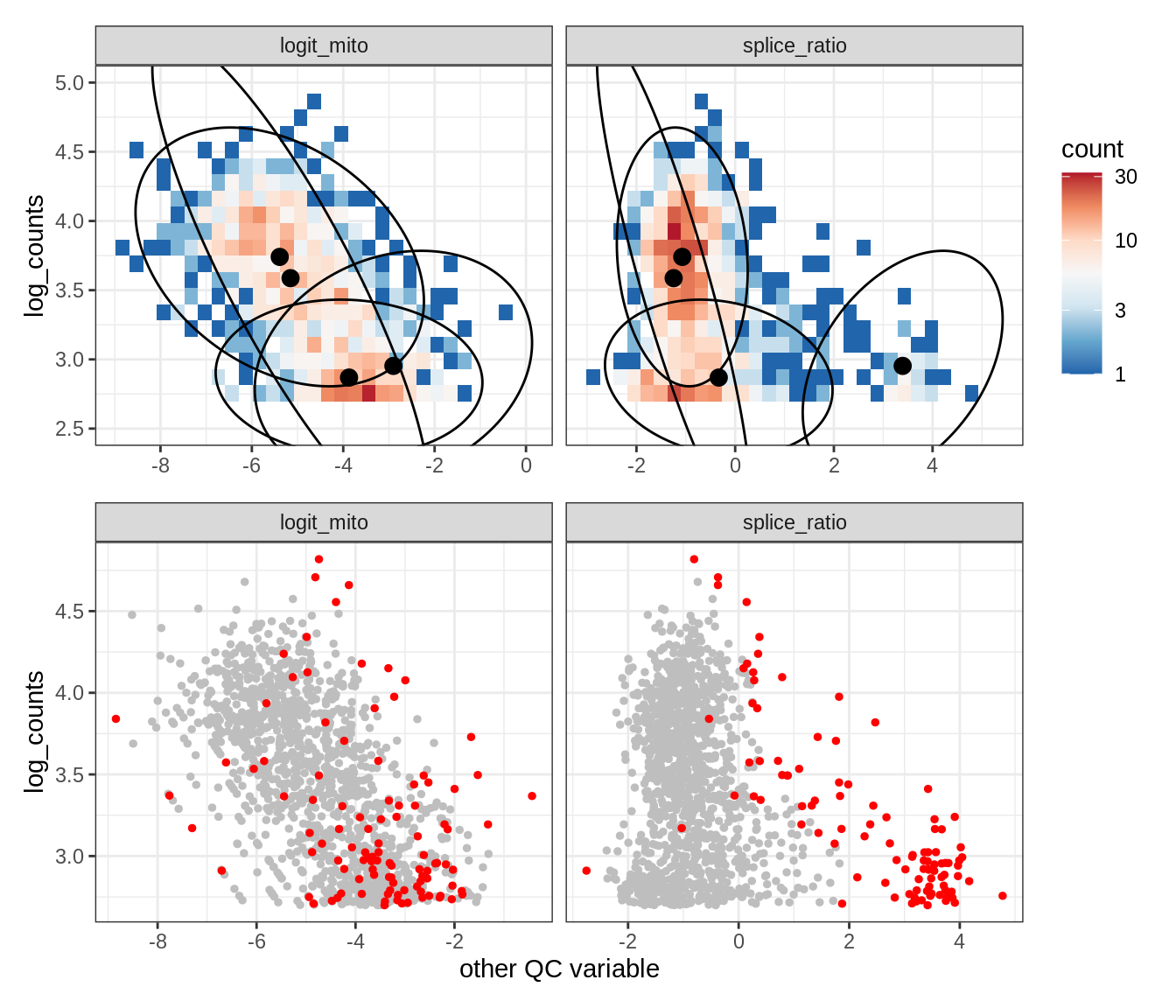
WM104
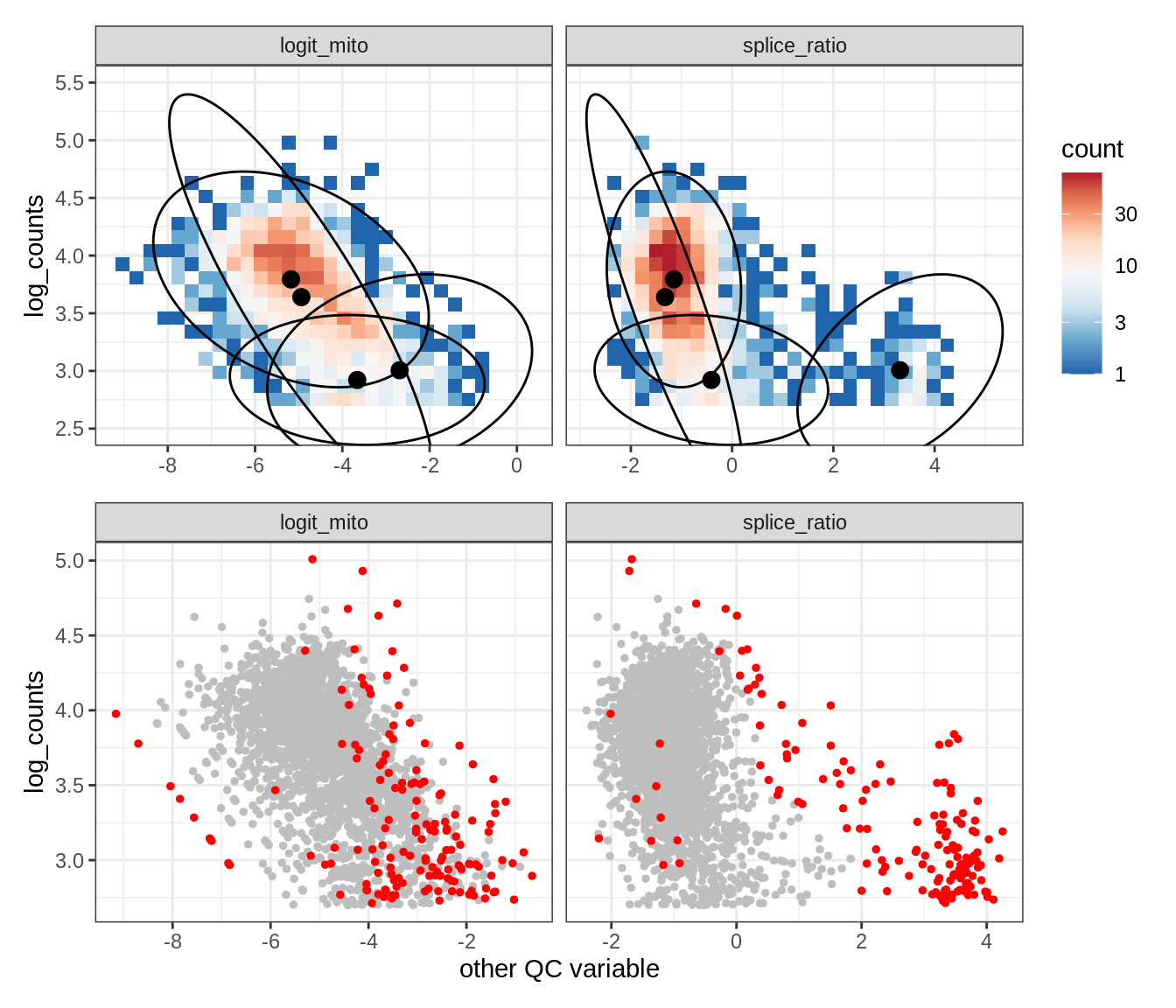
WM105
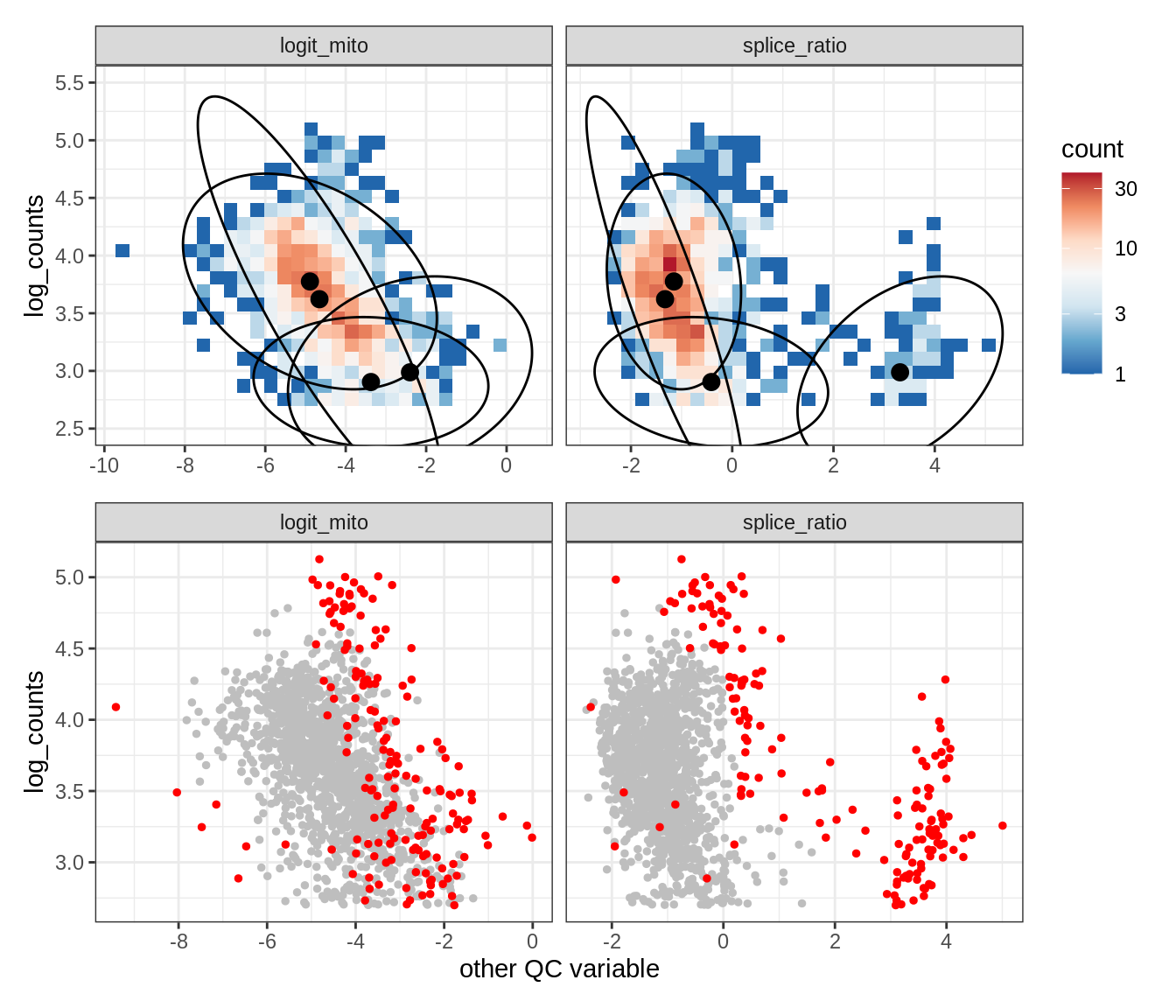
WM112
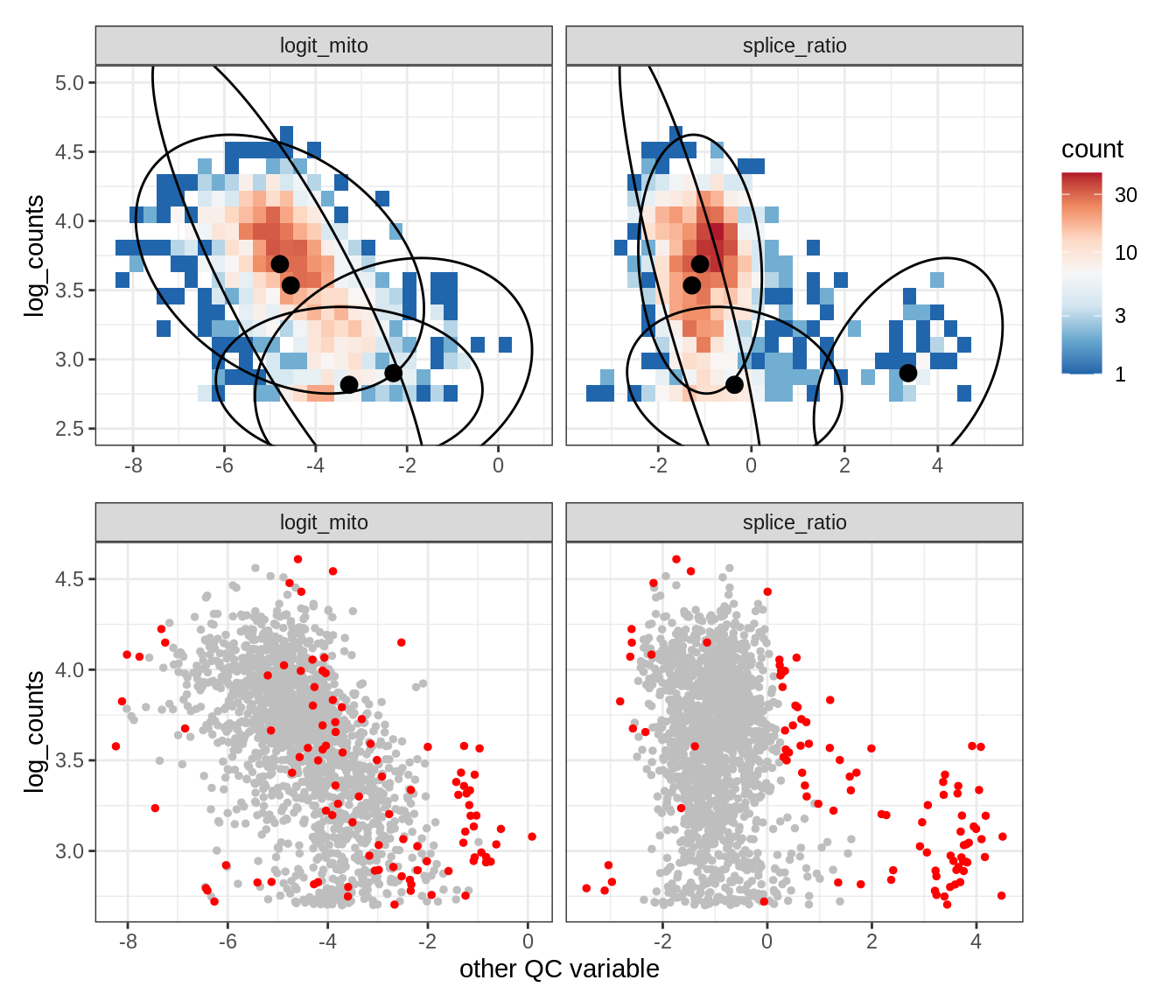
WM113
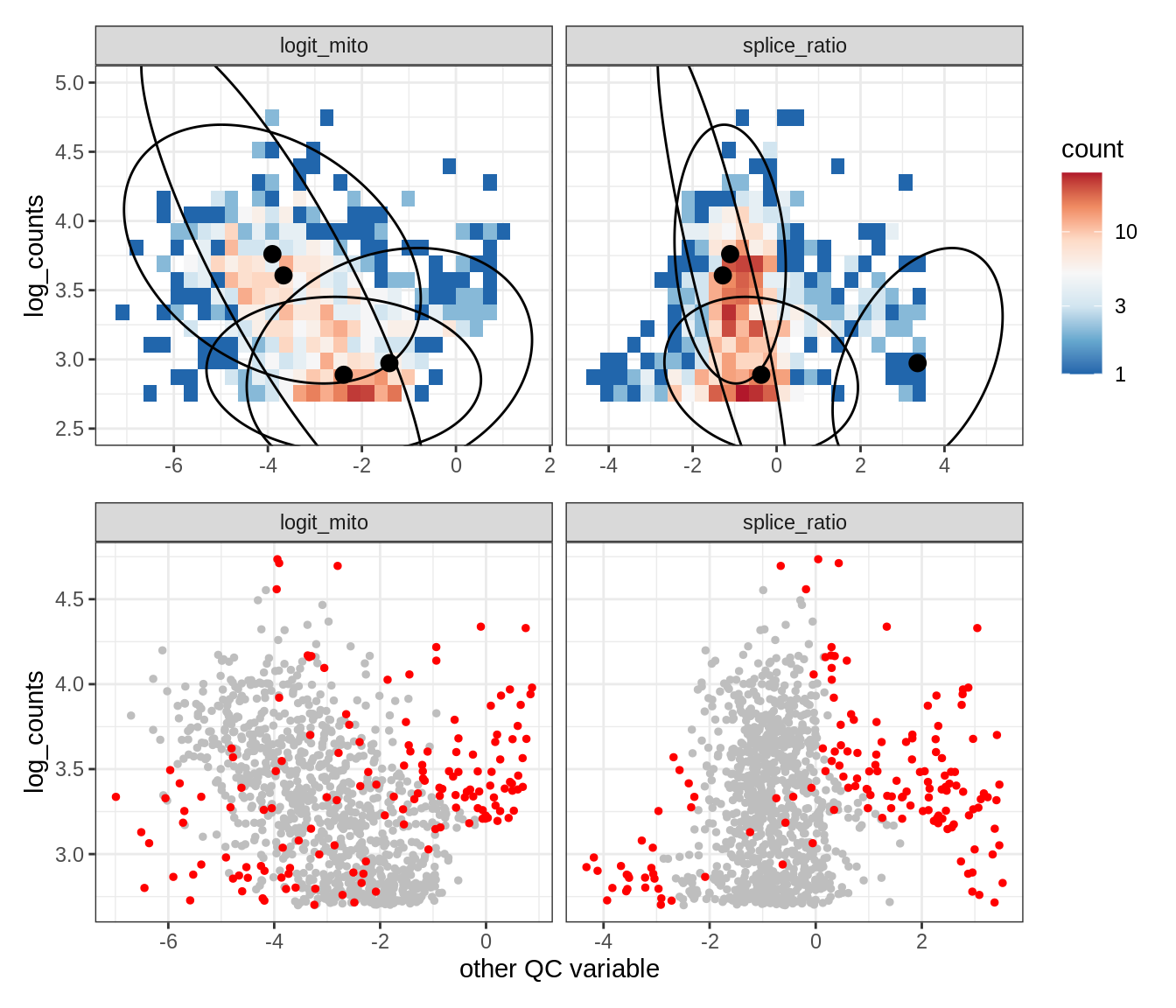
WM116
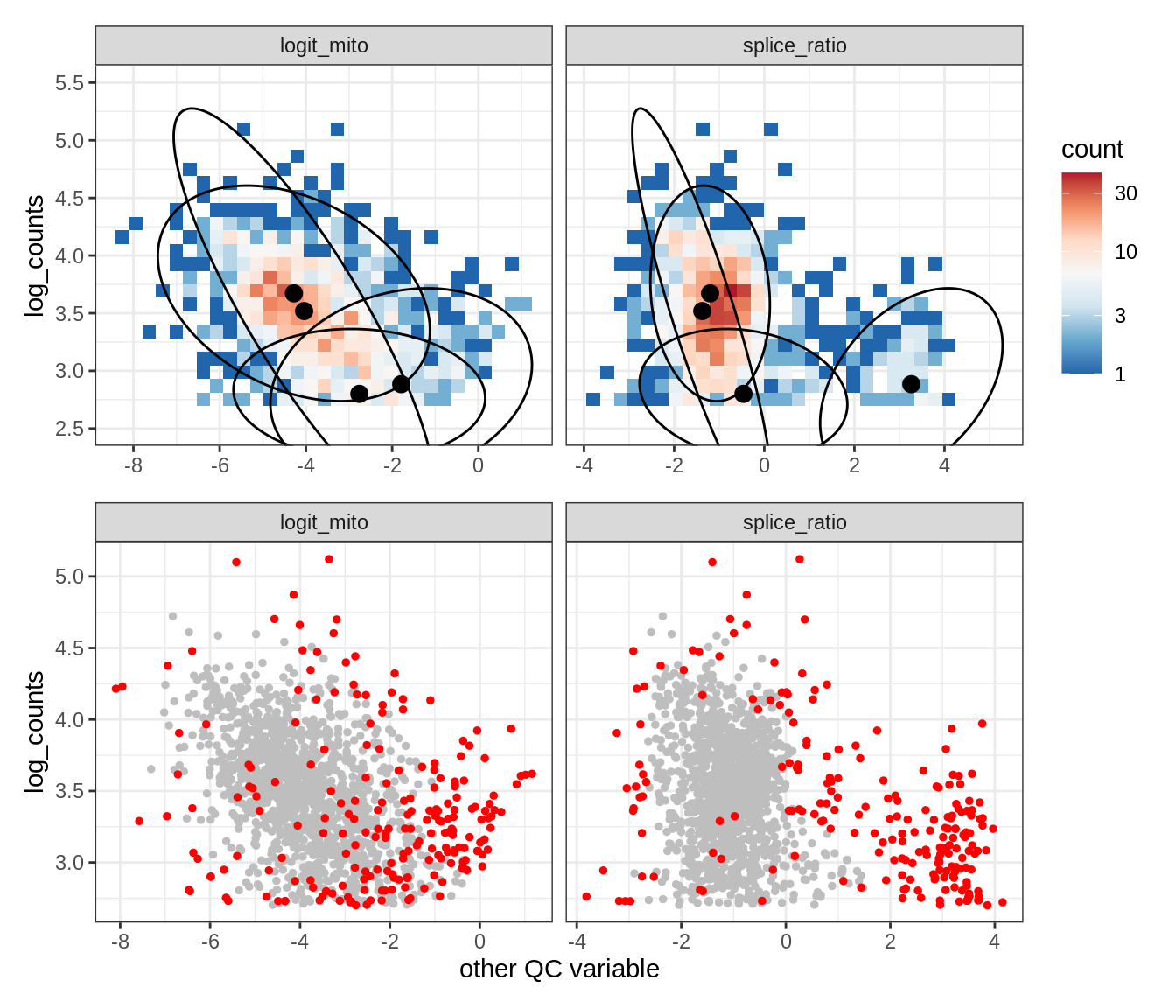
WM117
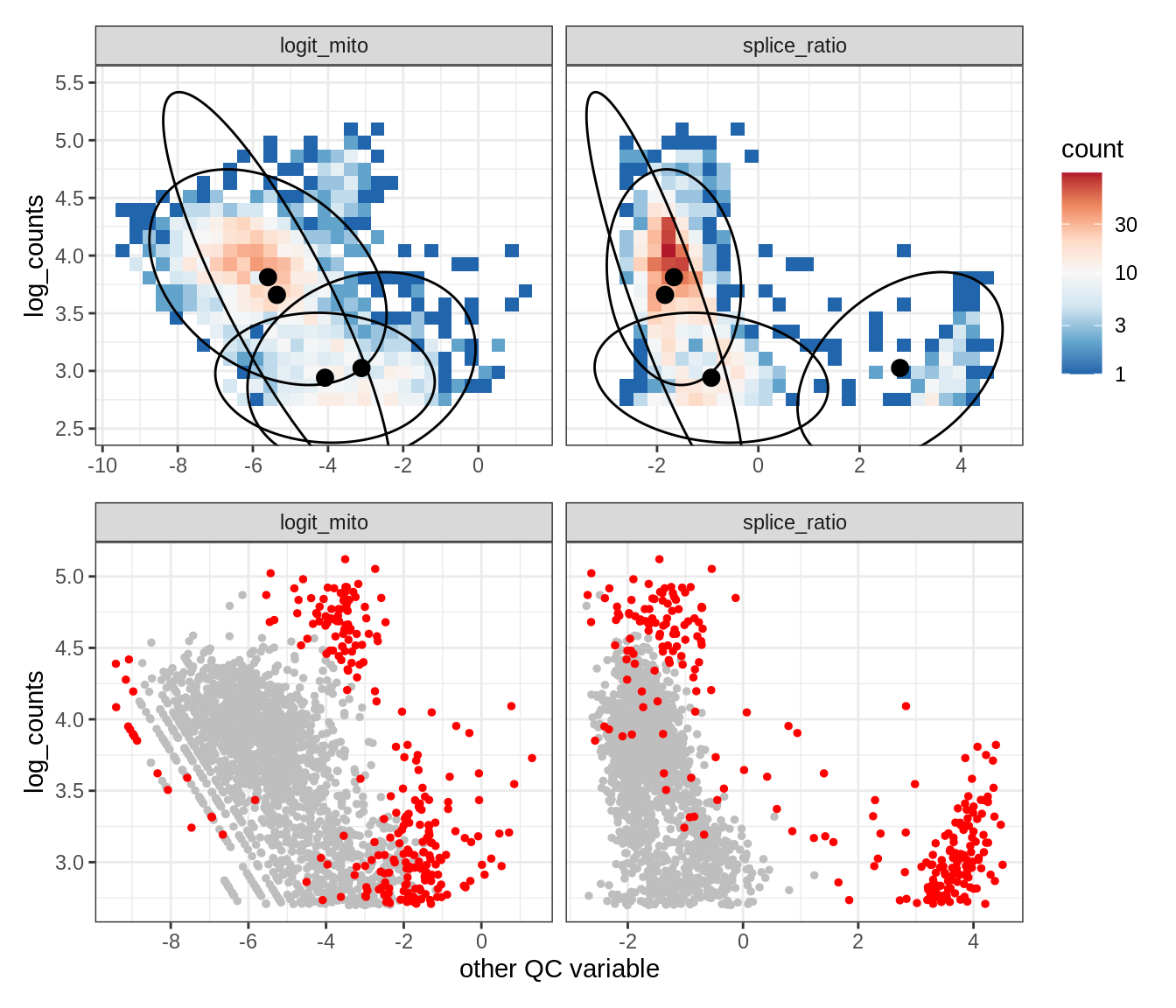
WM119
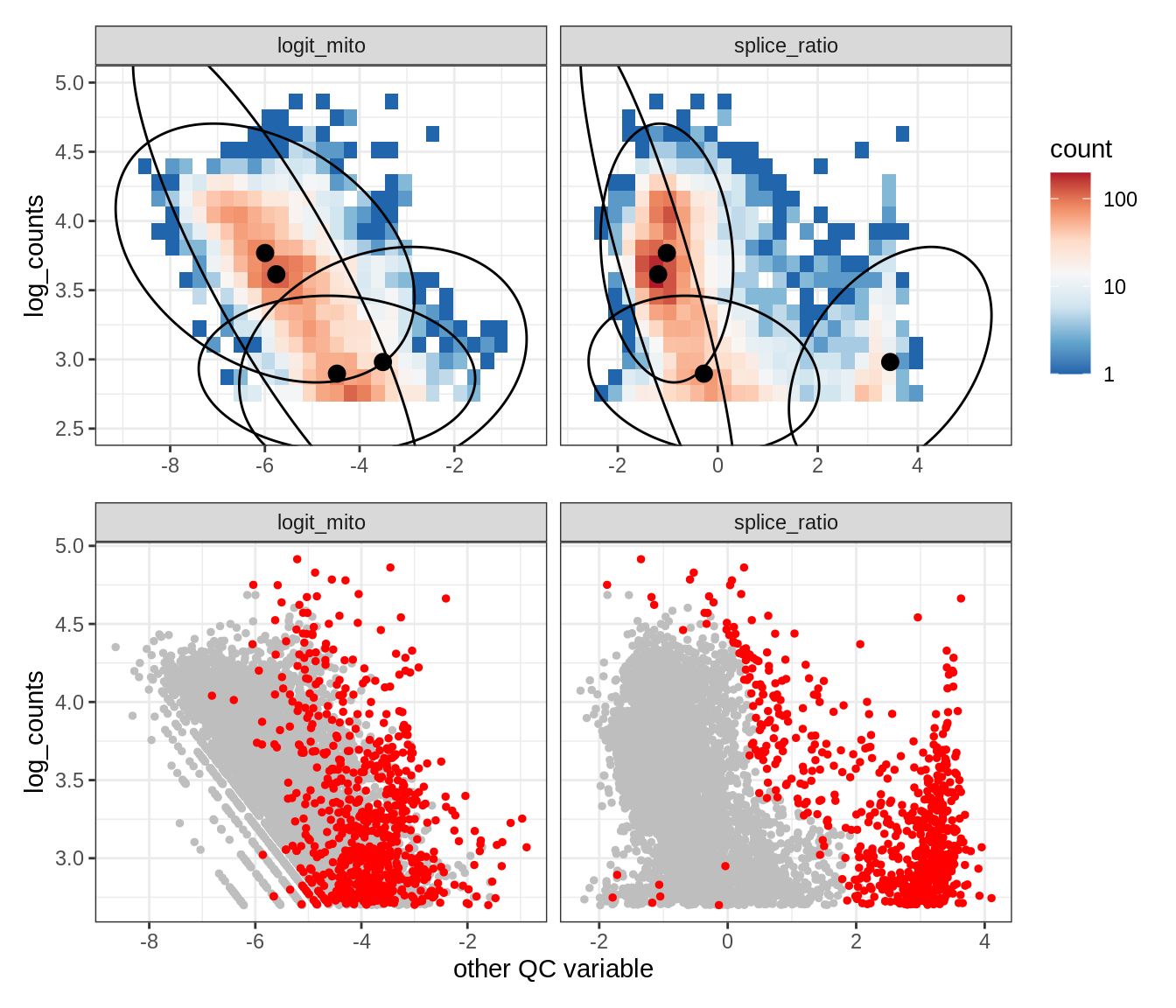
WM122
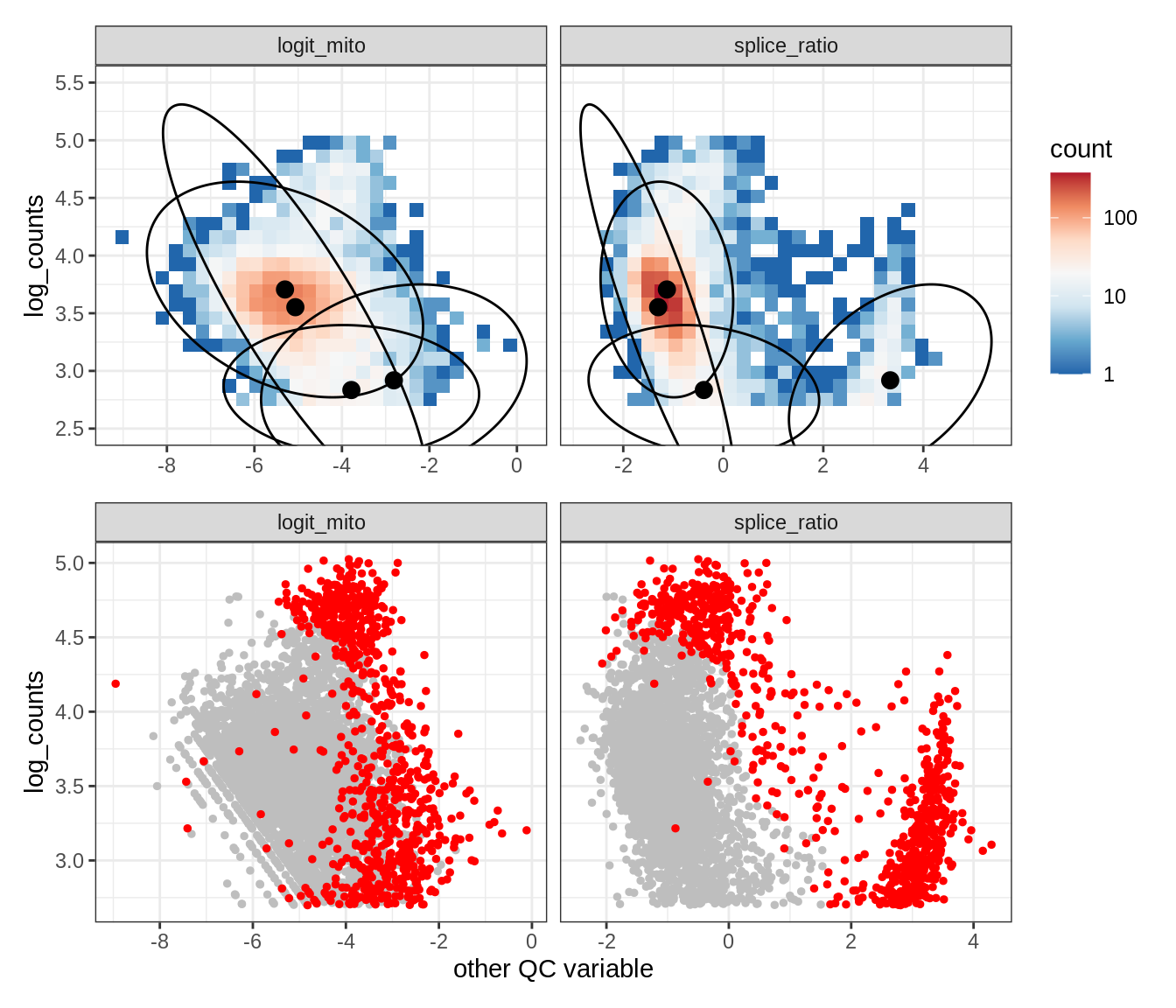
WM123
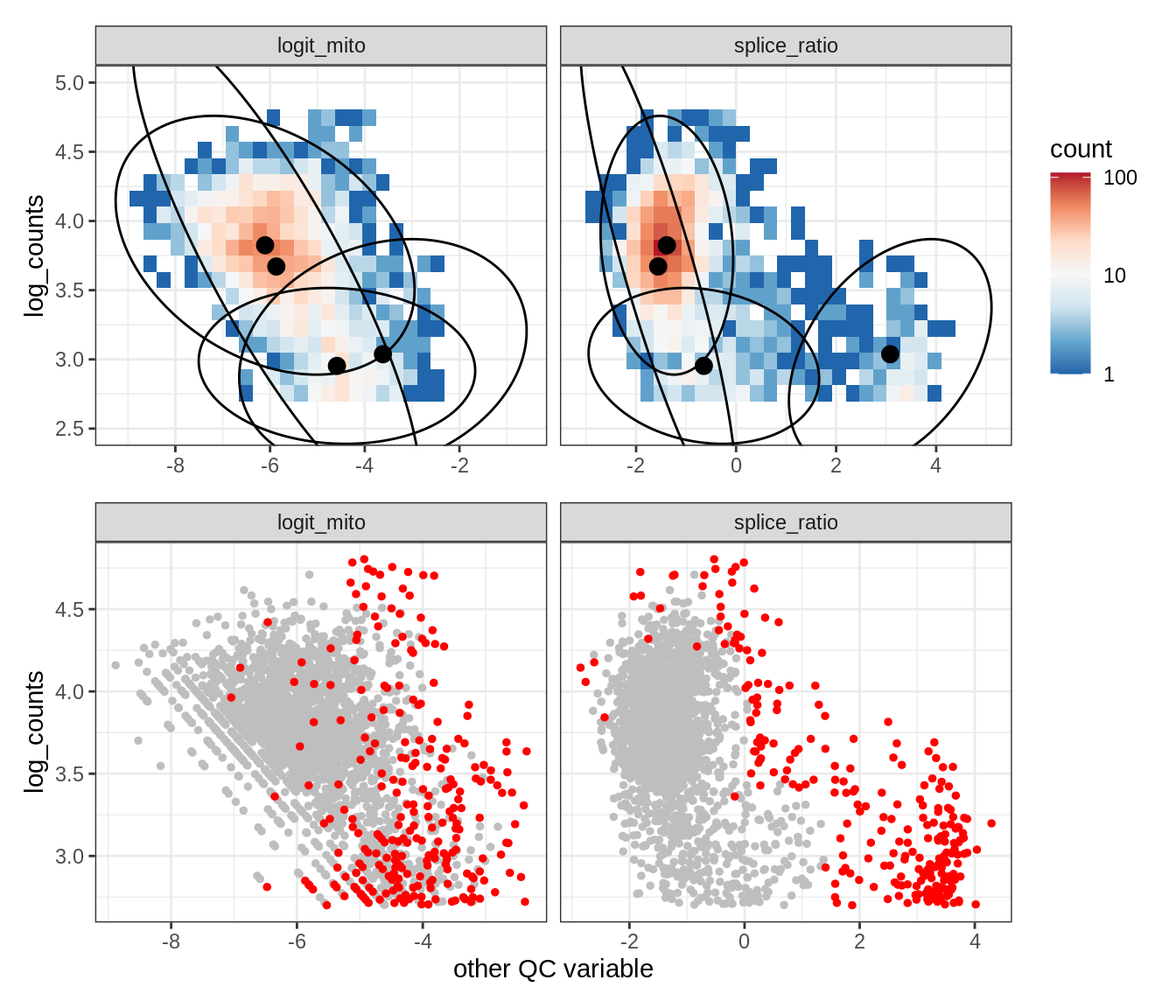
WM130
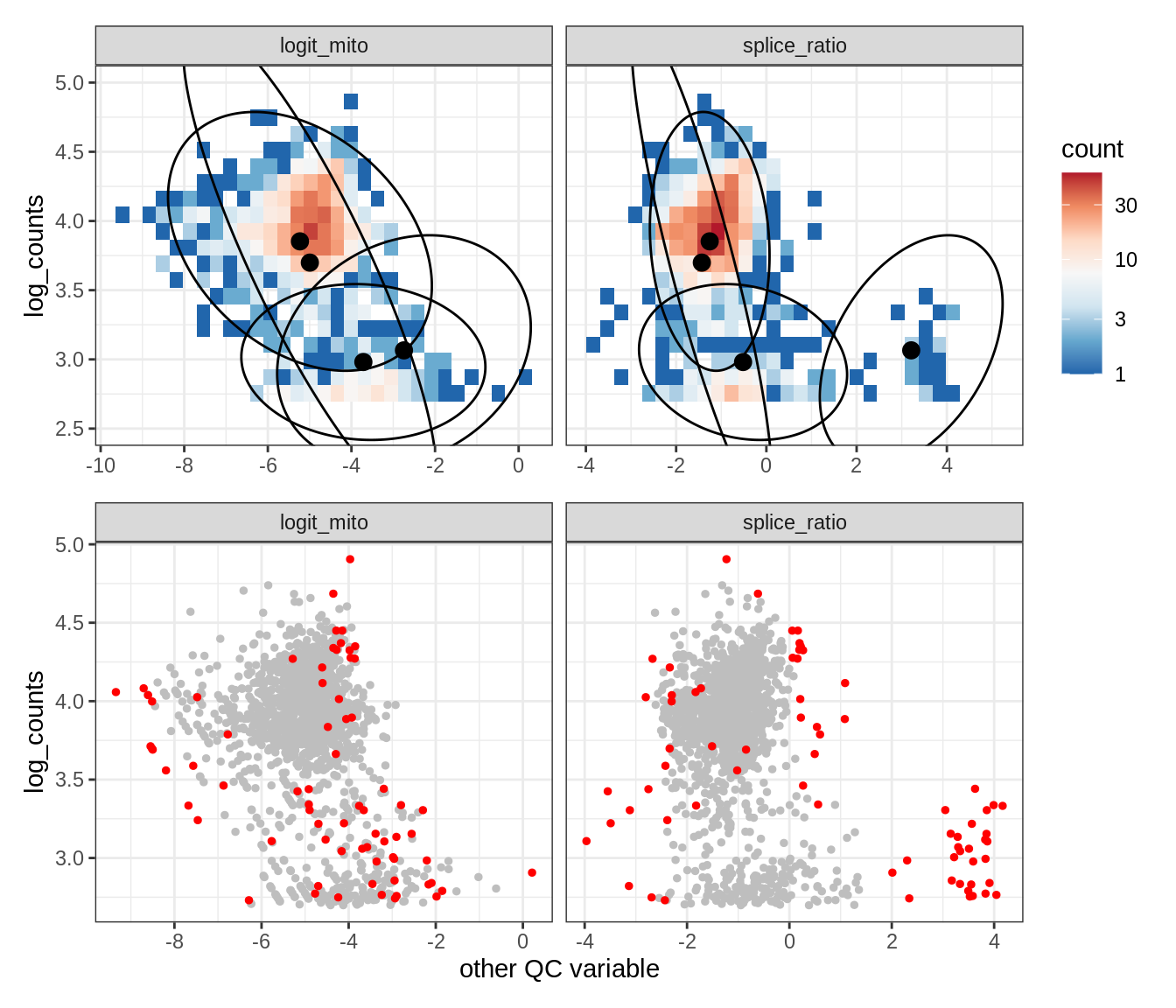
WM132
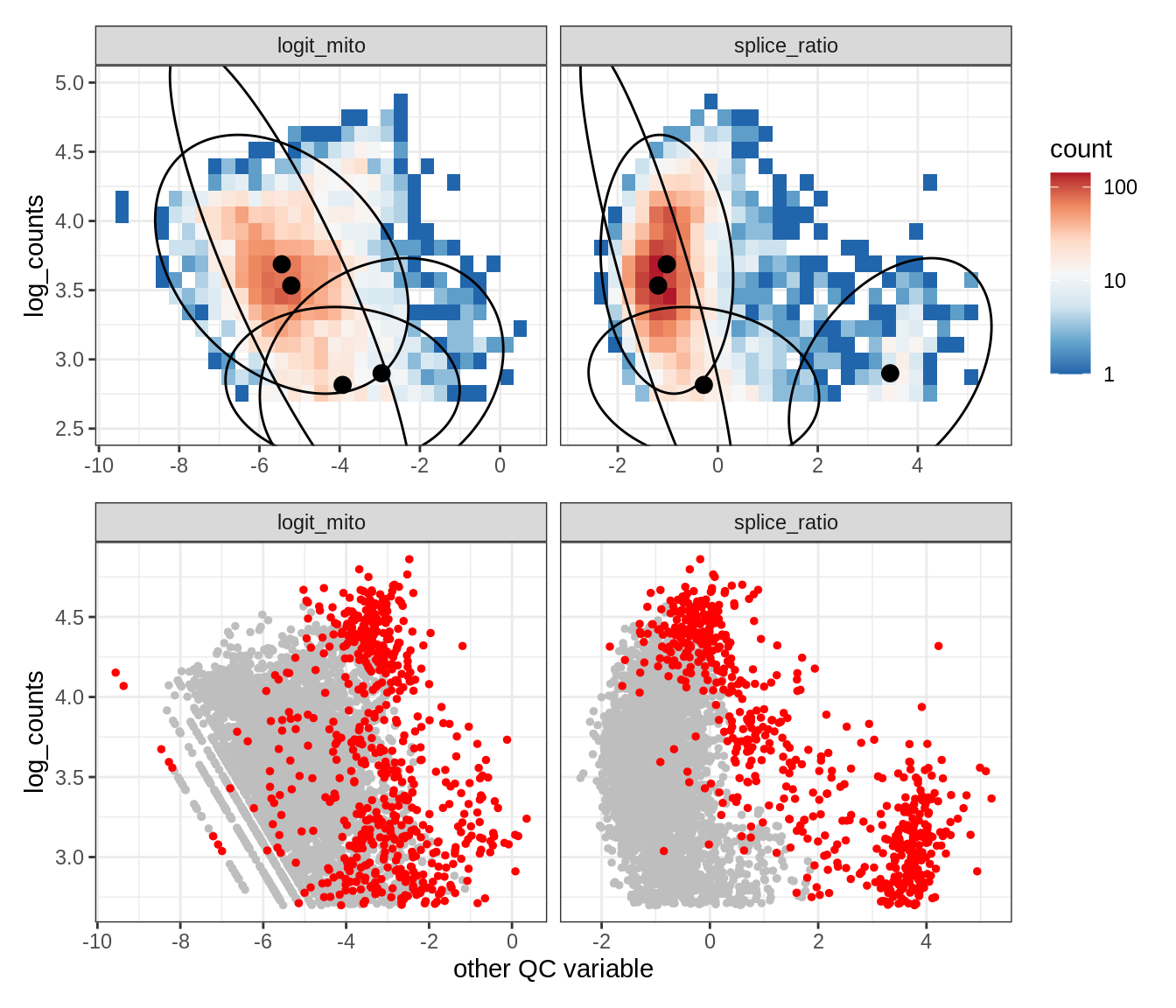
WM133
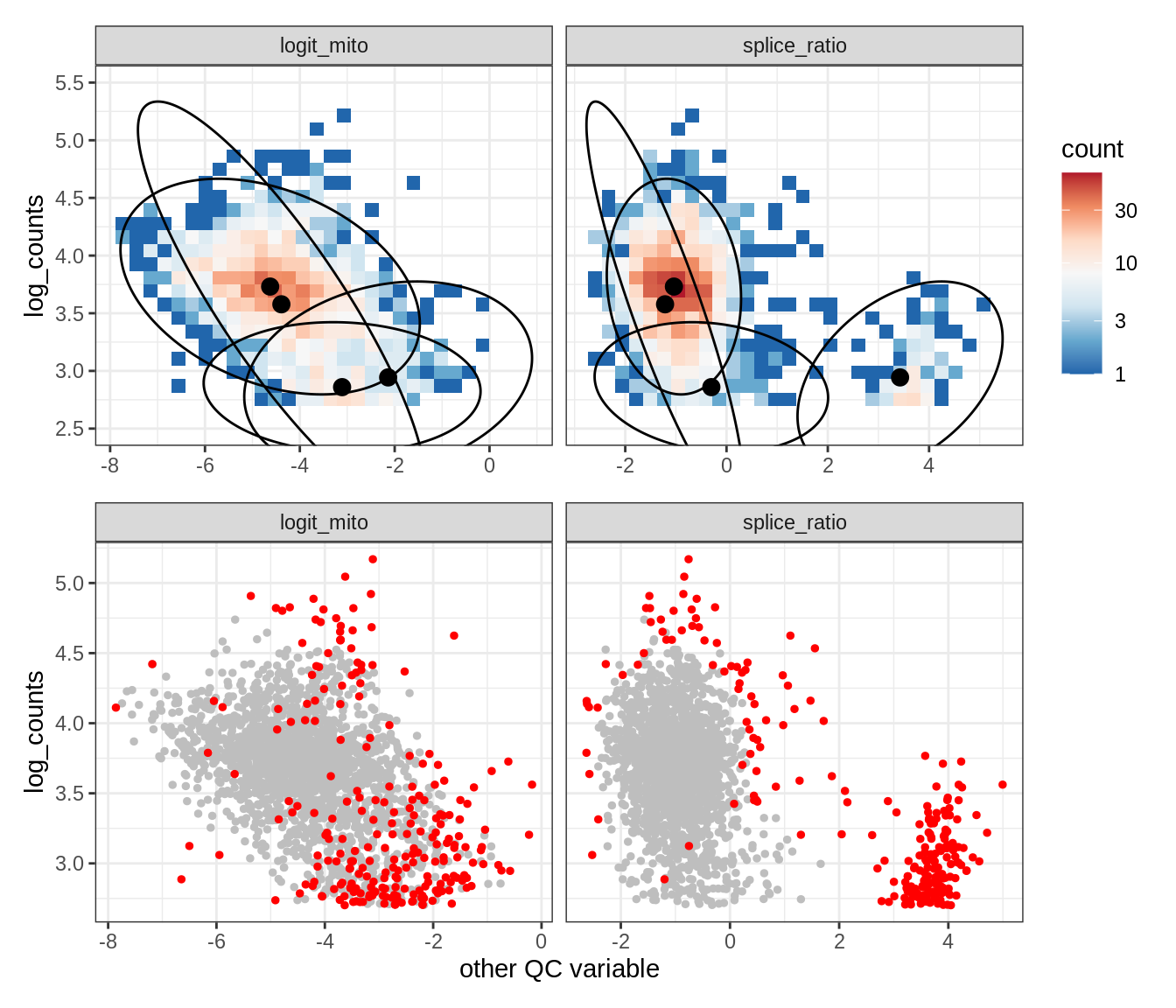
WM134
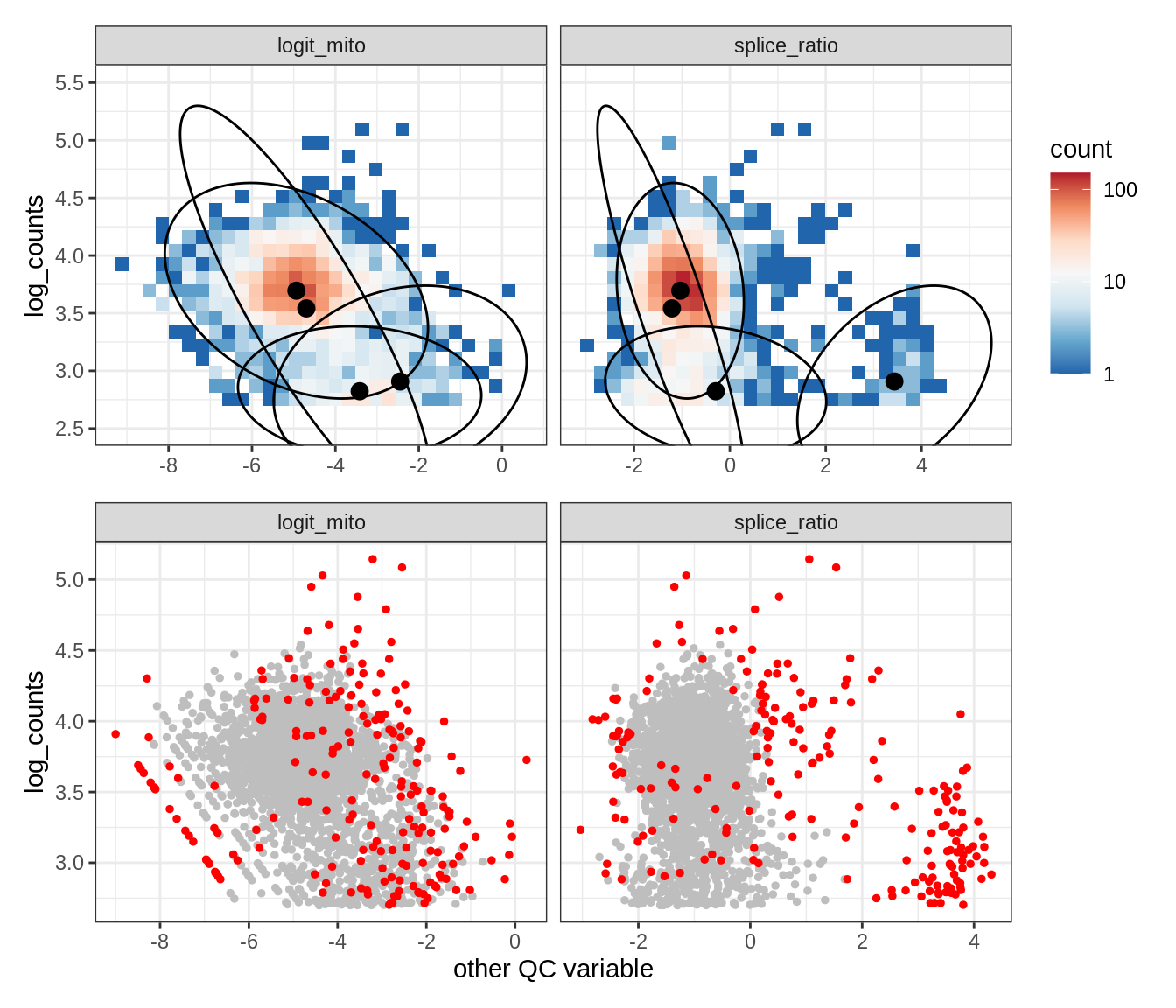
WM135
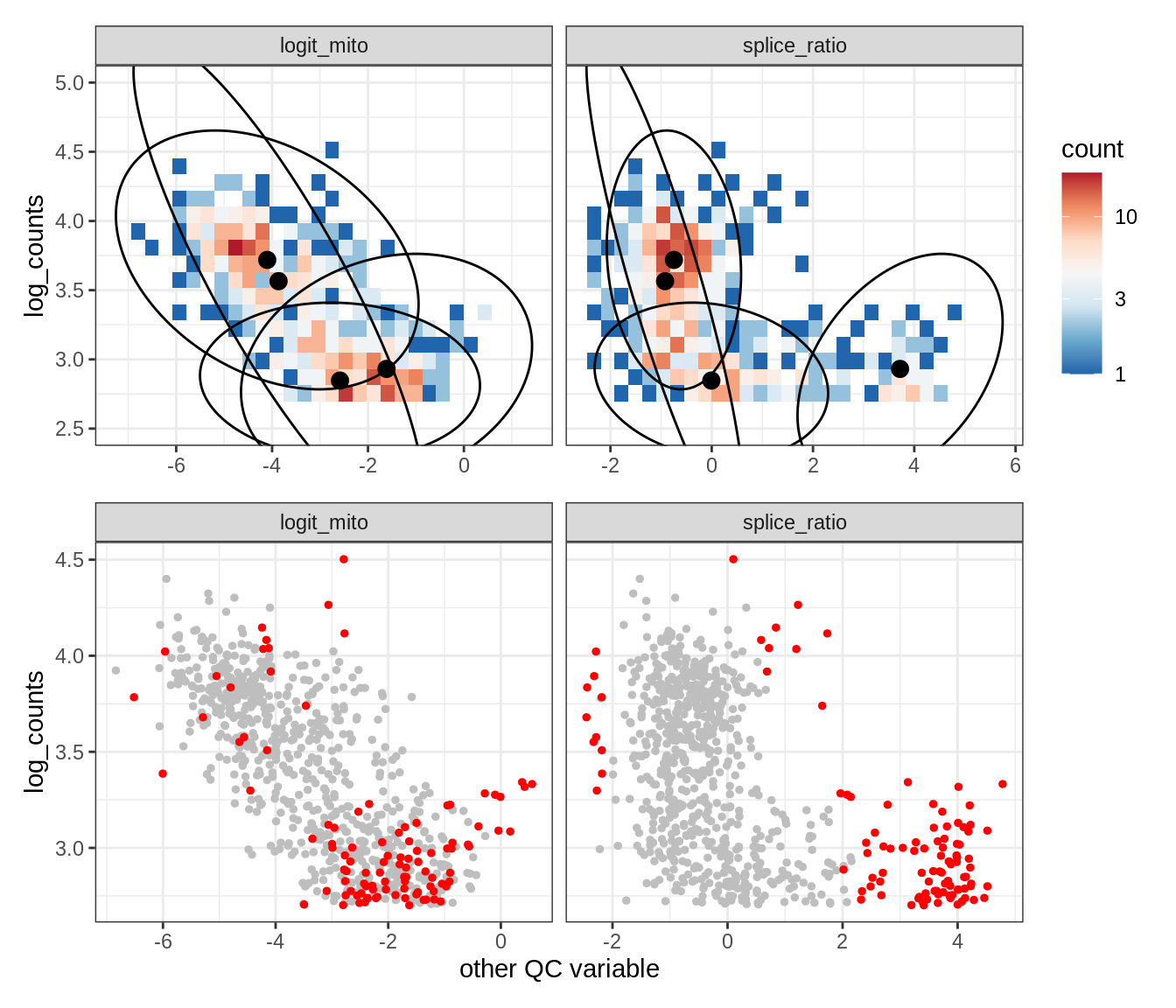
WM143
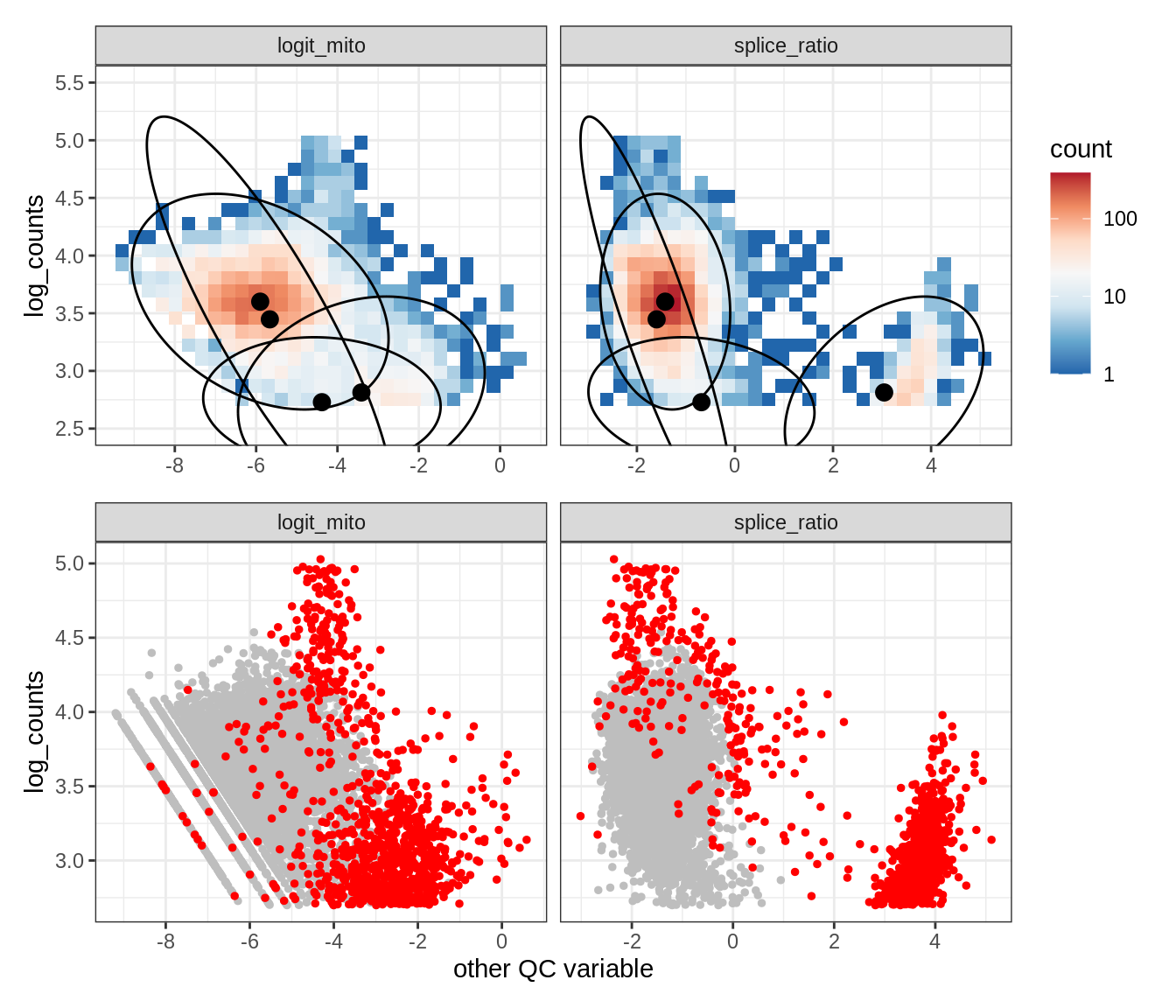
WM147
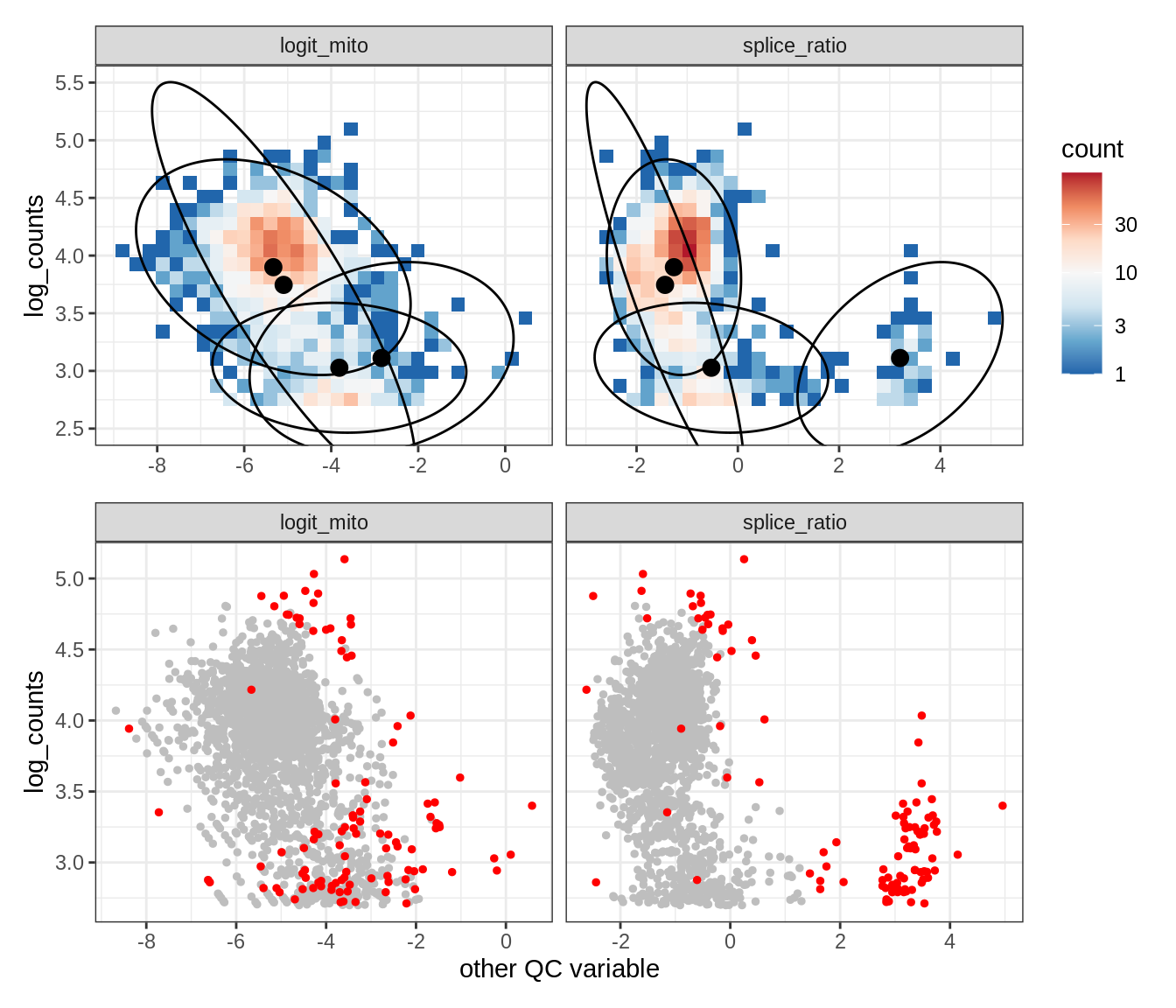
WM153
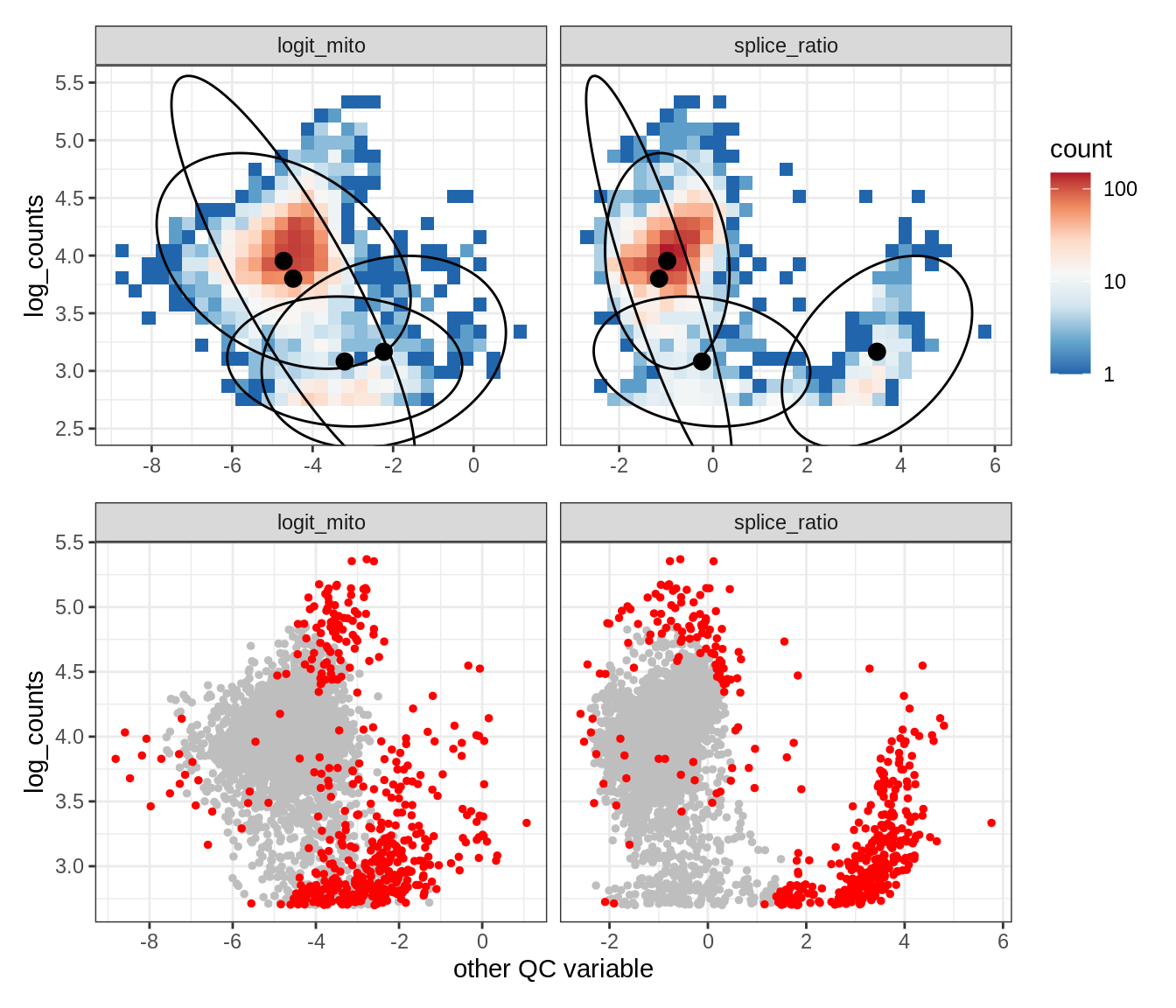
WM156
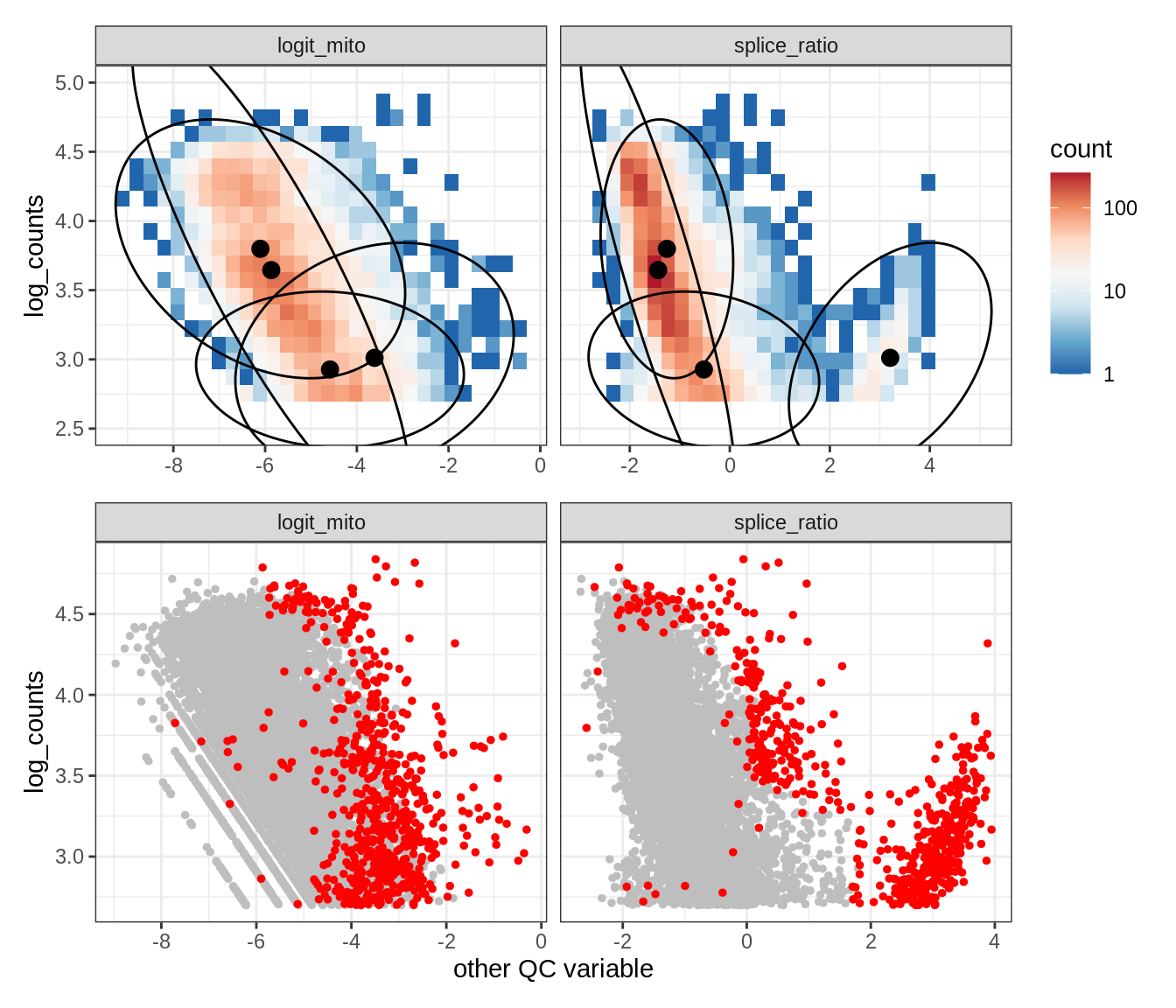
WM157
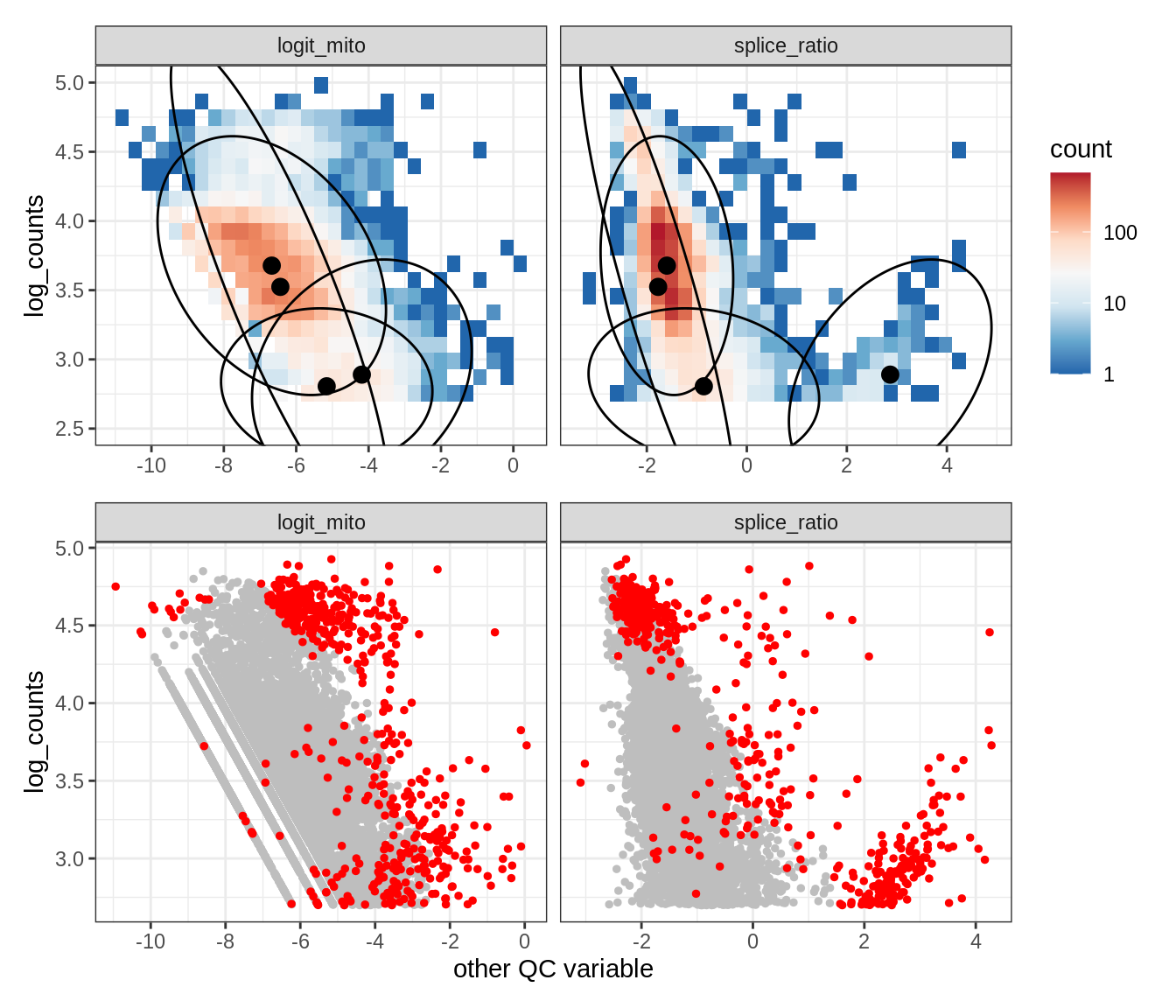
WM158
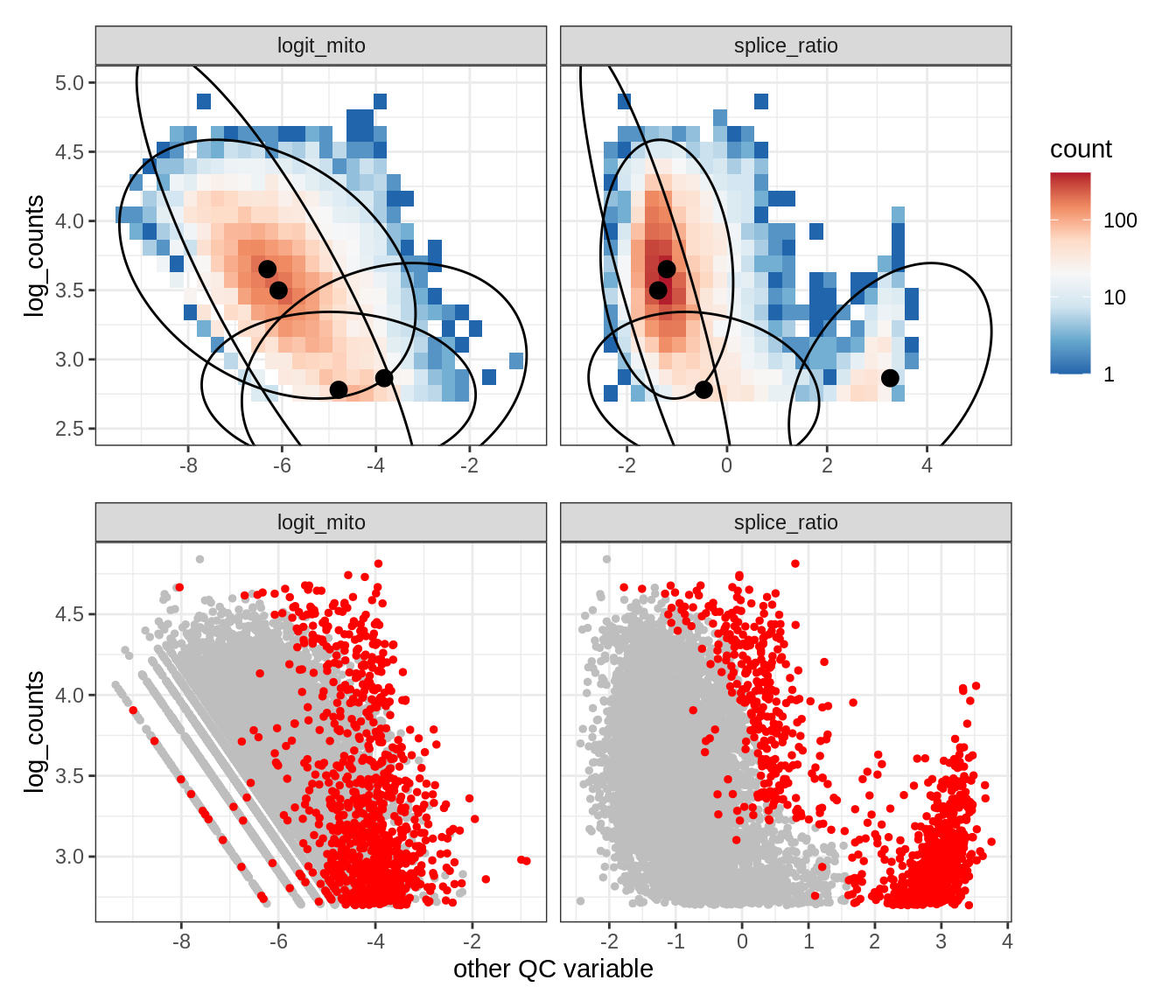
WM162
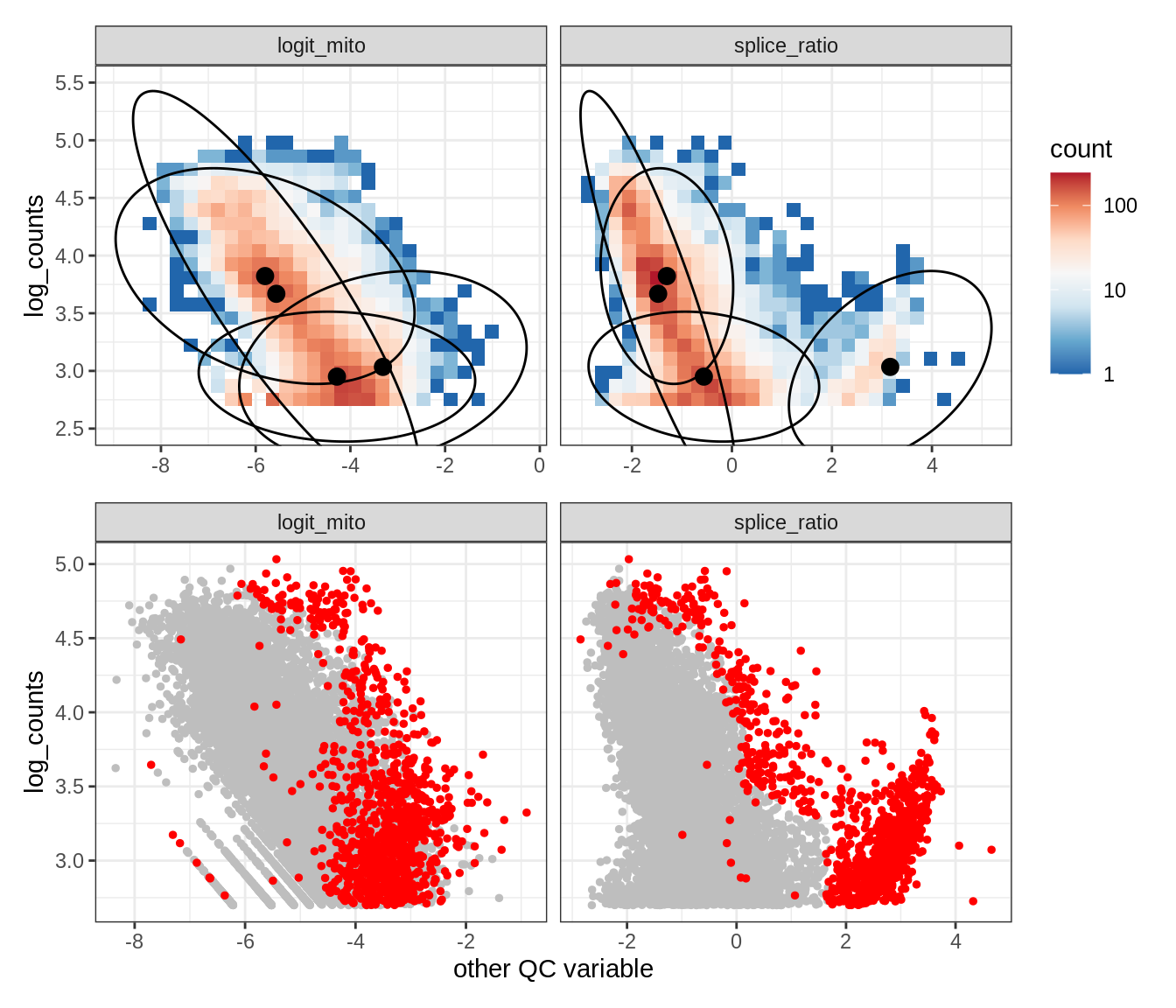
WM164
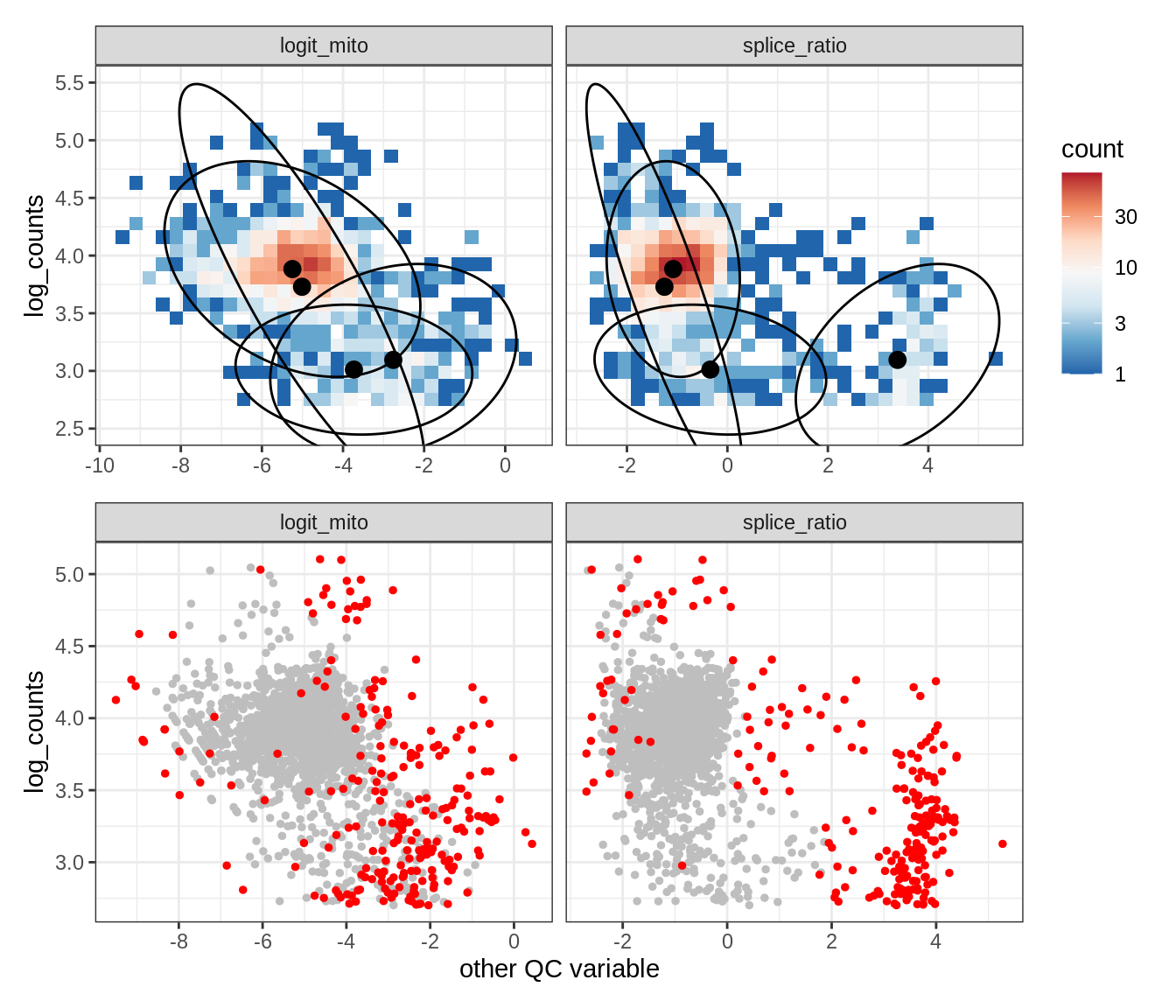
WM168
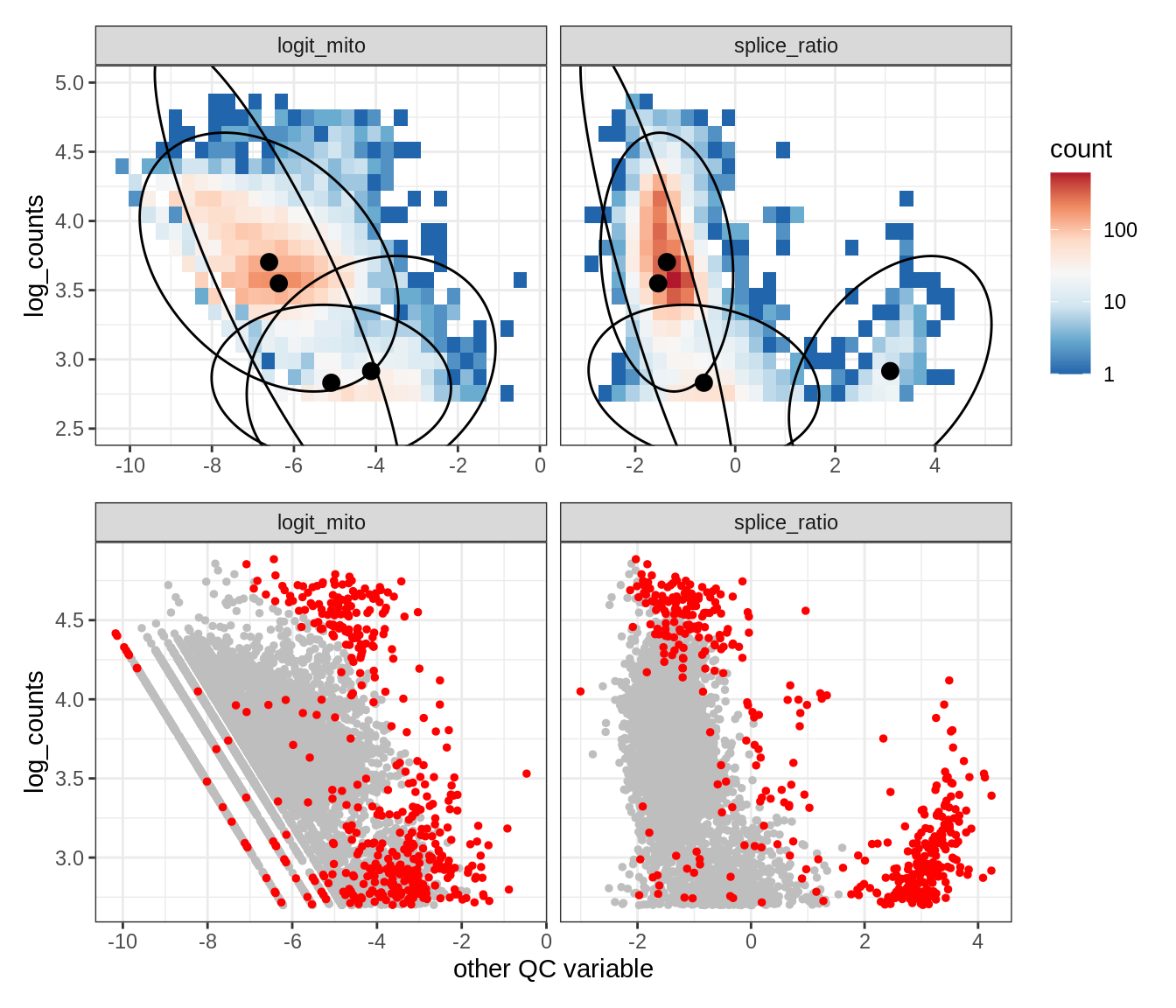
WM169
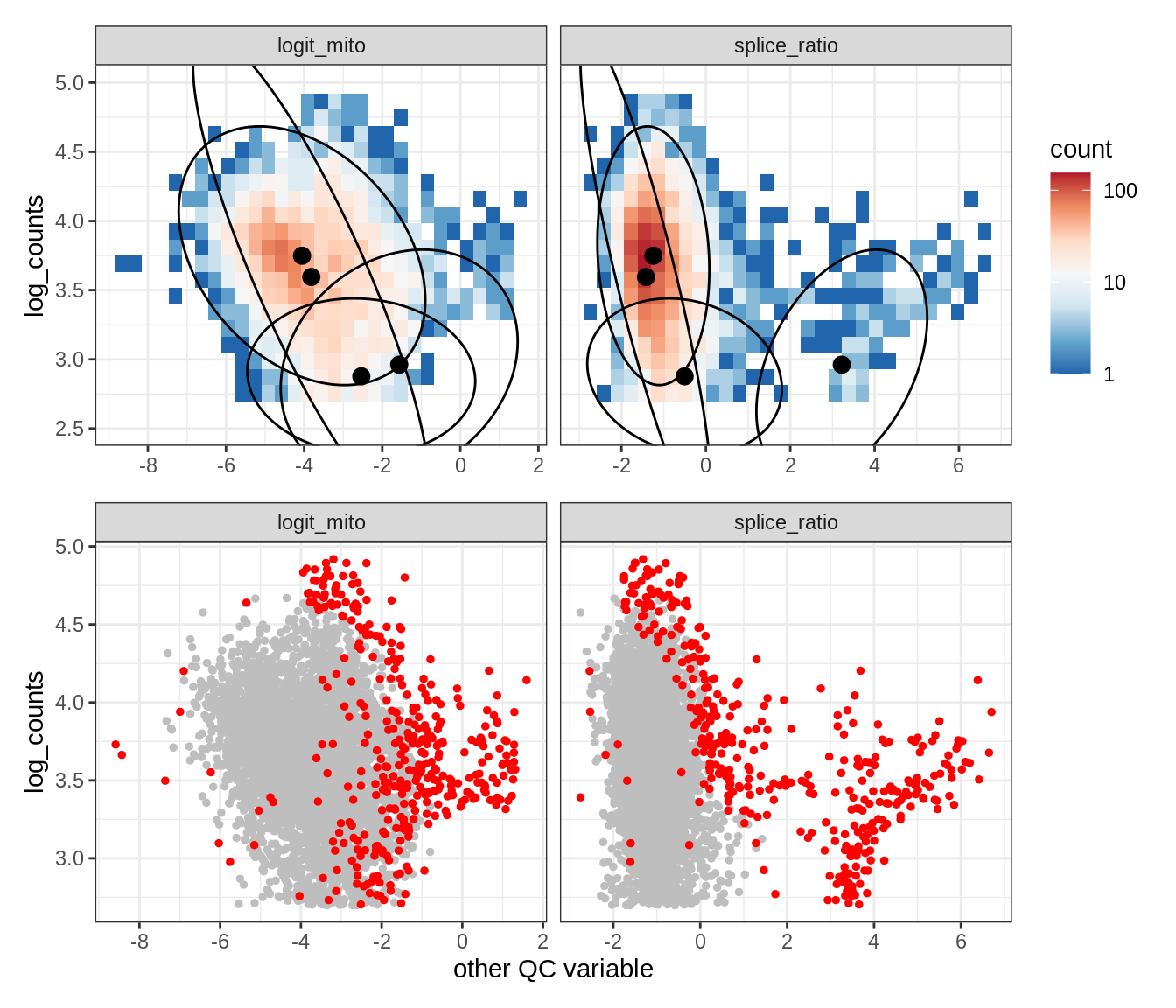
WM170
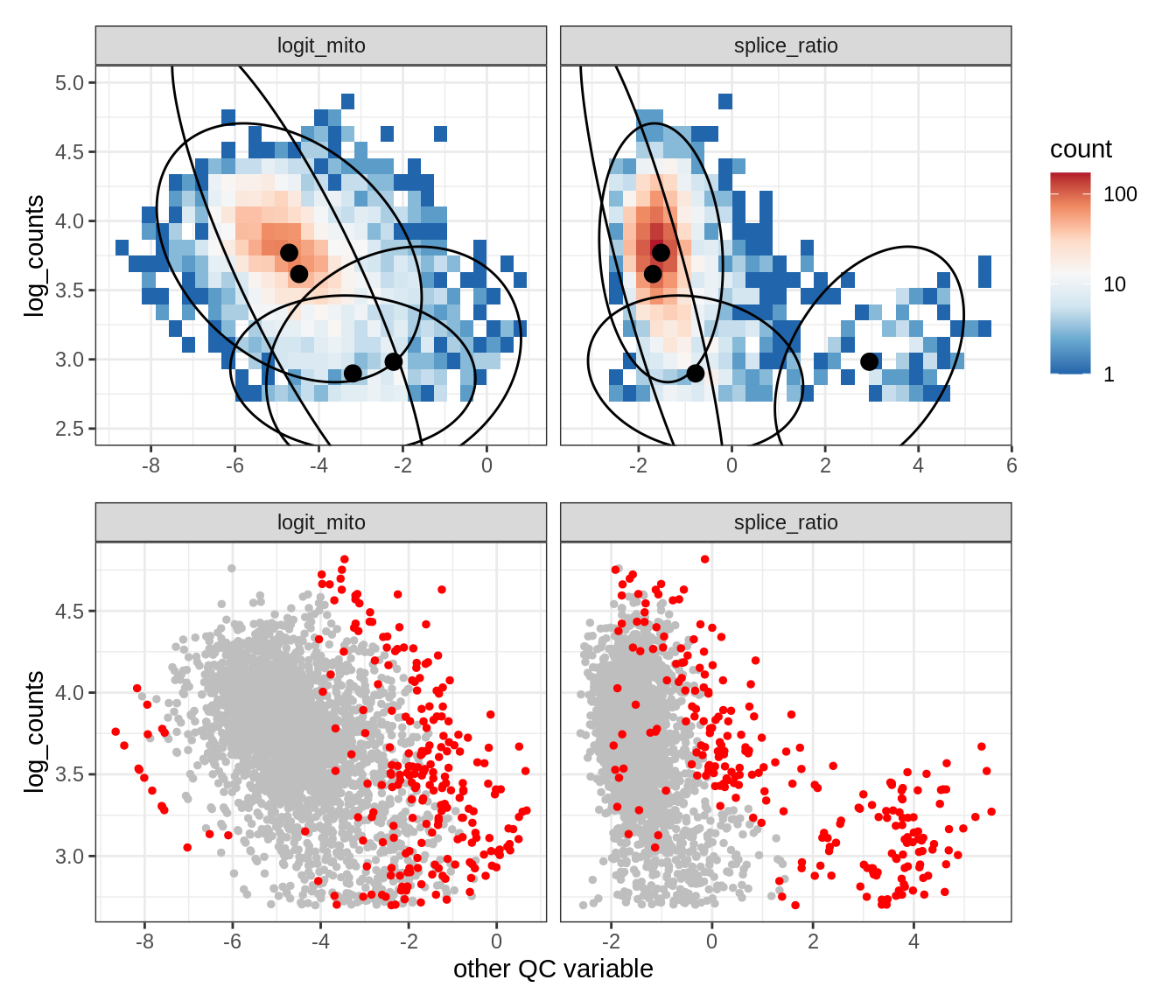
WM175
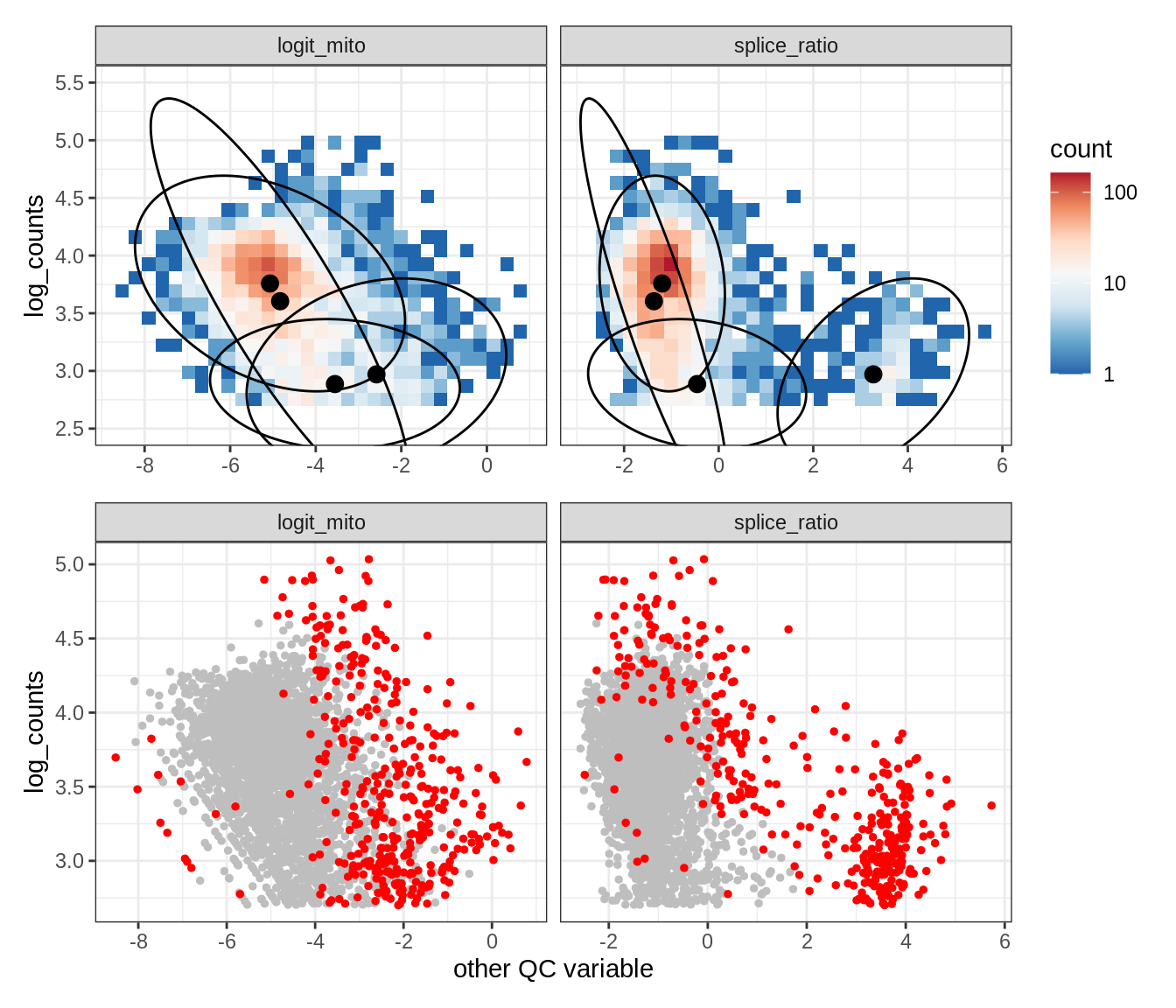
WM177
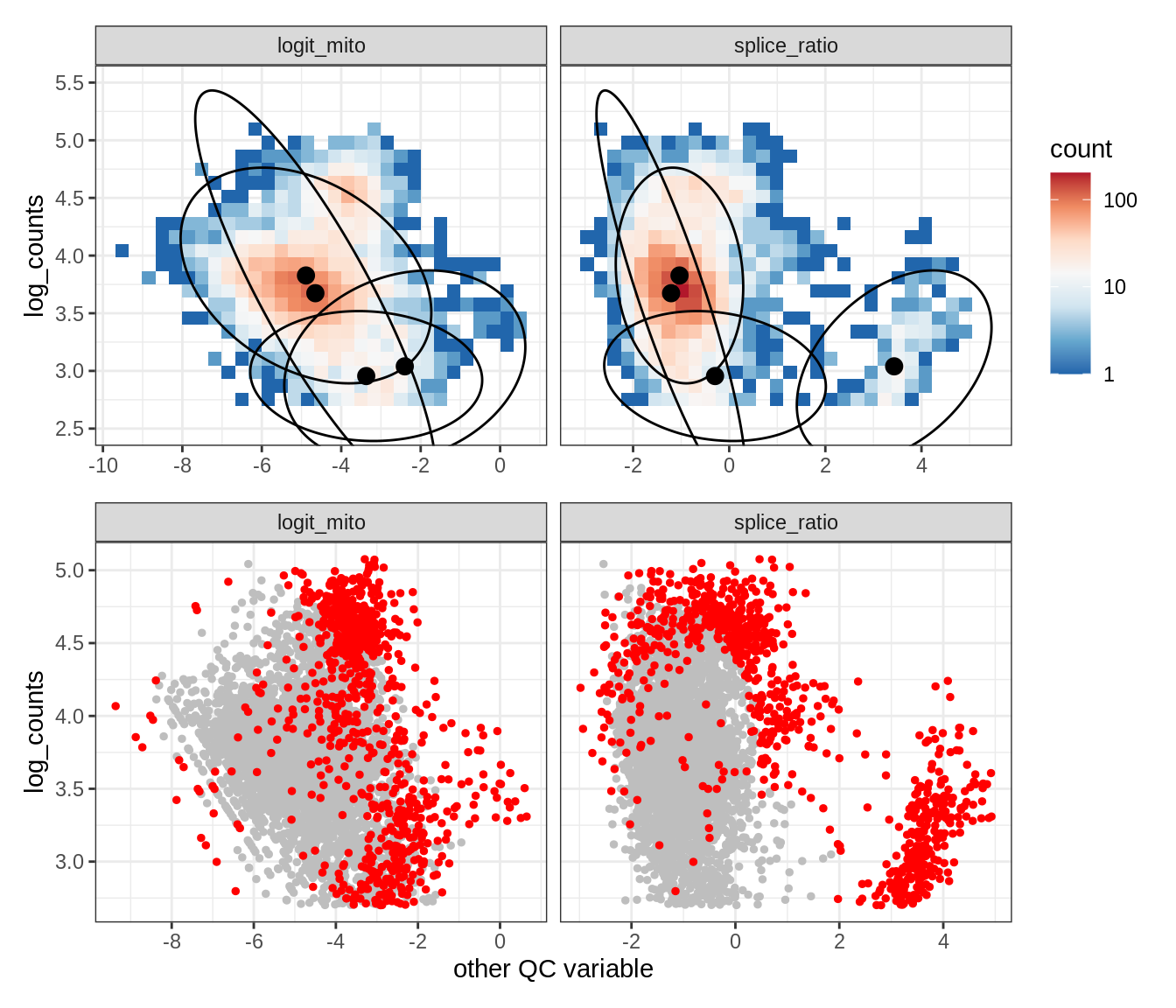
WM184
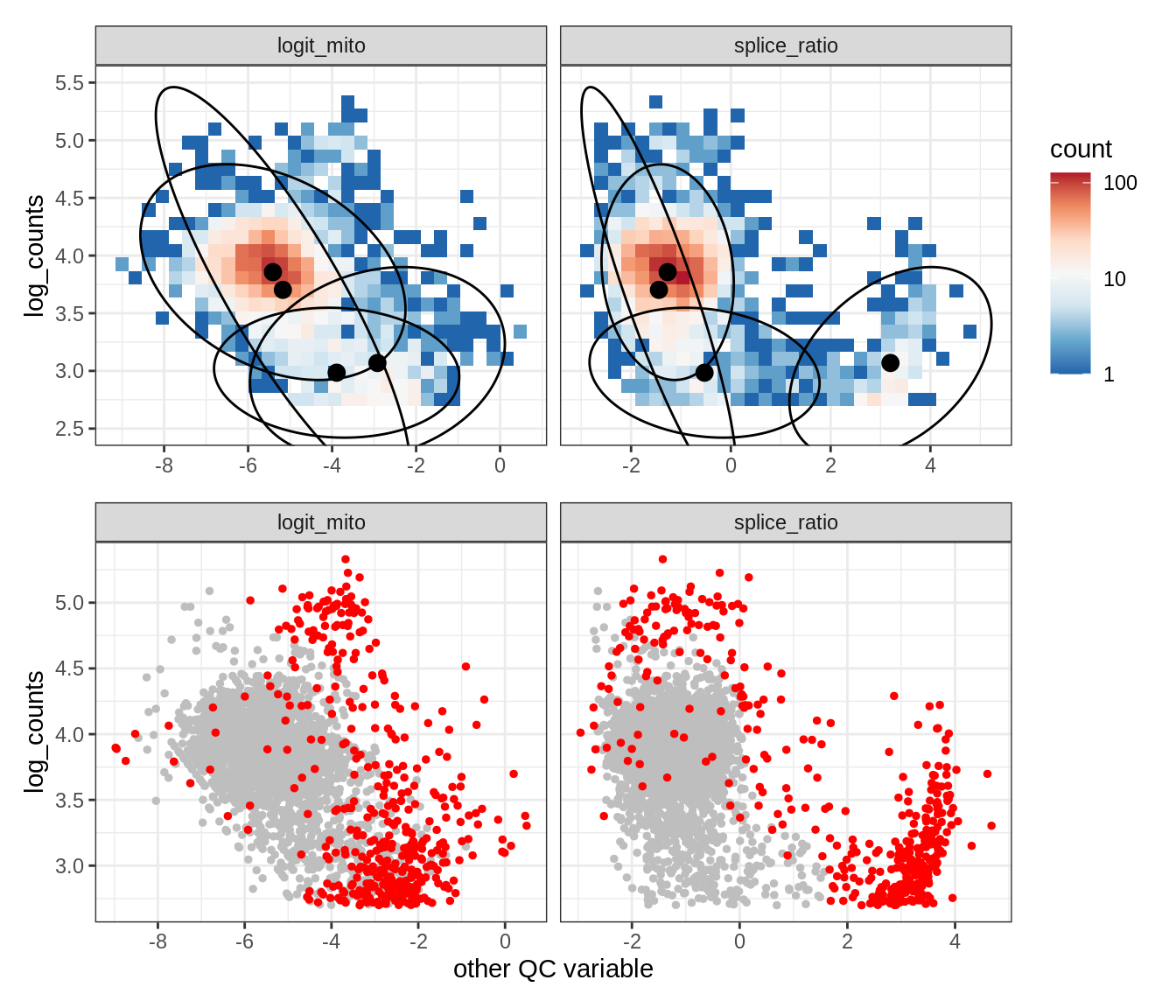
WM190
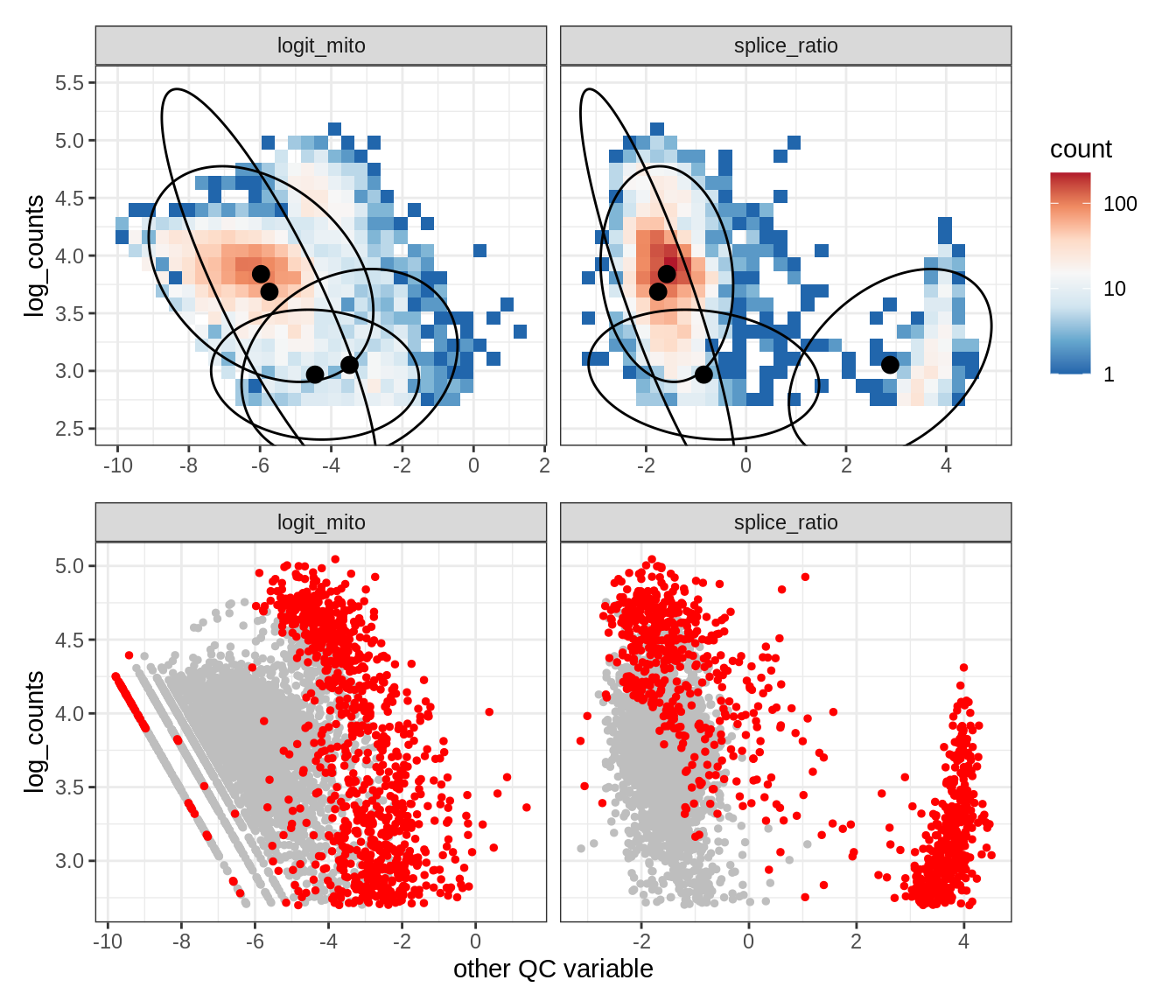
WM195
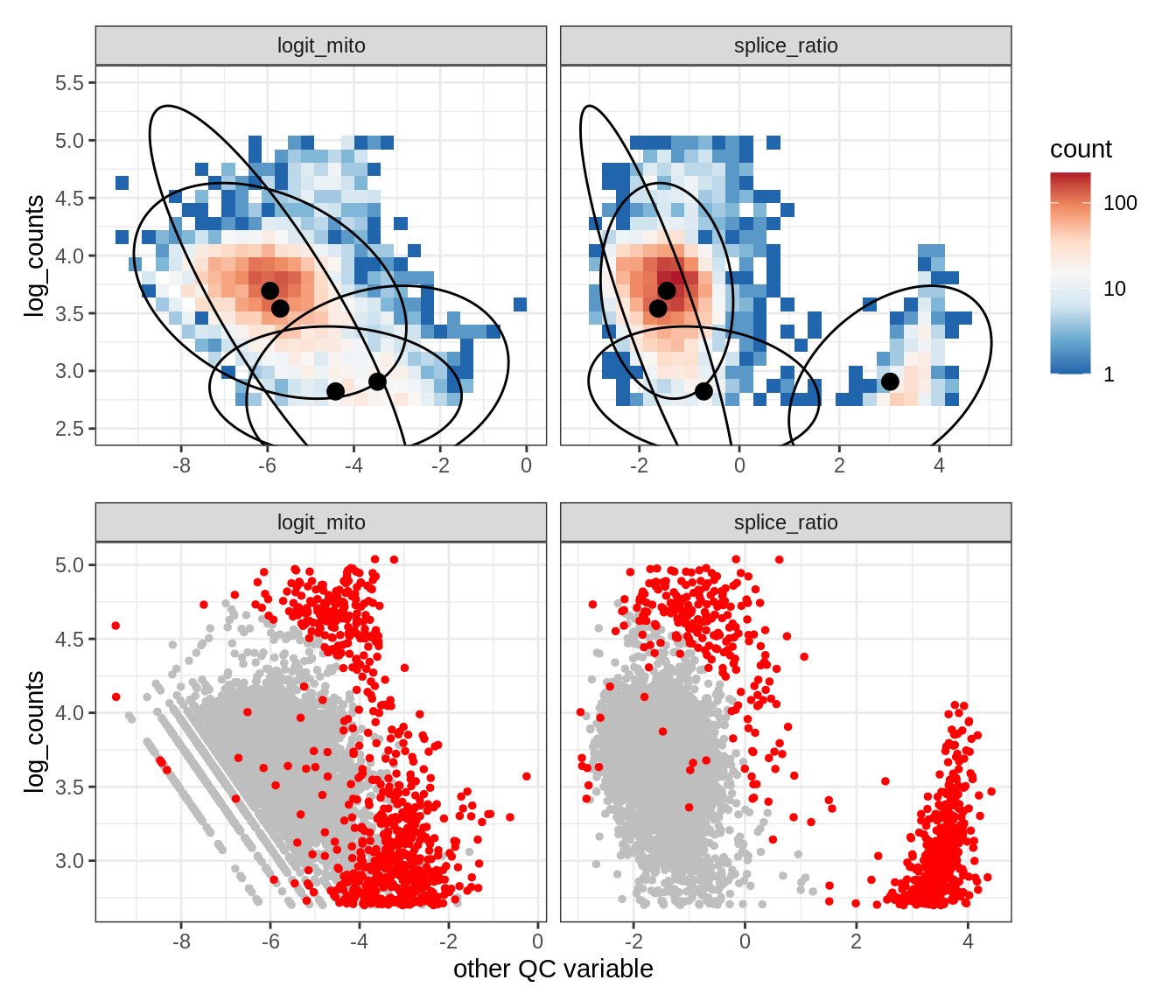
WM196
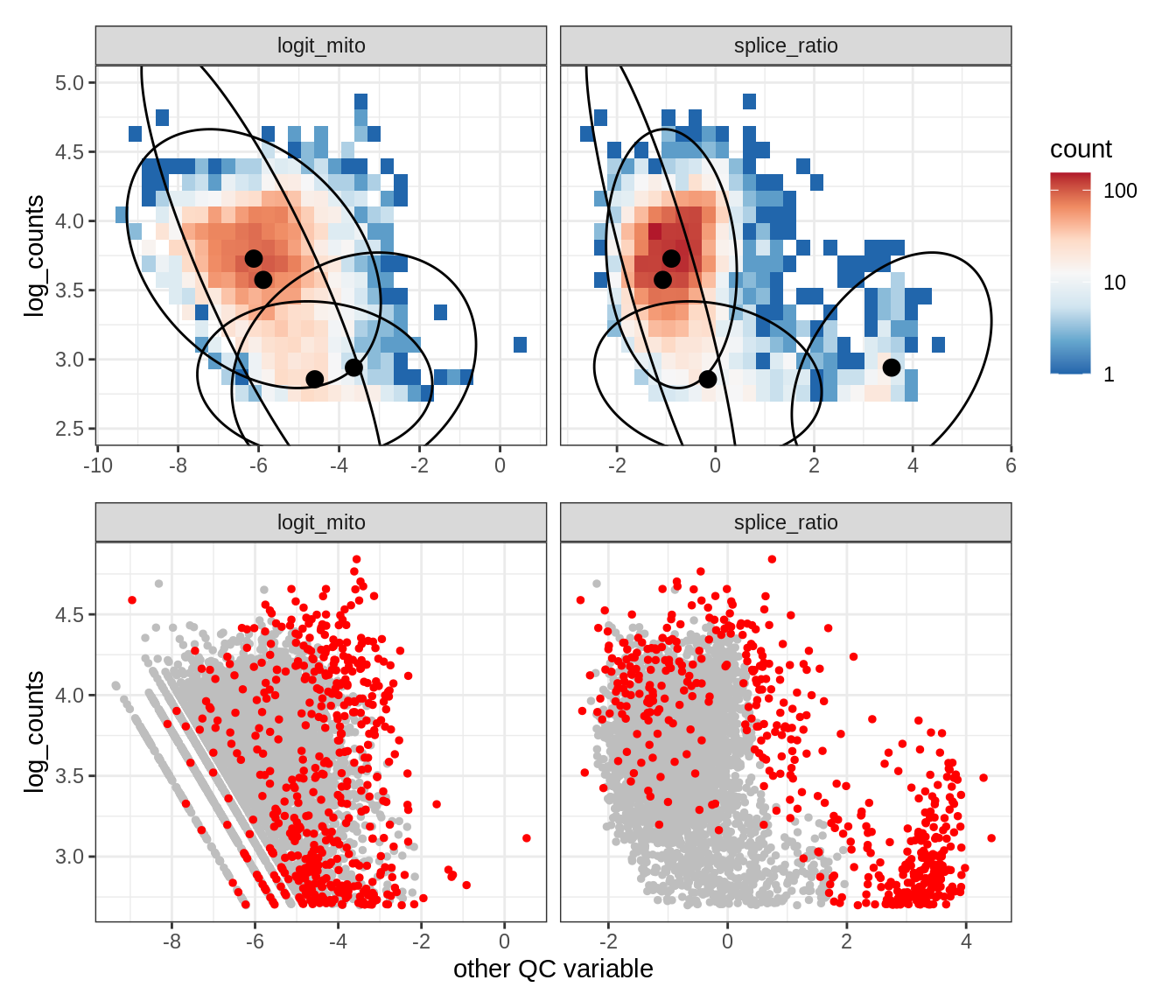
SG2
EU009
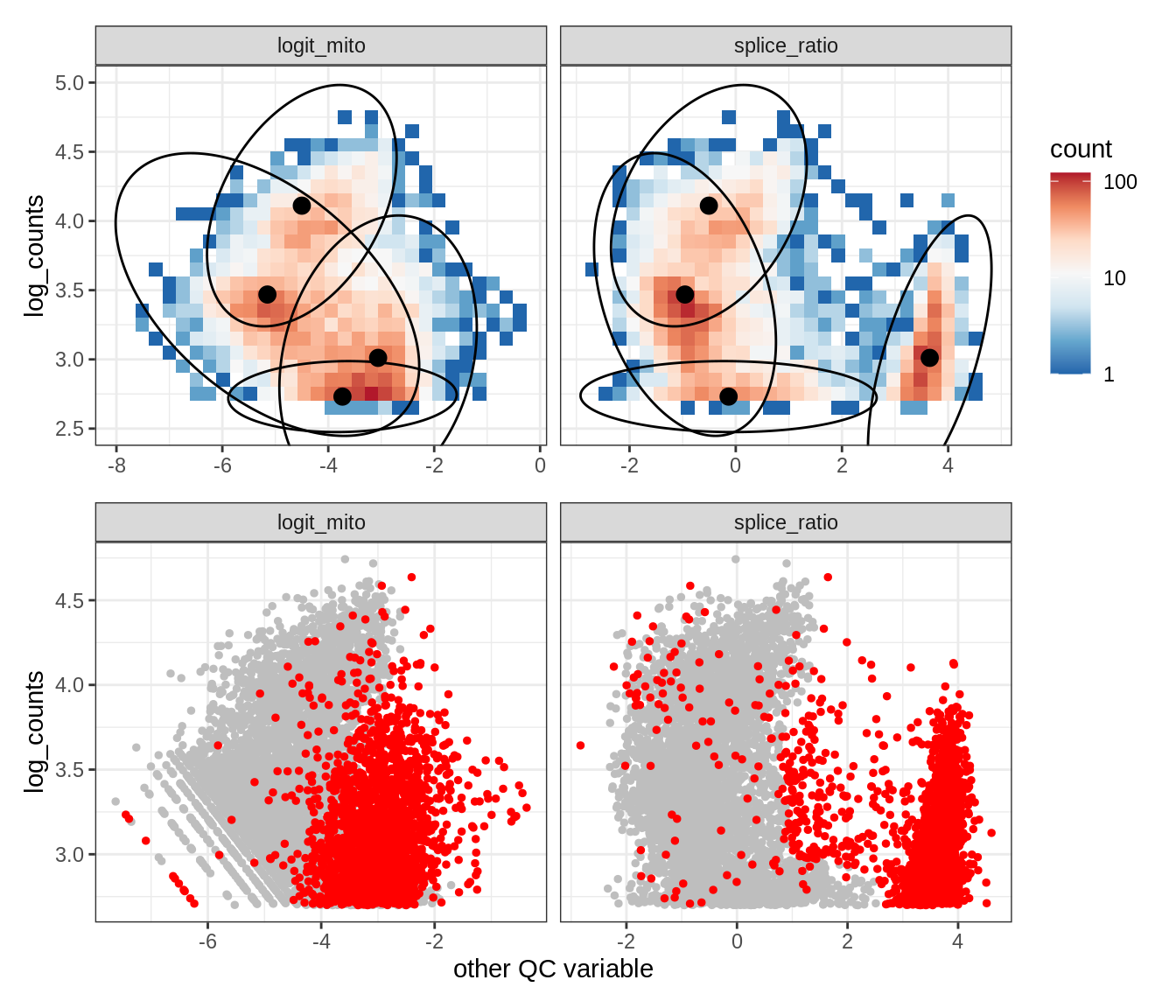
EU012
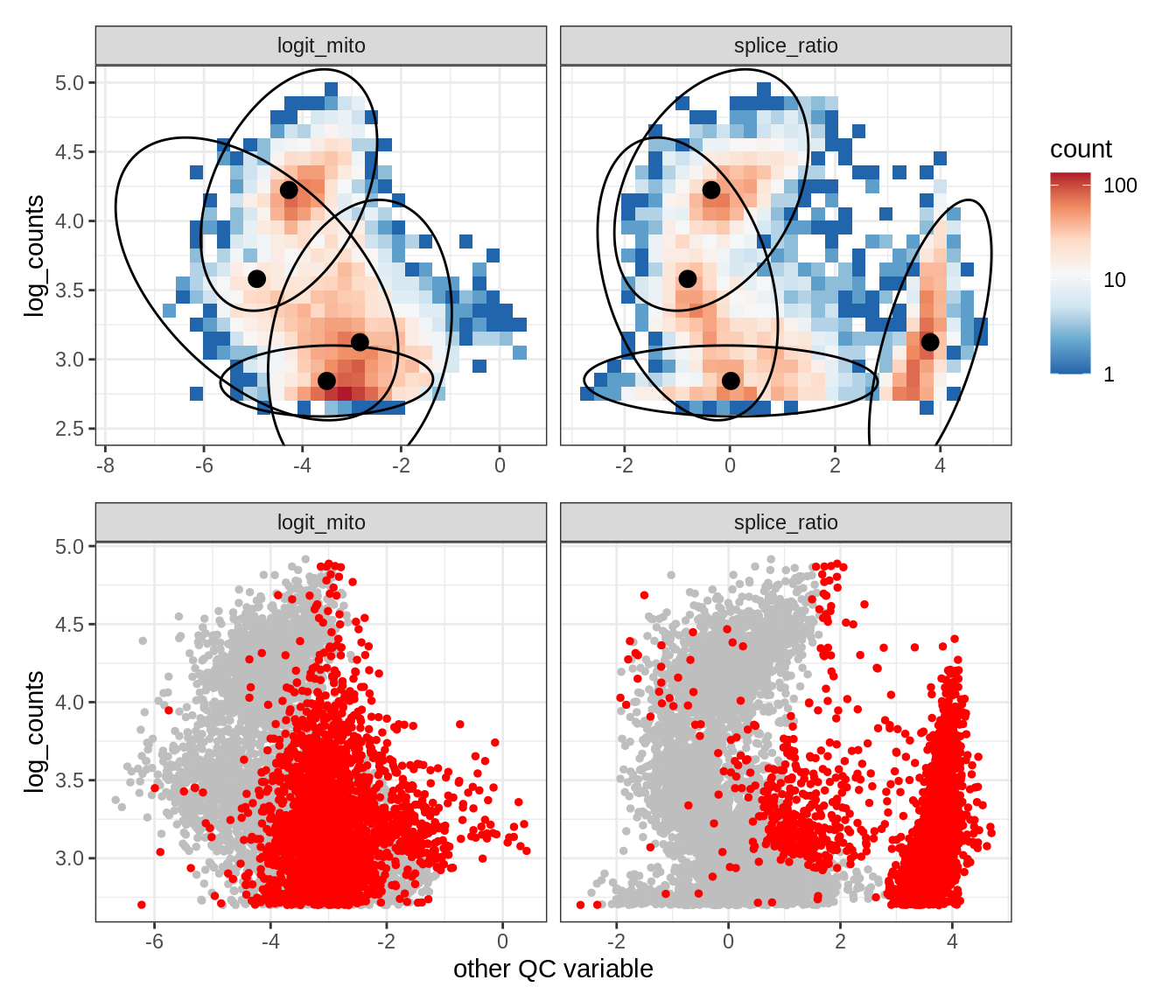
EU014
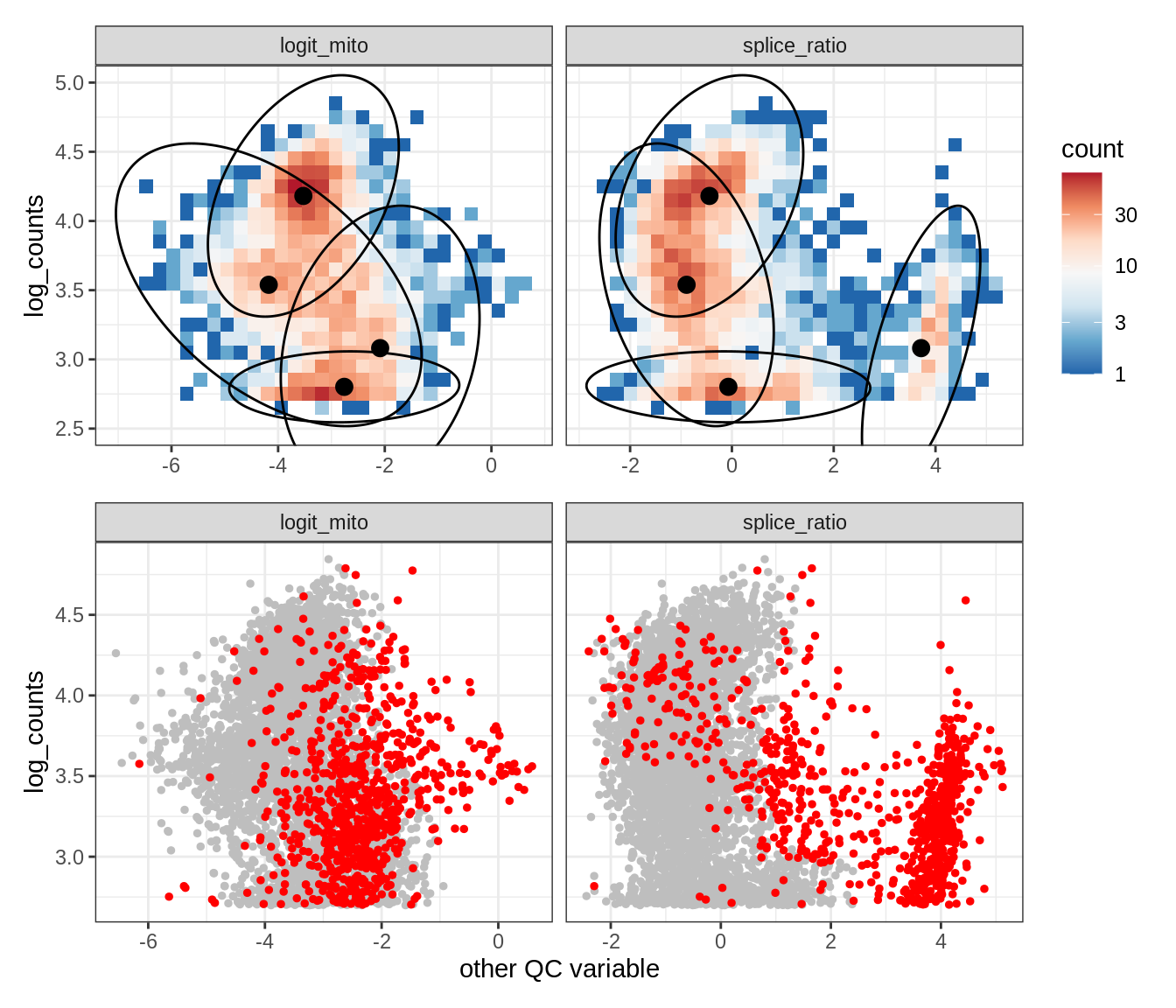
EU020
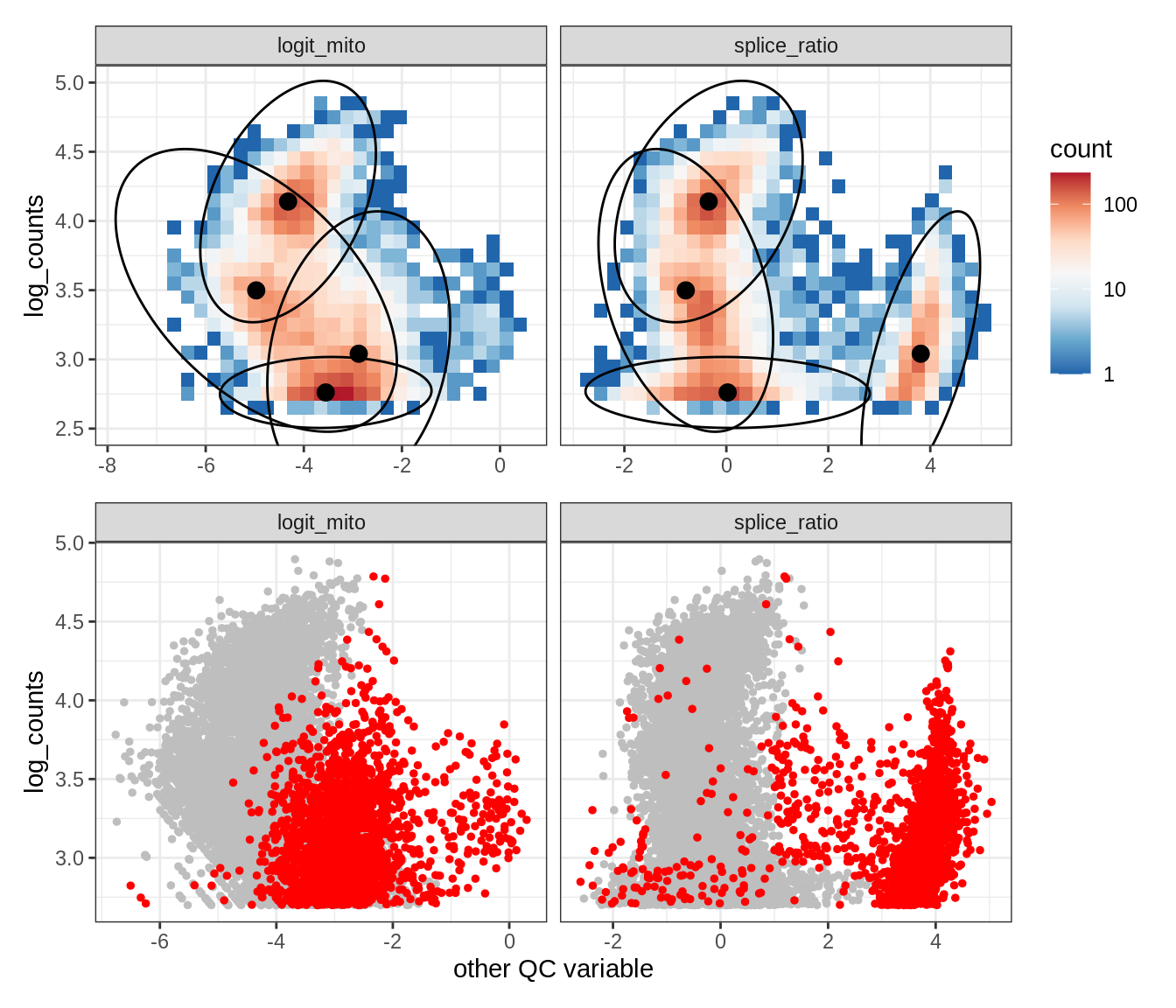
EU021
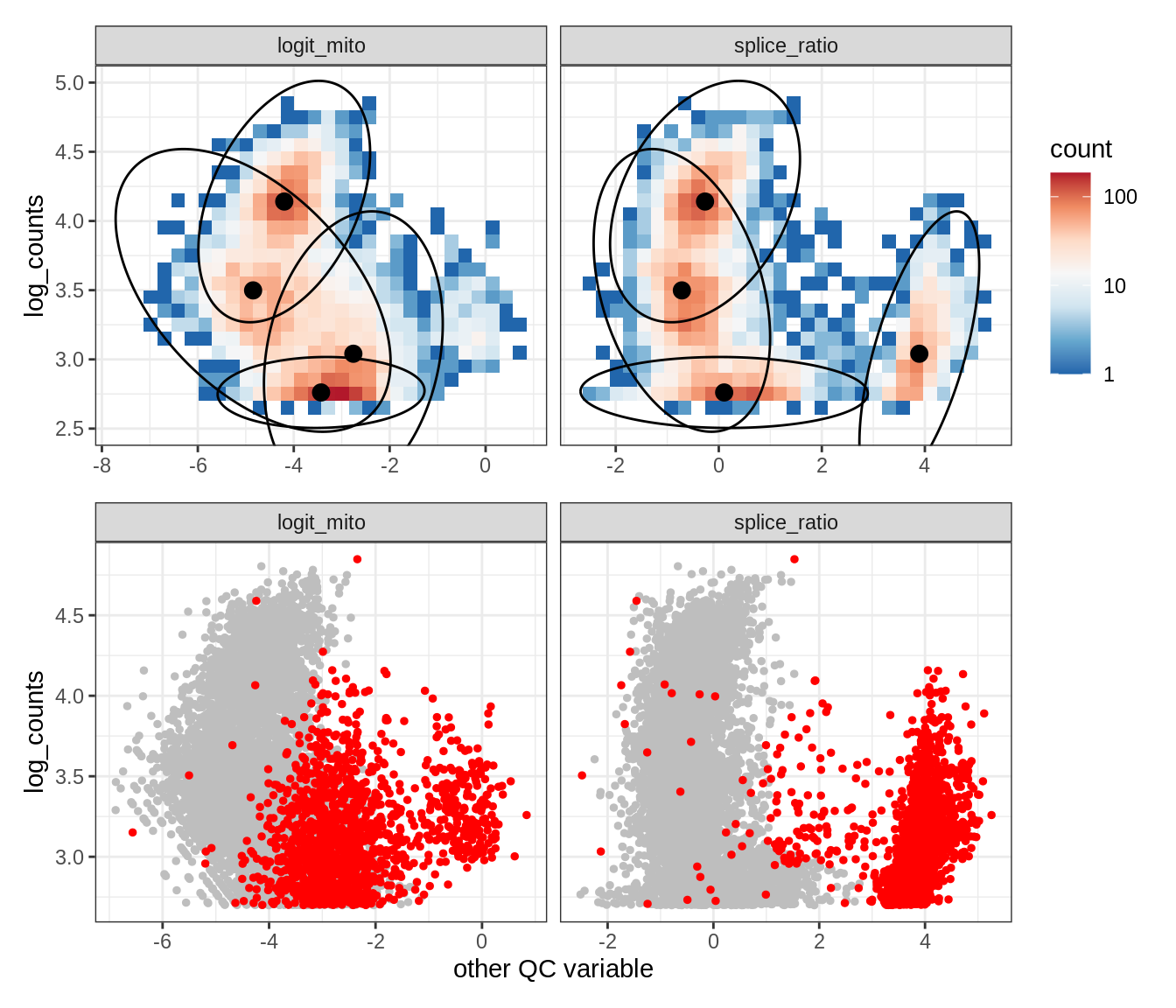
EU026
EU027
EU051
EU052
EU053
EU054
EU057
EU059
EU060
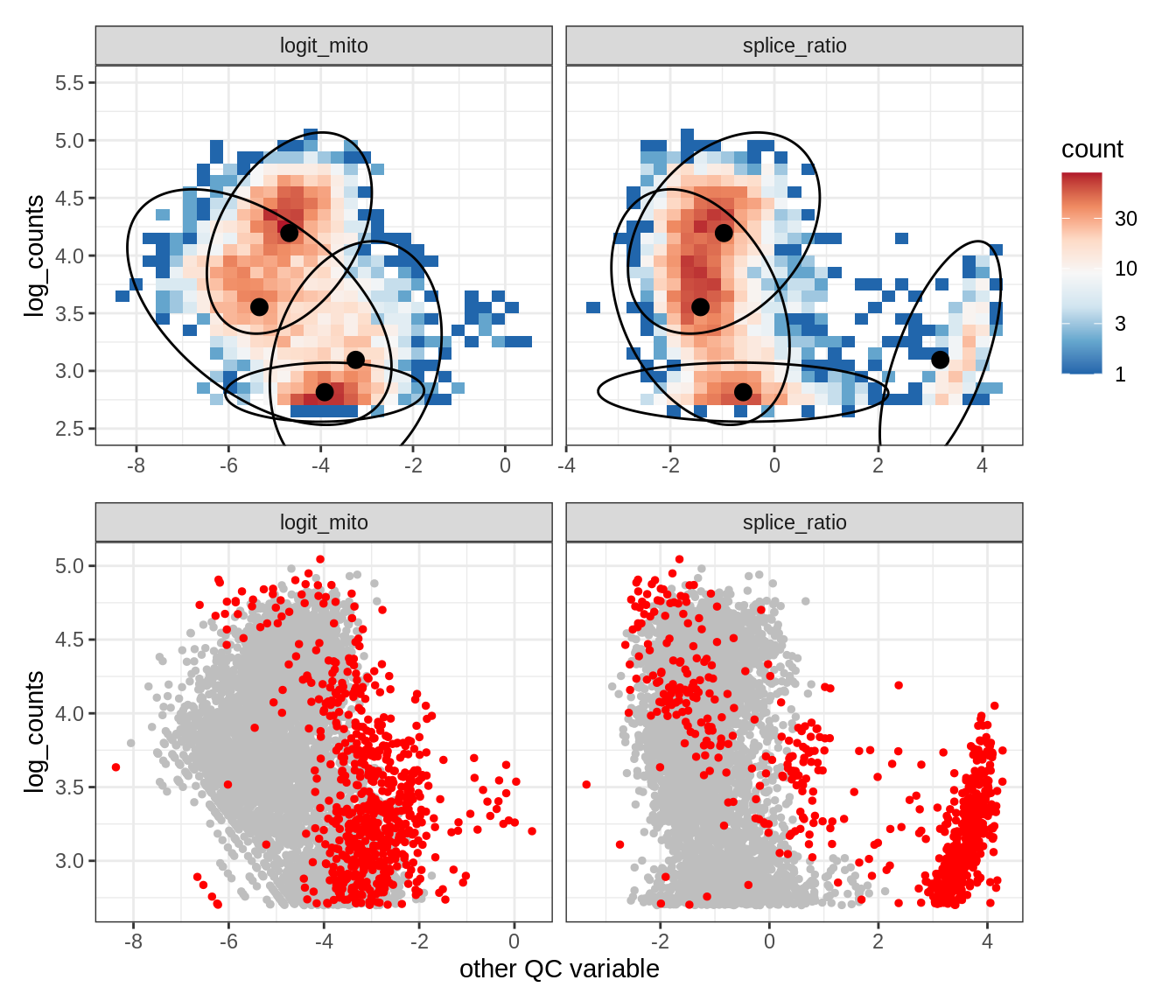
EU061
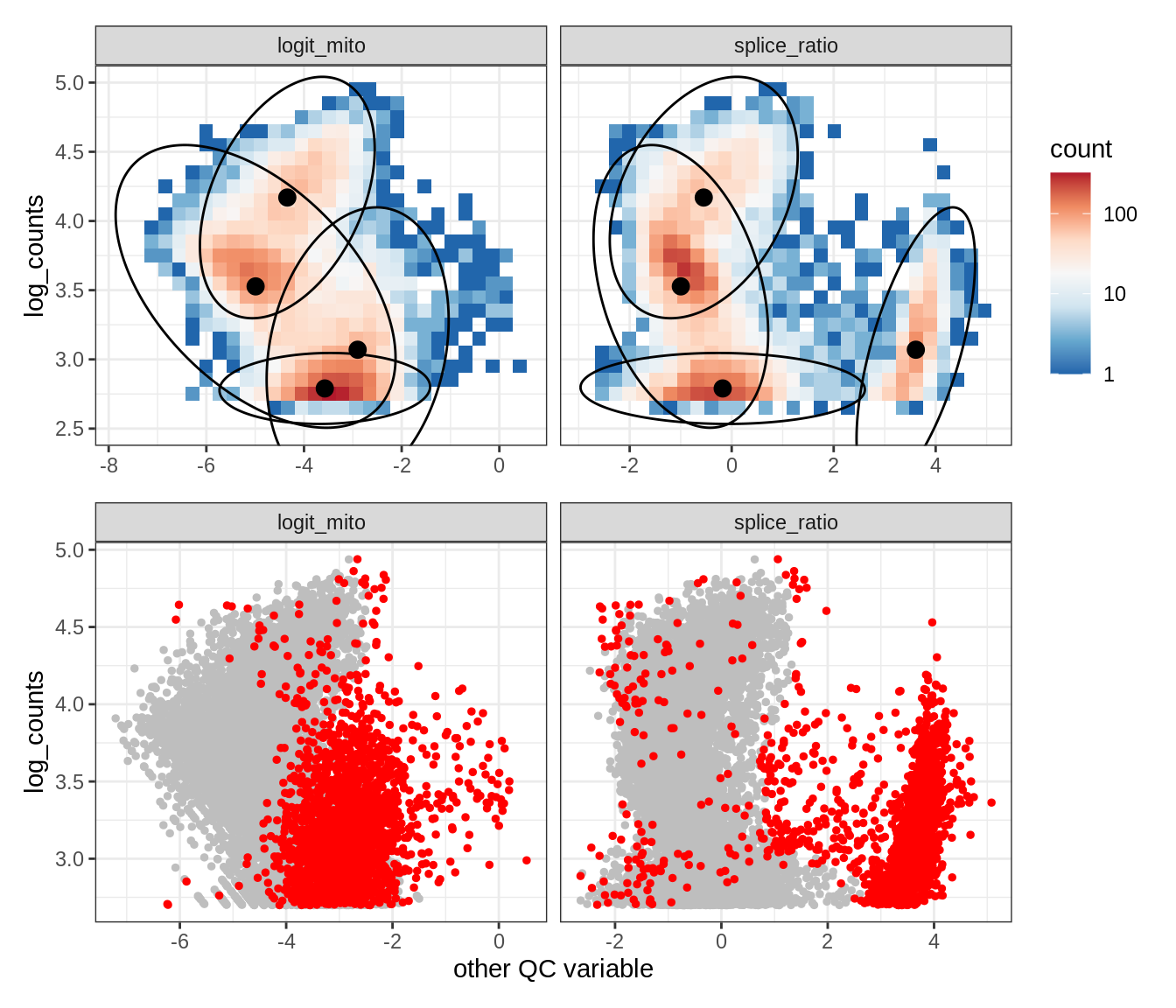
EU062
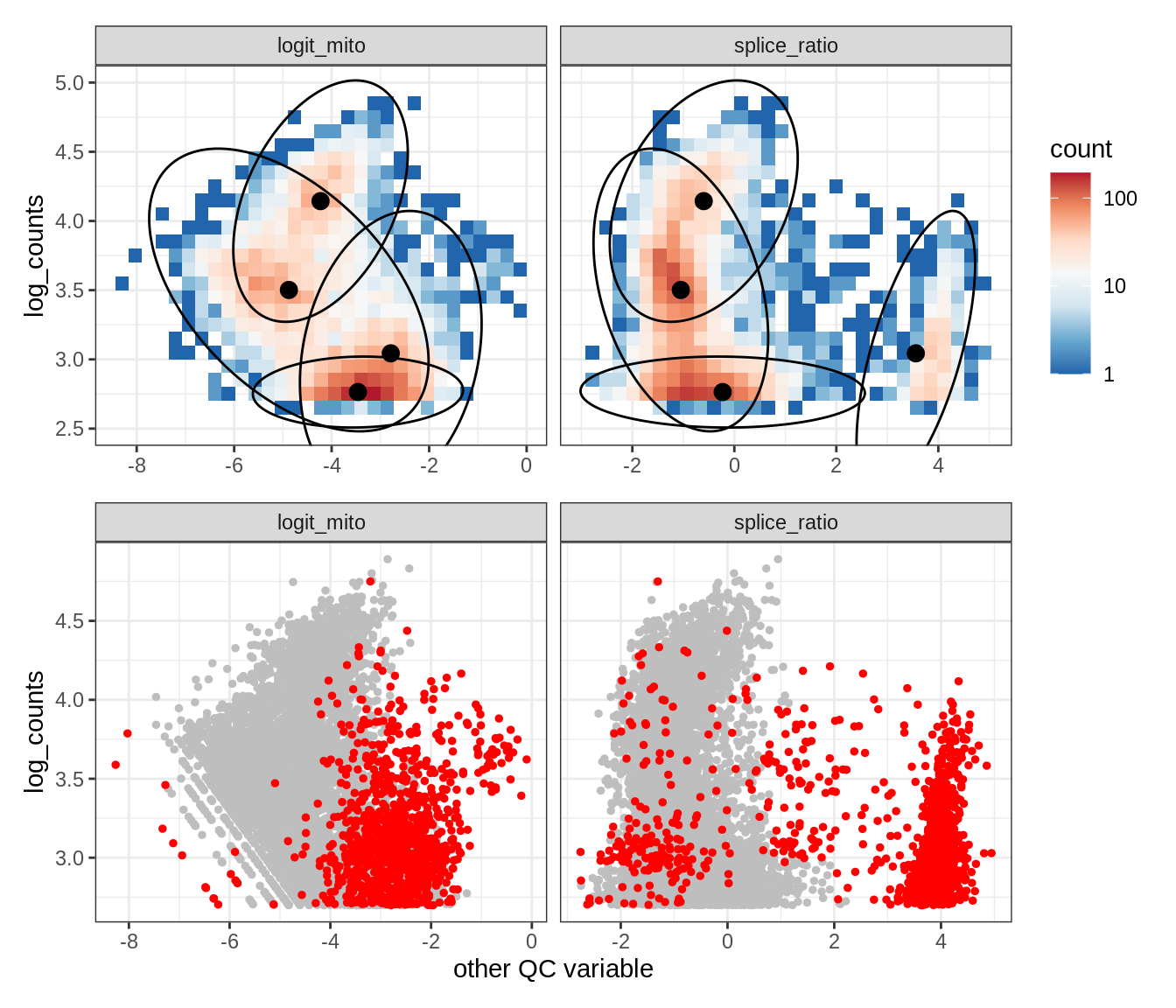
EU063
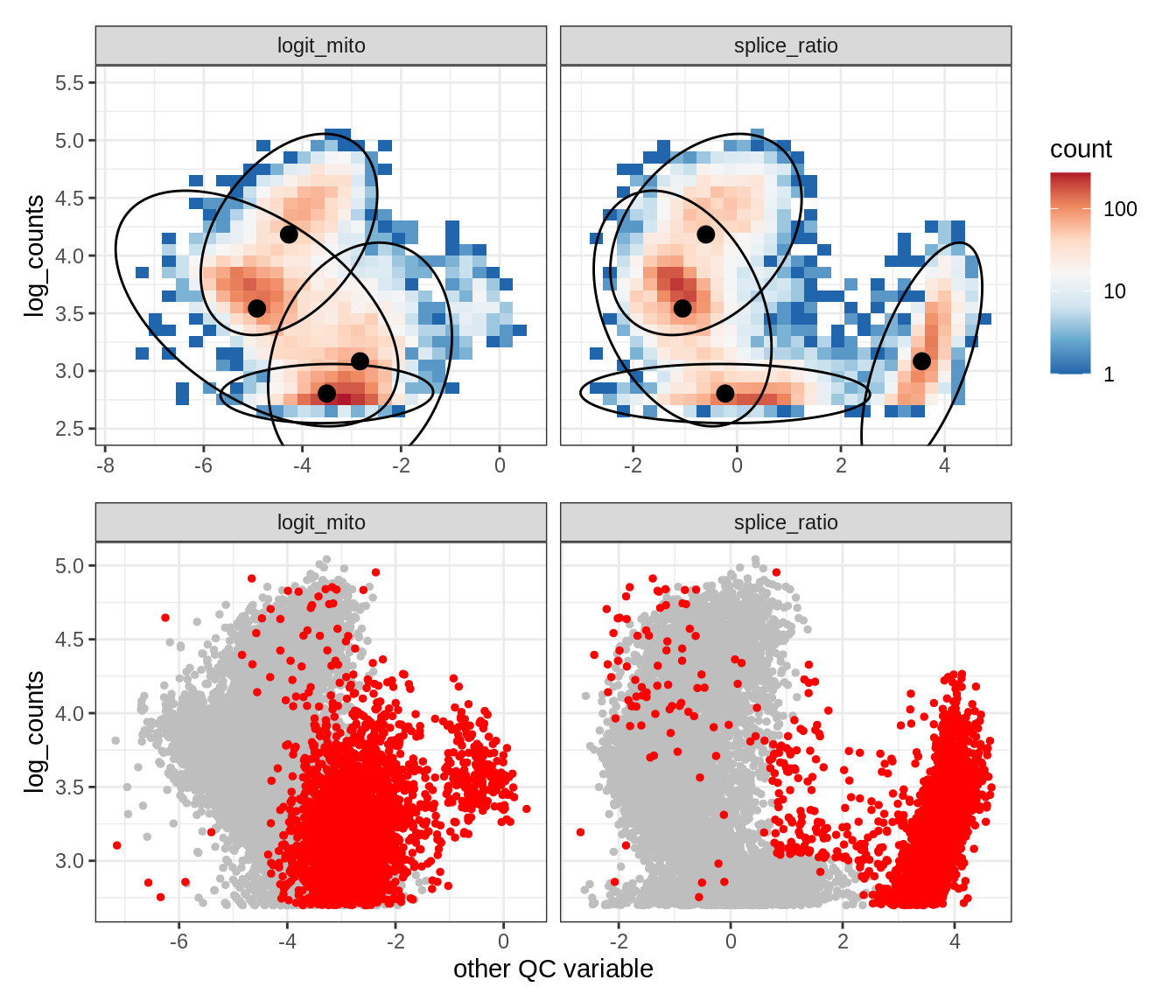
EU065
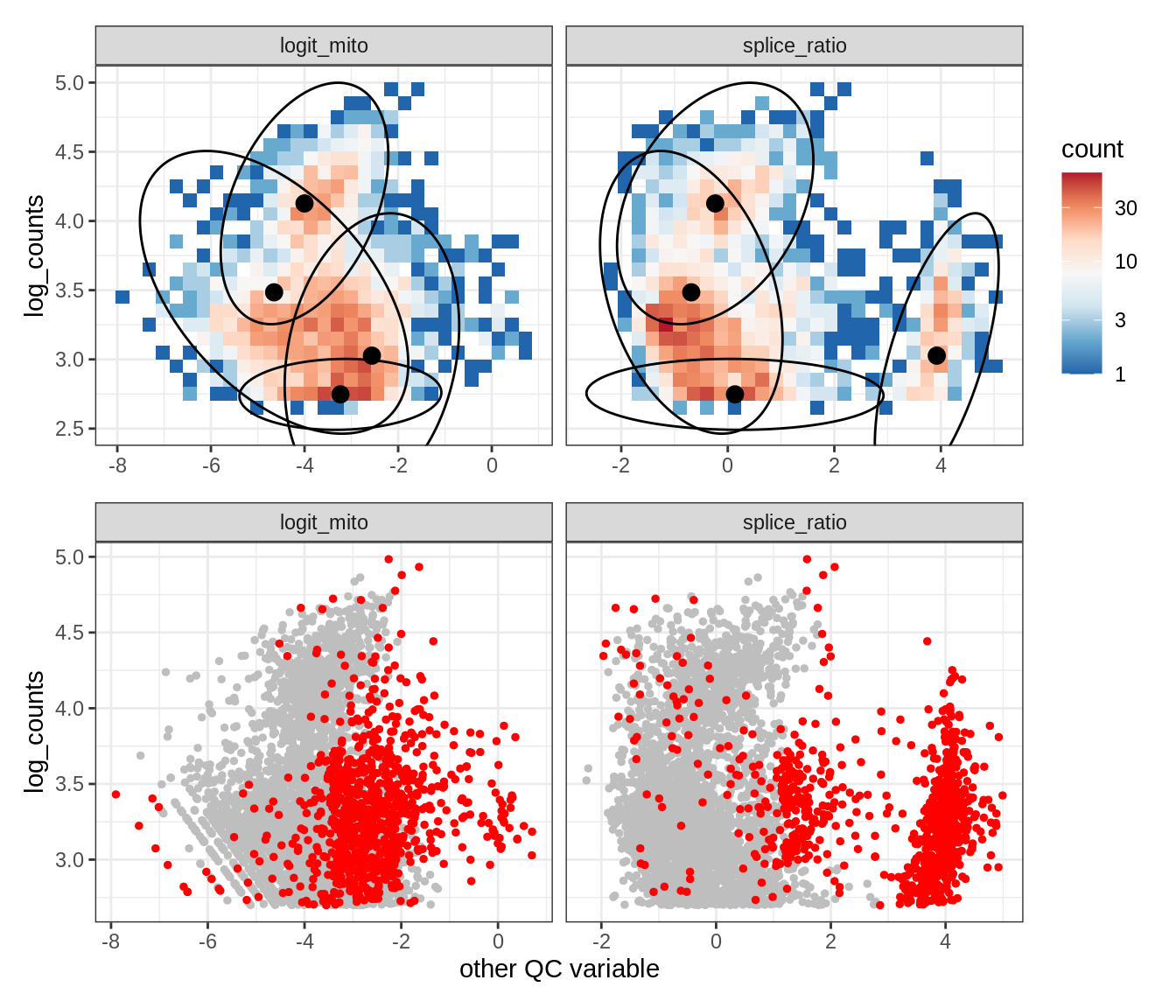
EU074
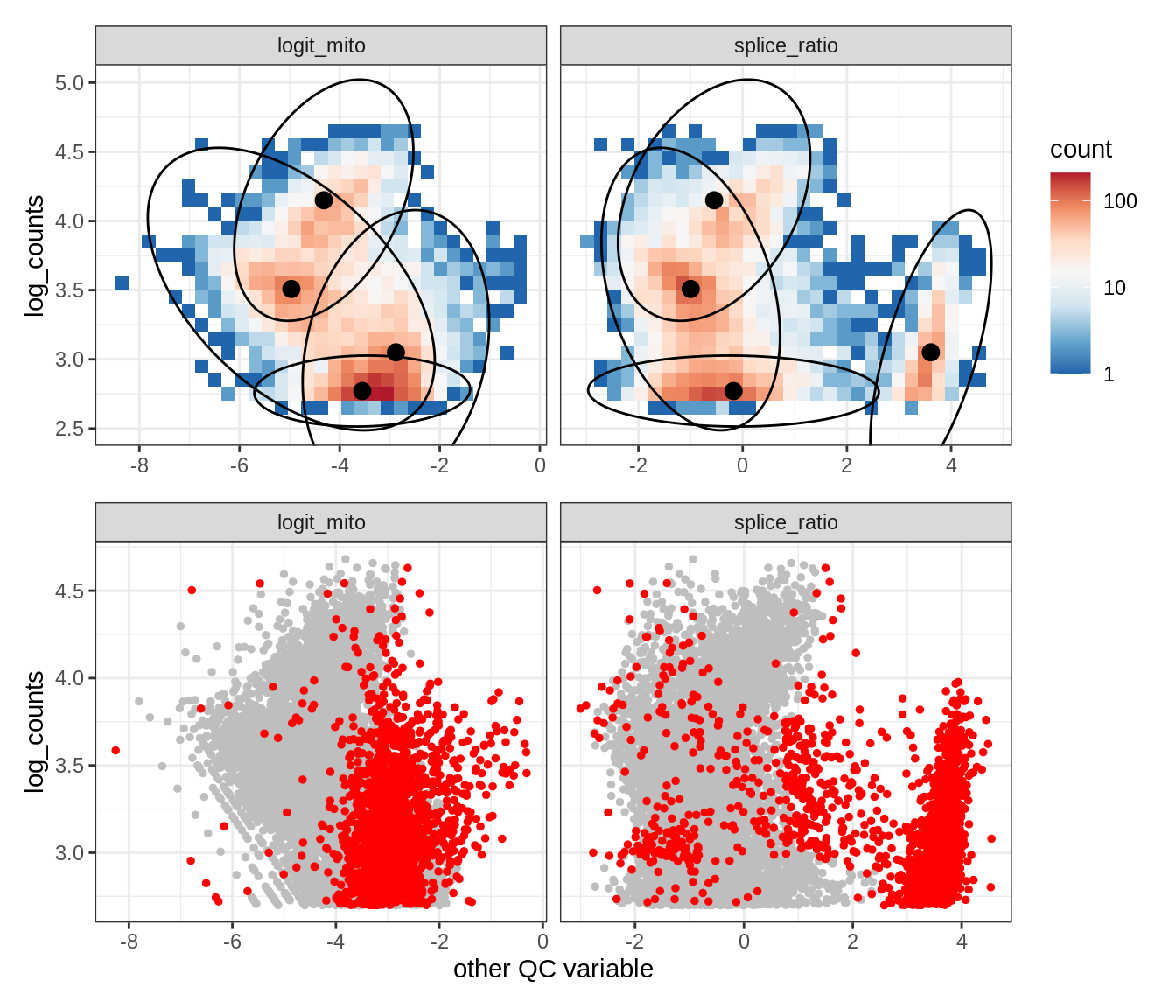
WM109
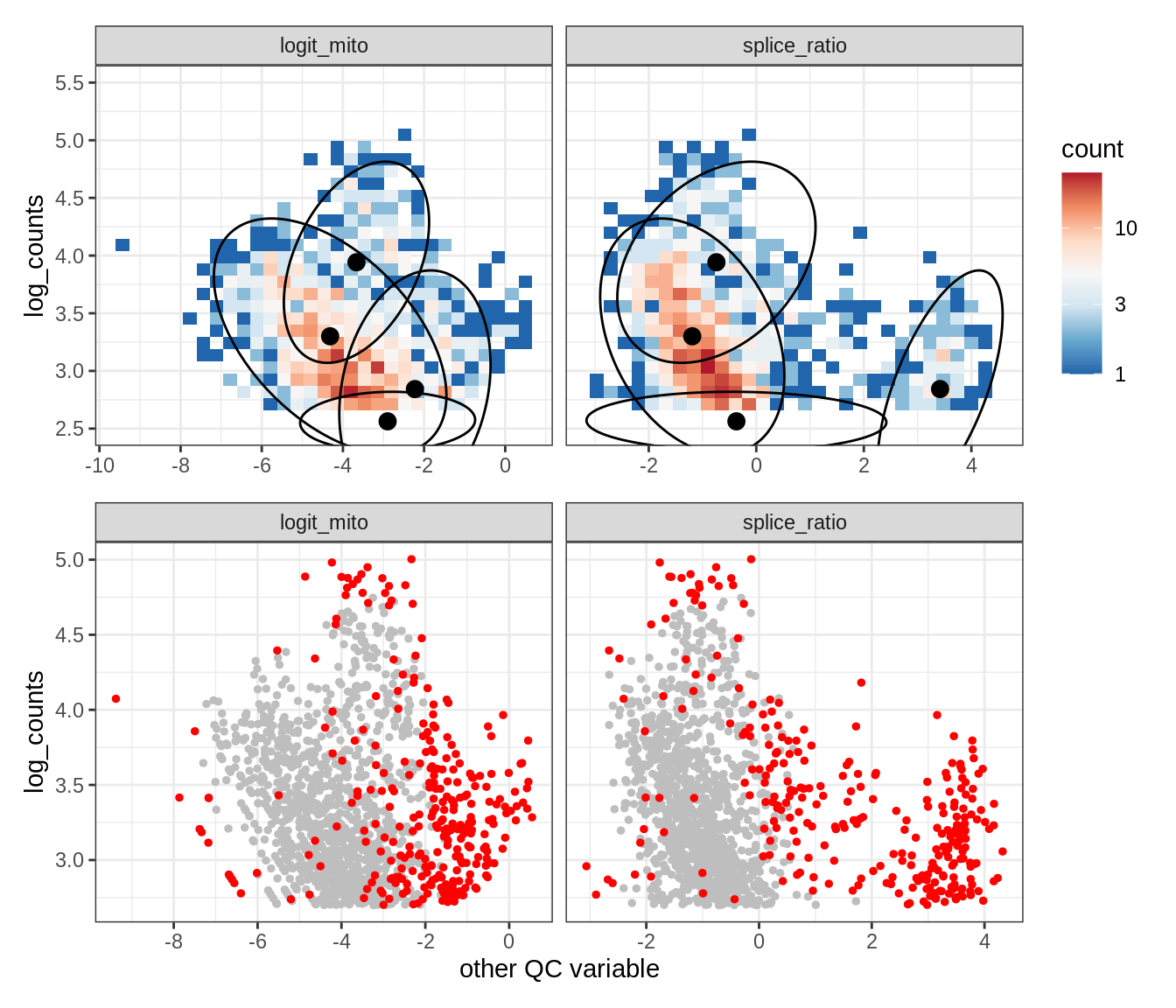
WM124
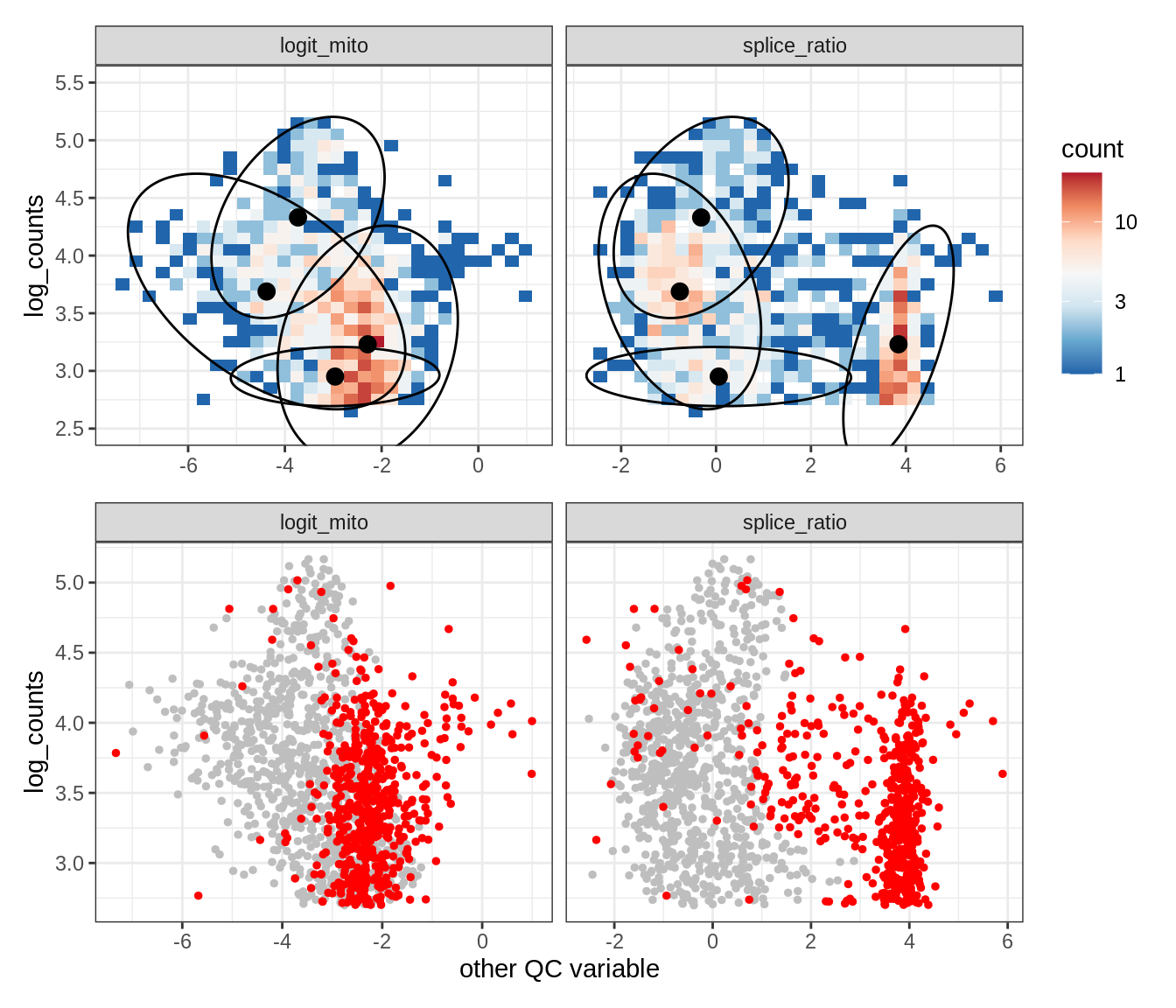
WM131
WM139
WM151
SG3
EU056
WM107
WM108
WM118
WM120
WM121
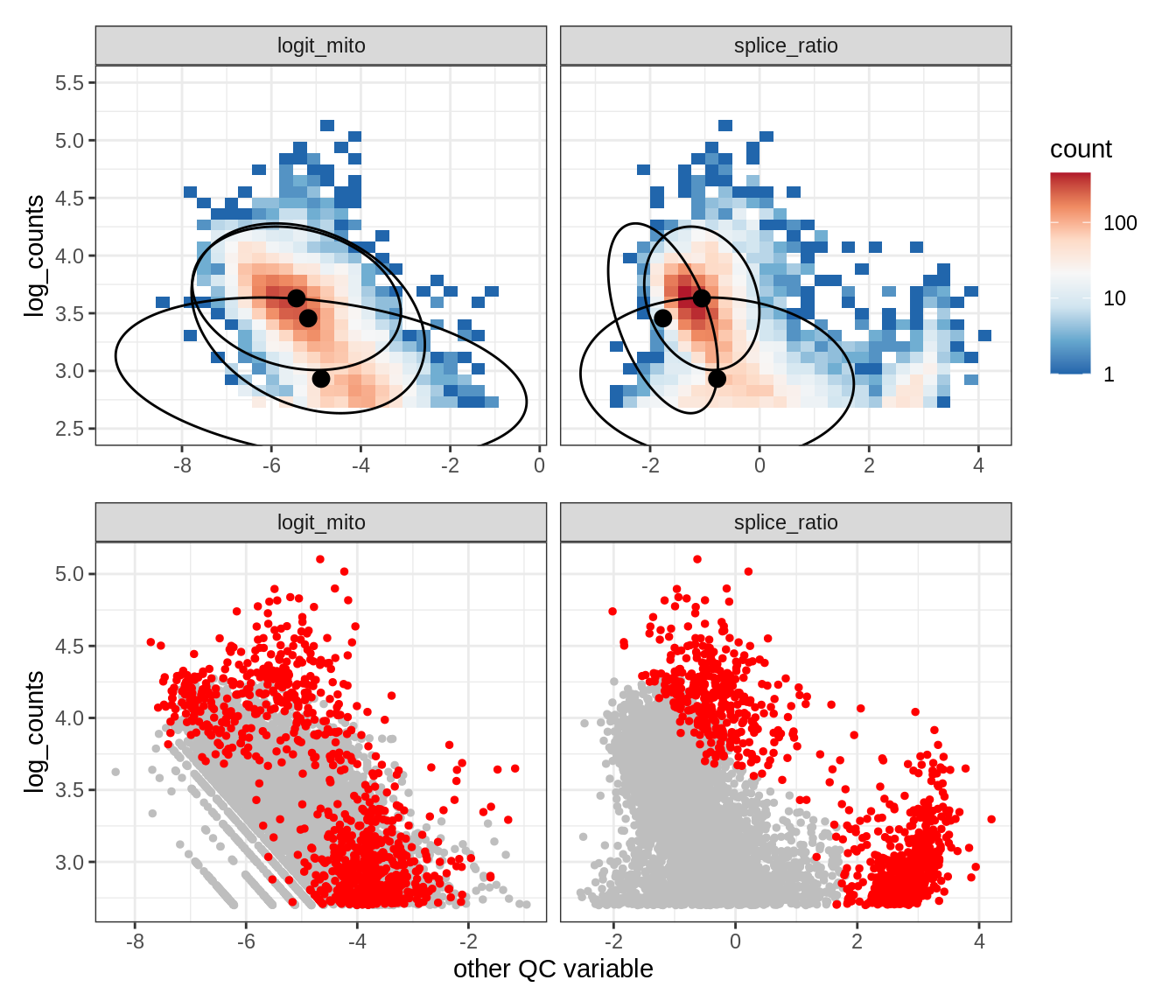
WM137
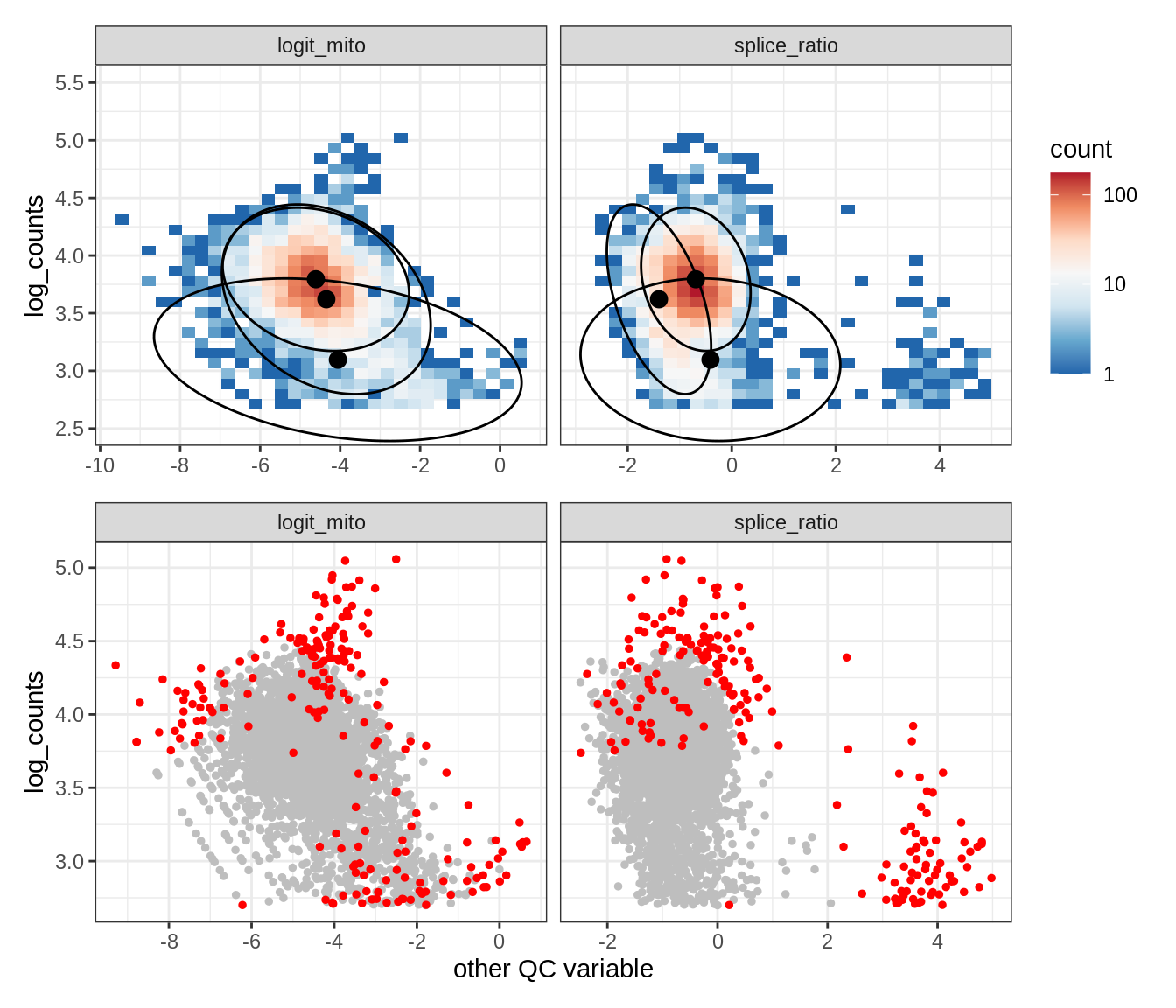
WM152
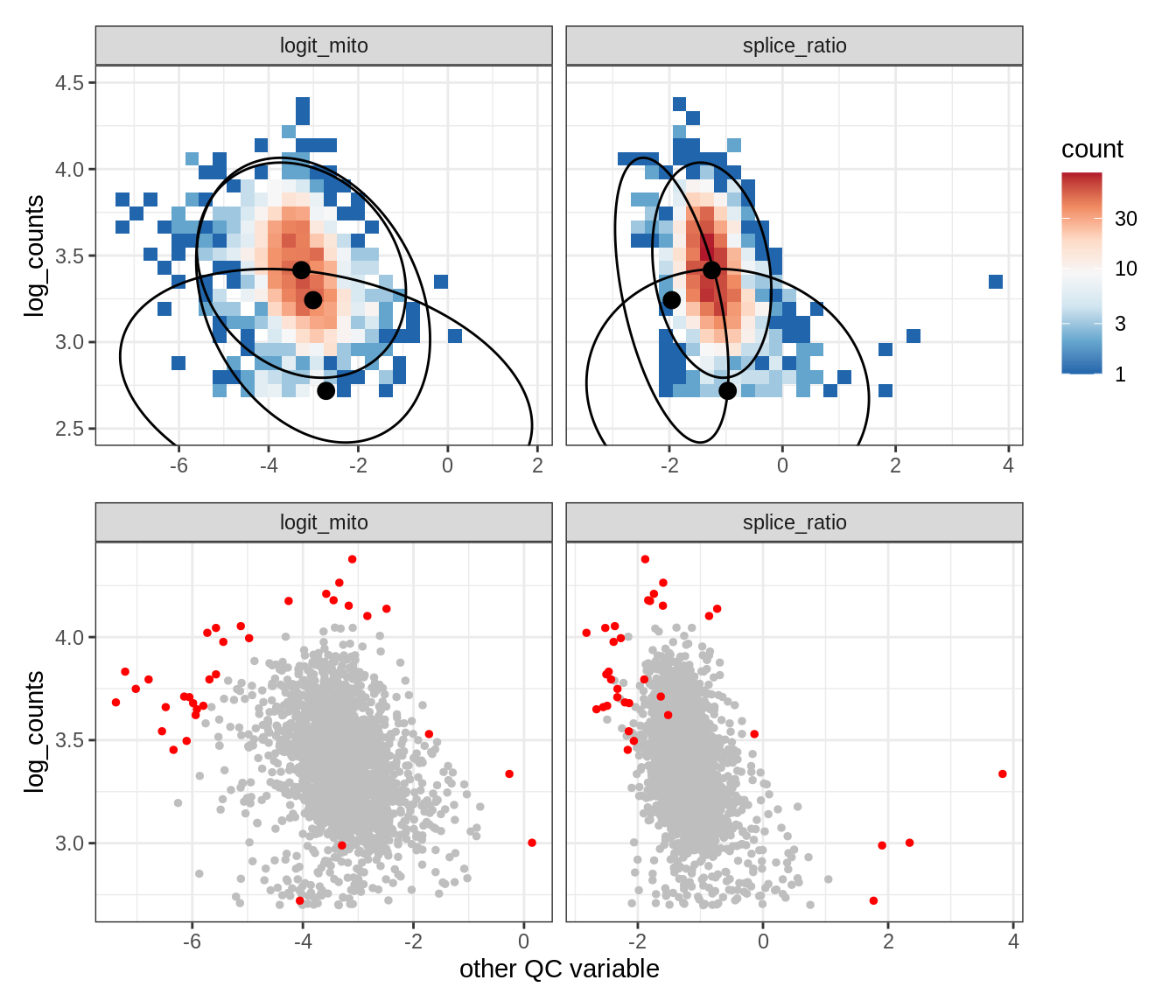
WM154
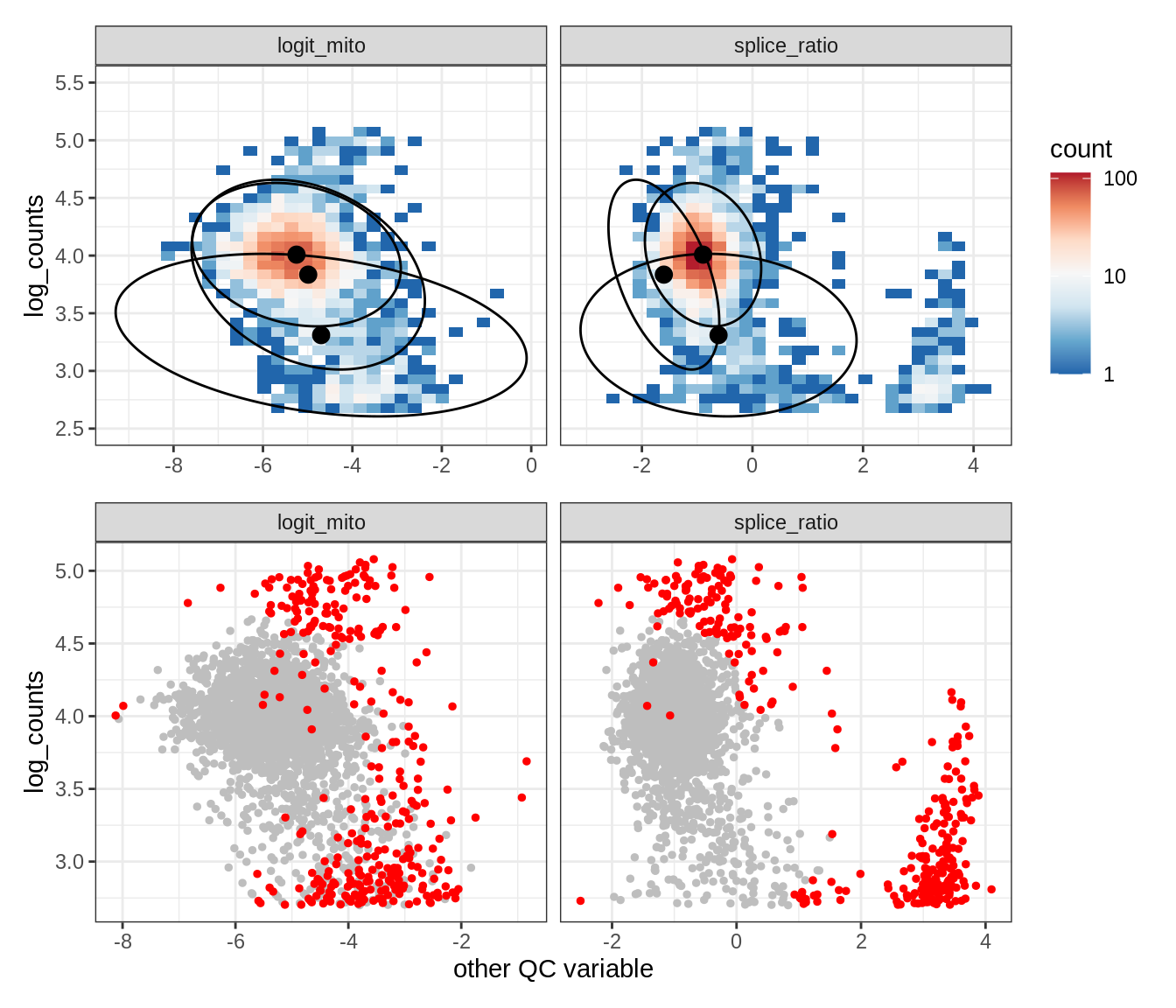
WM155
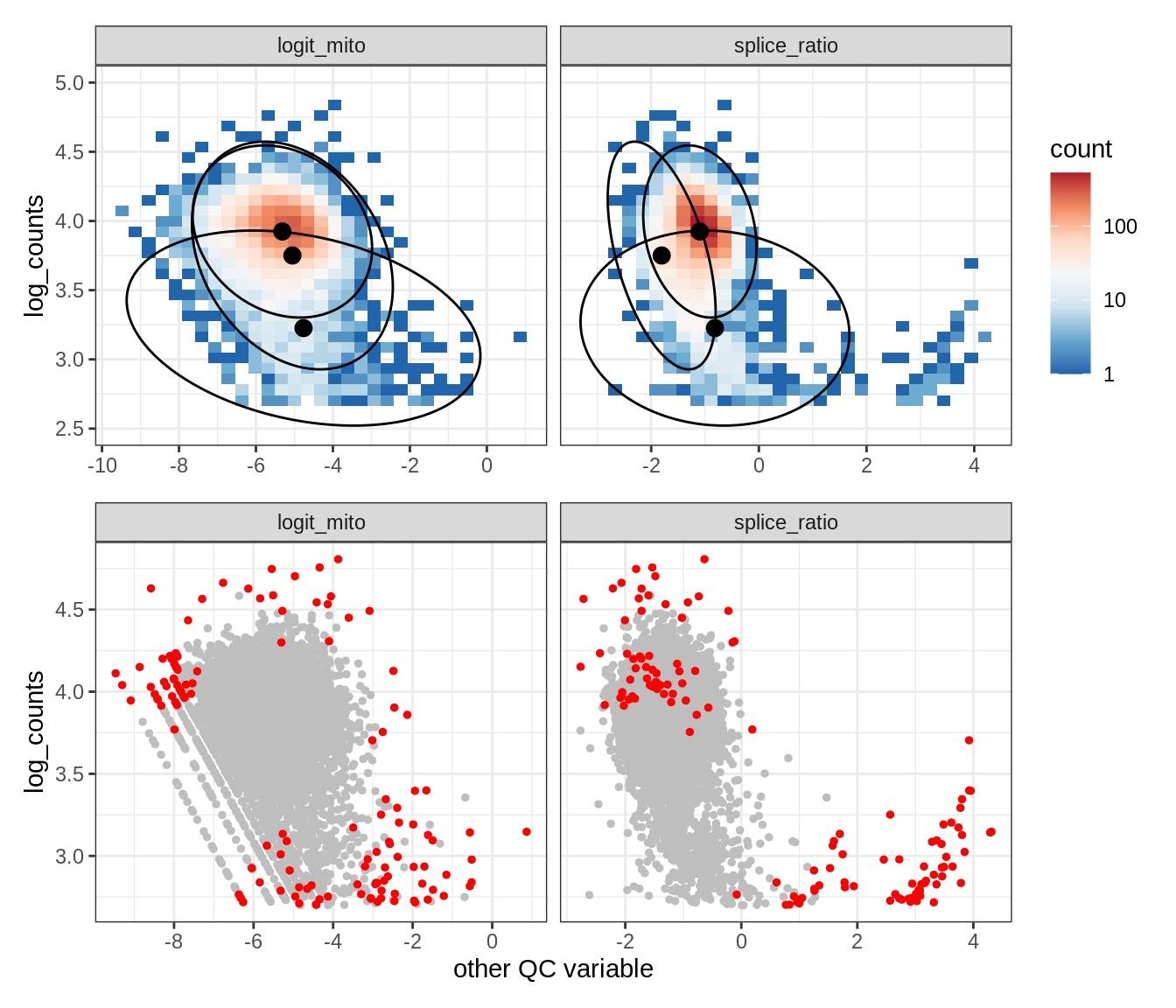
WM160
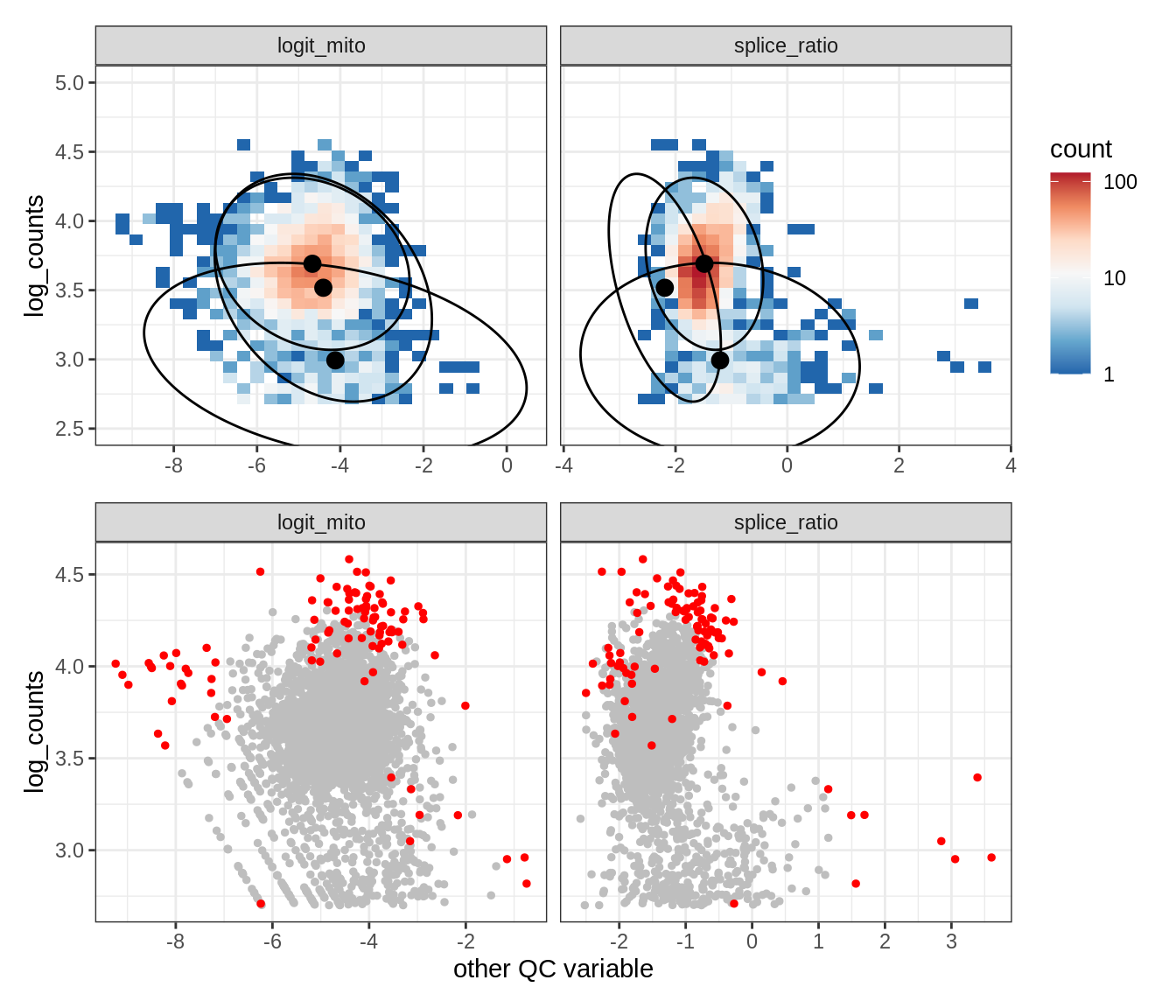
WM163
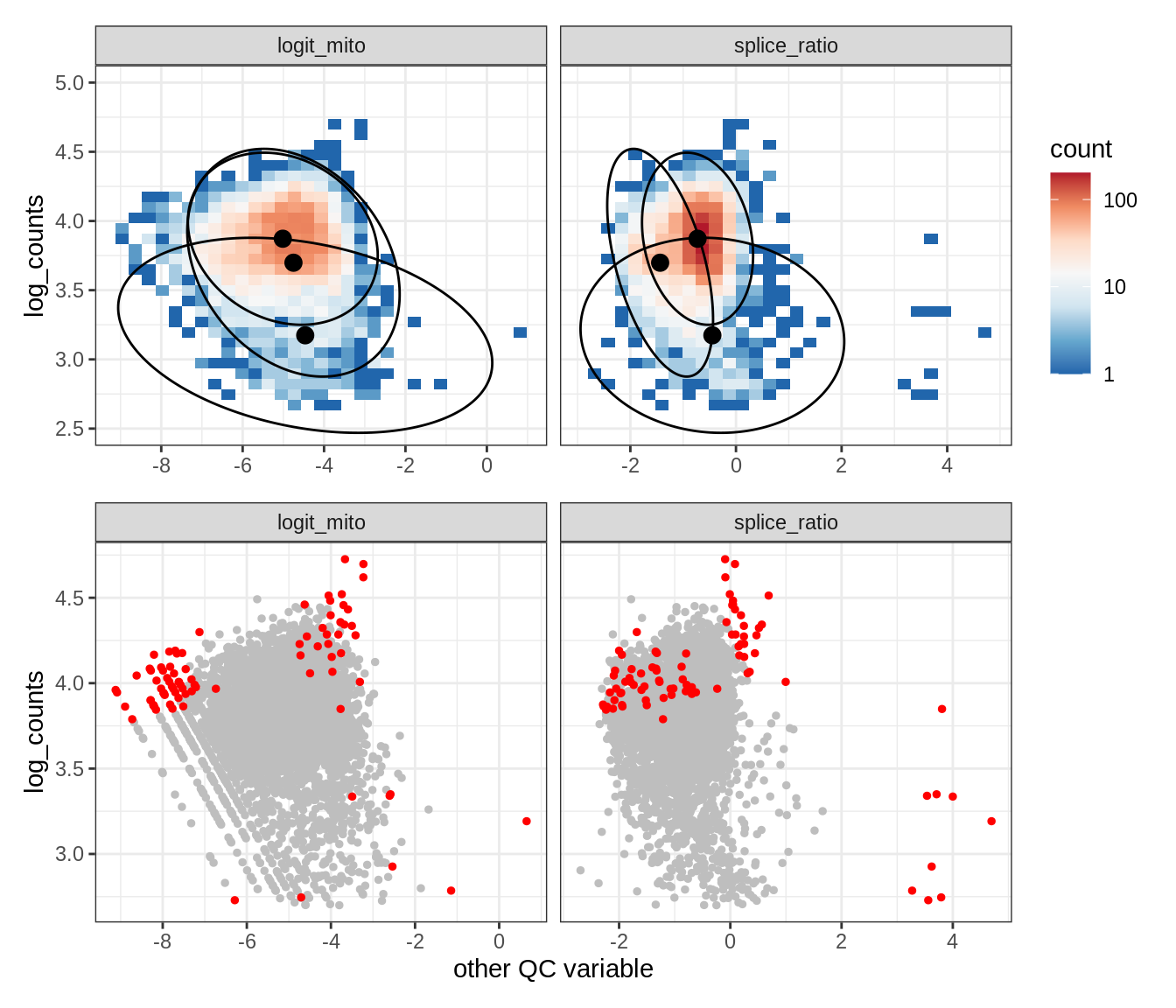
WM171
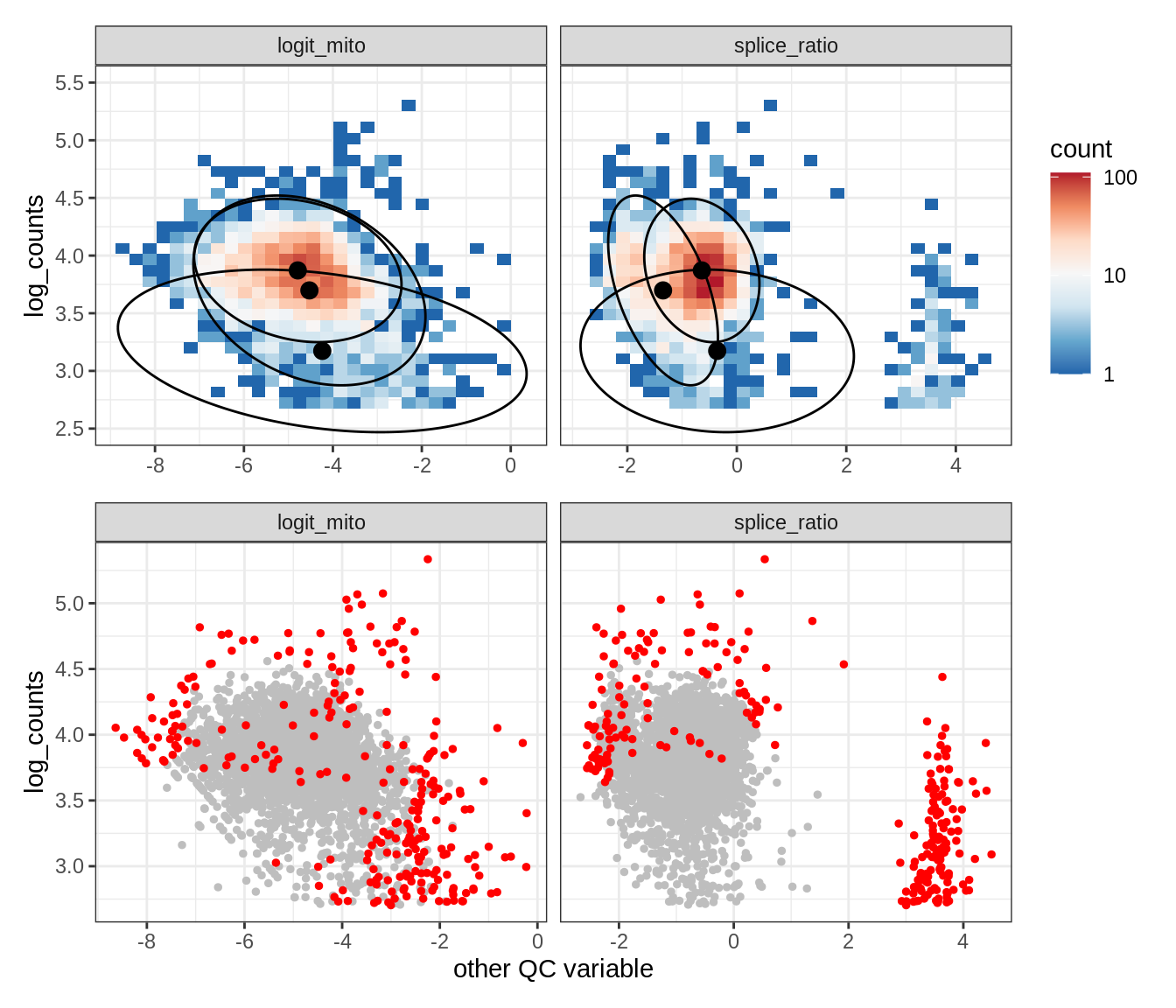
WM179
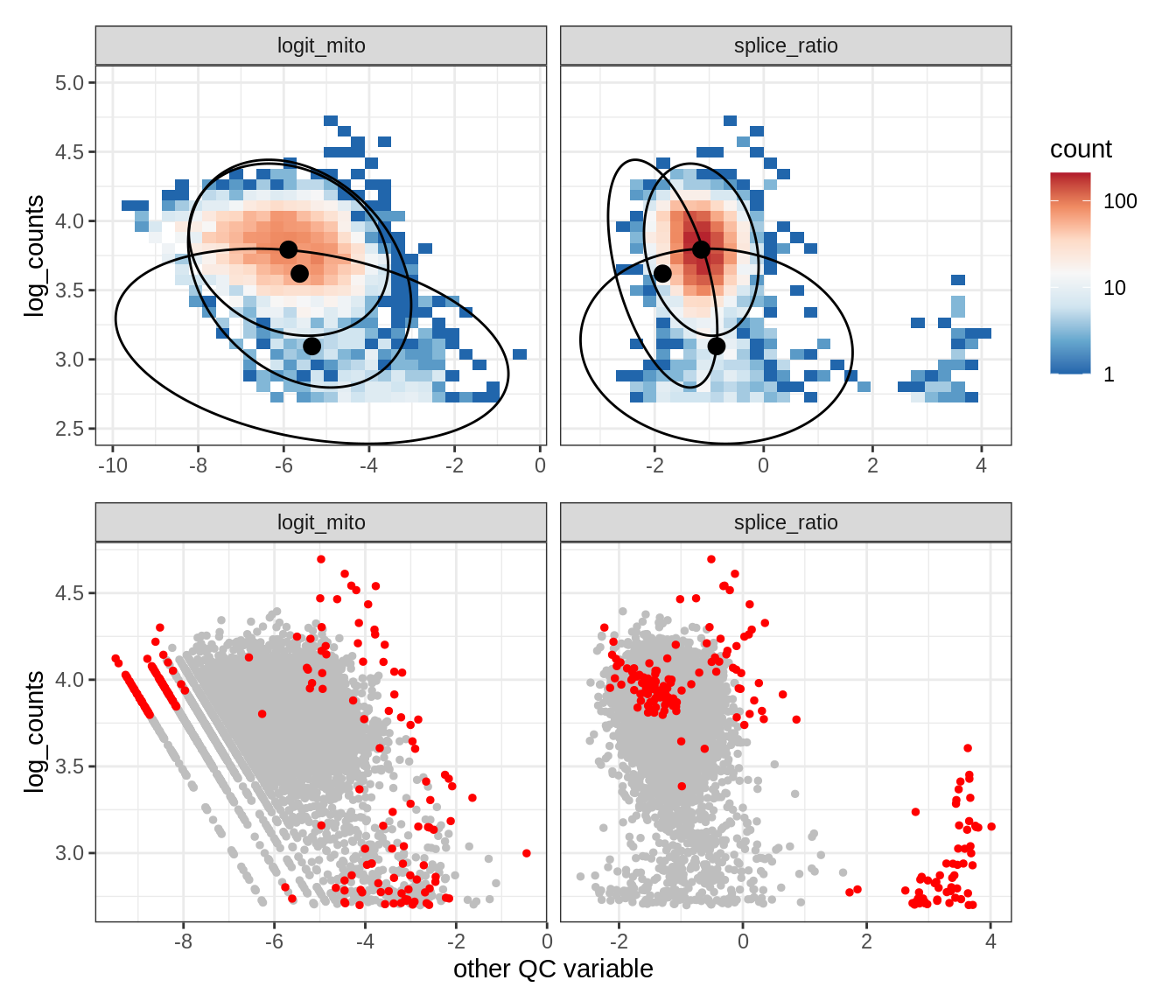
WM182
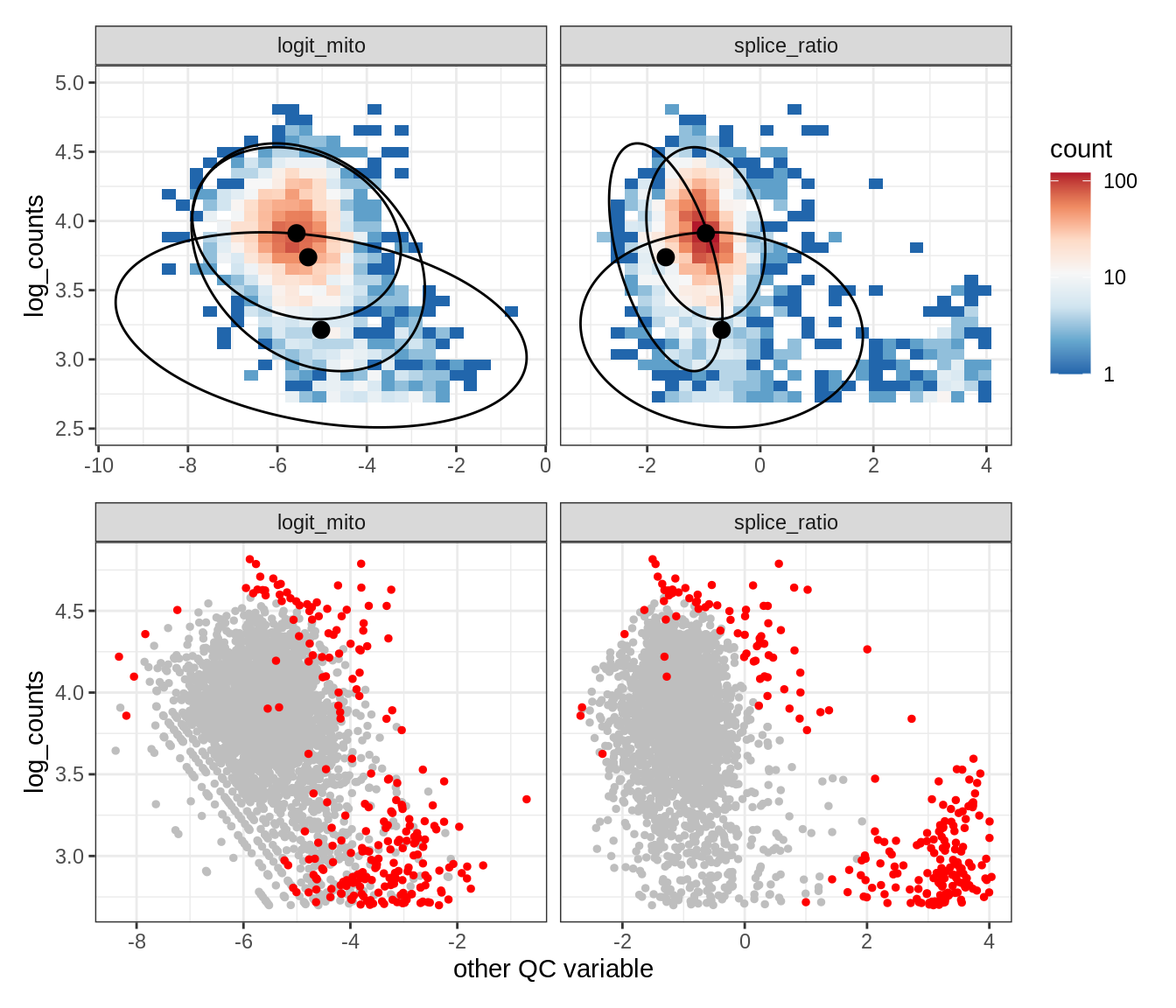
WM183
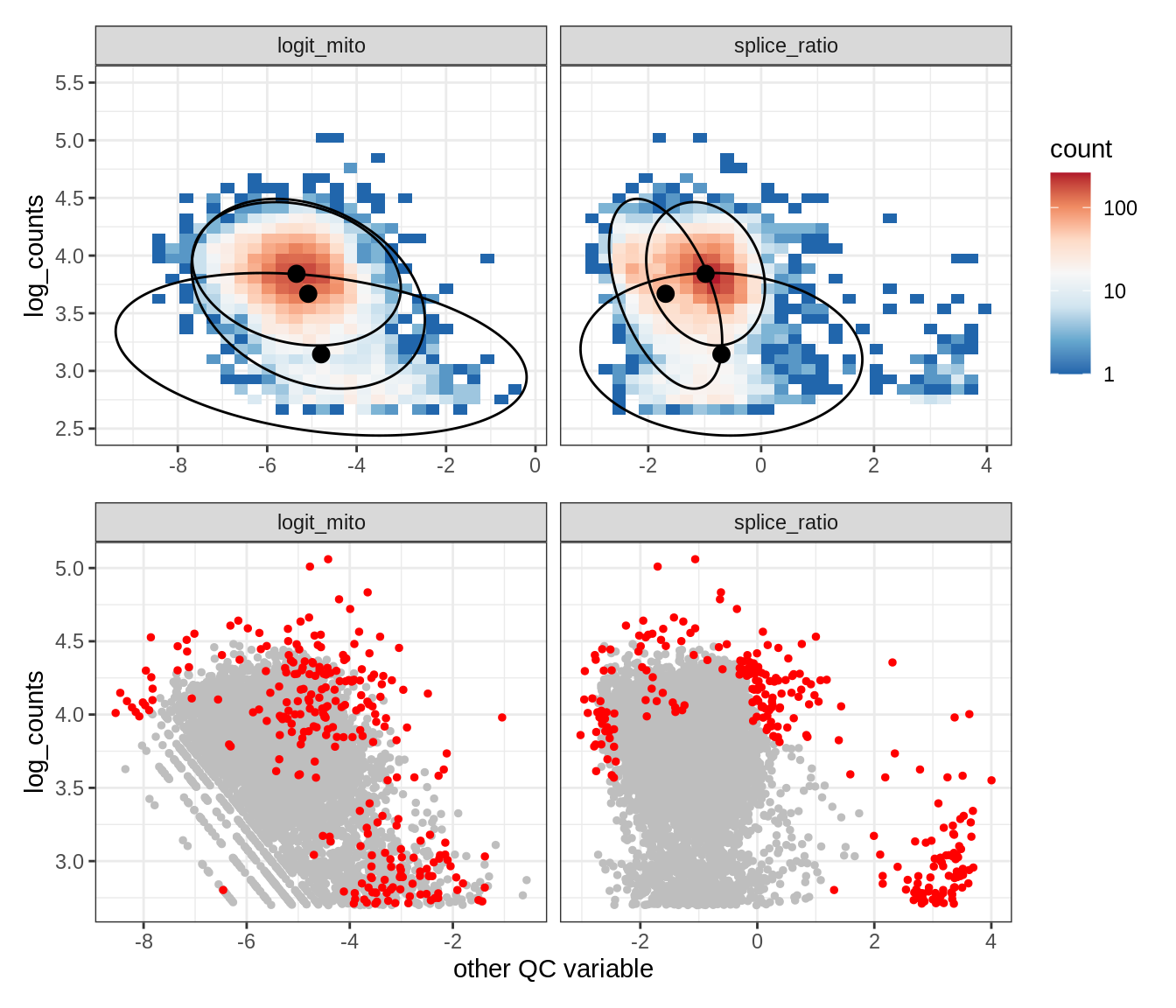
WM185
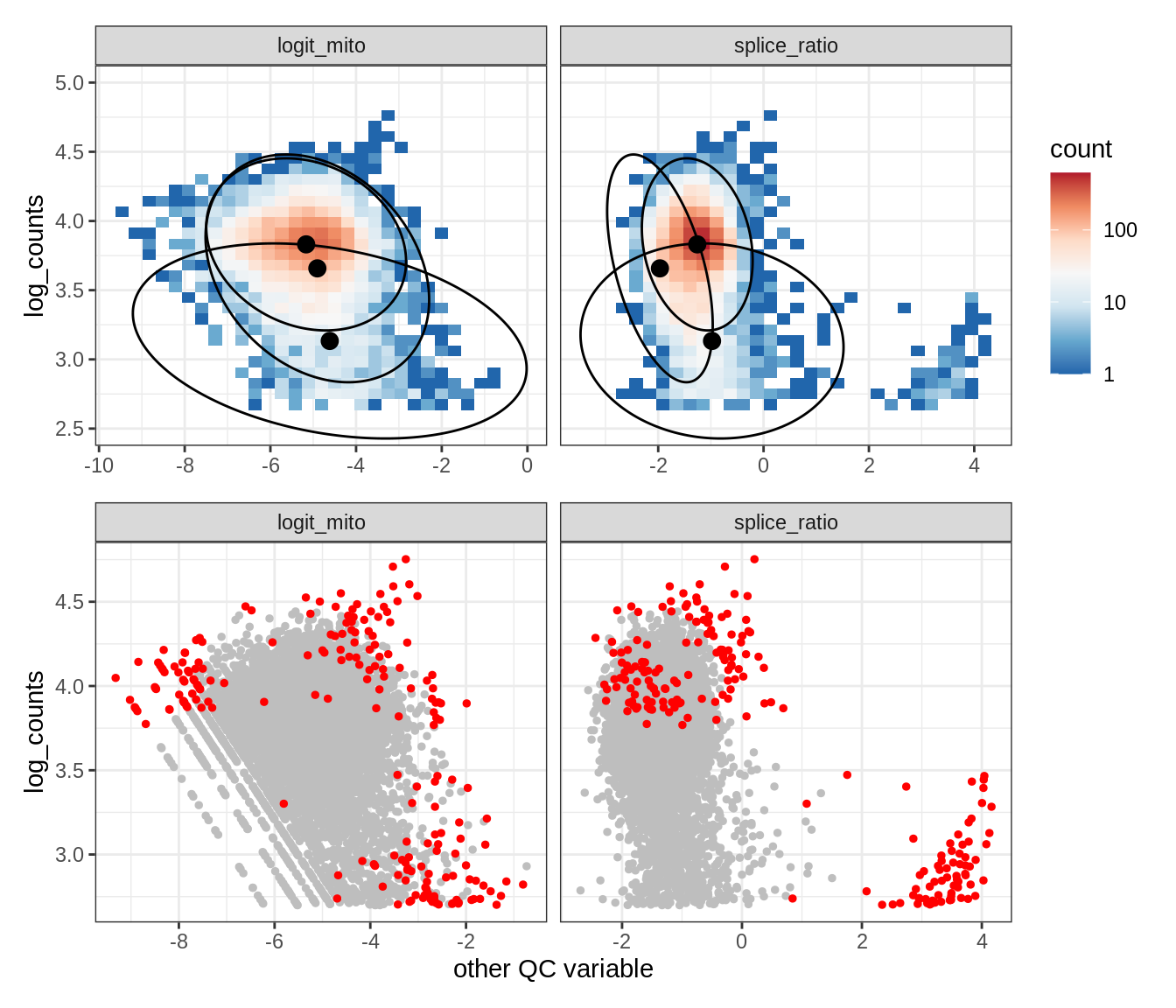
WM186
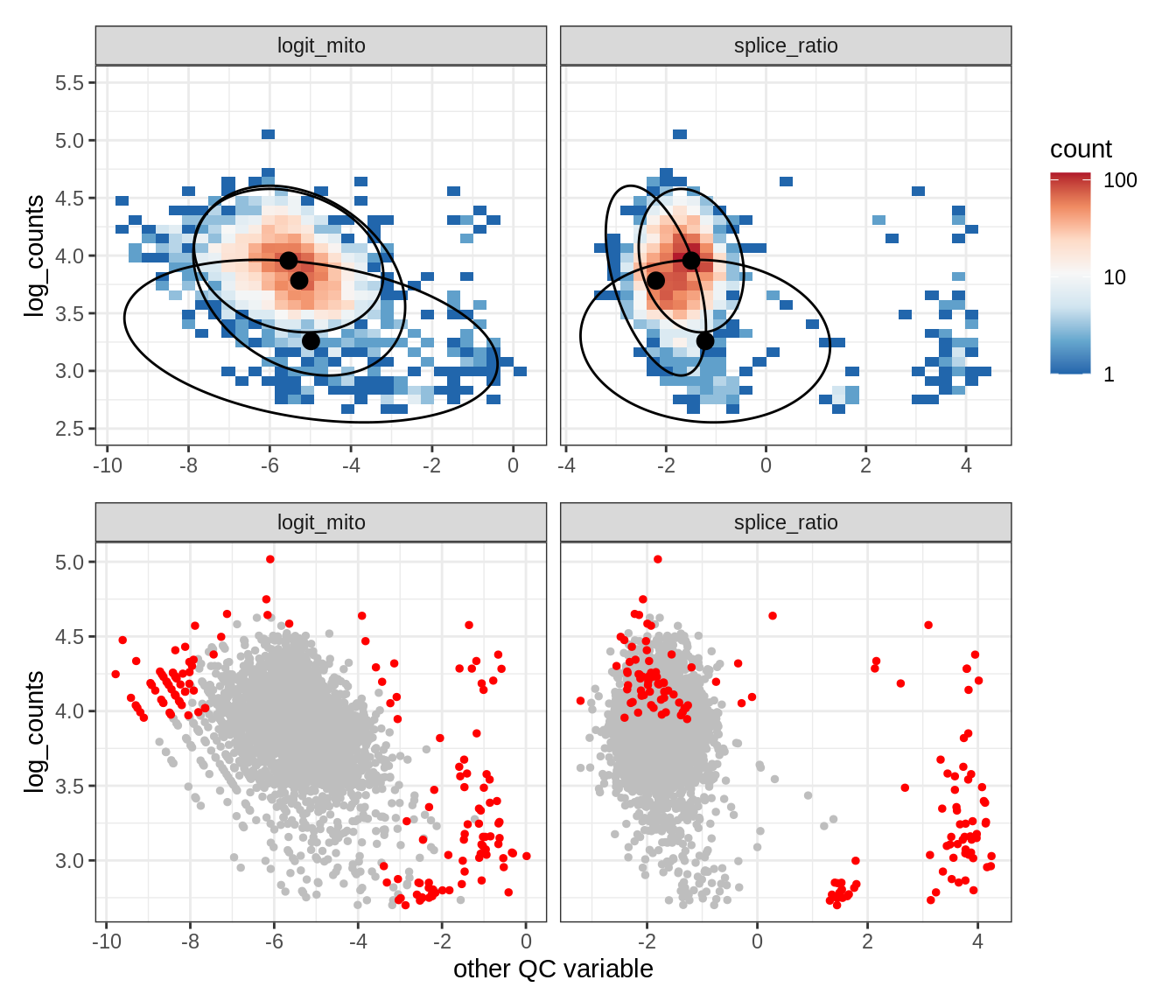
WM187
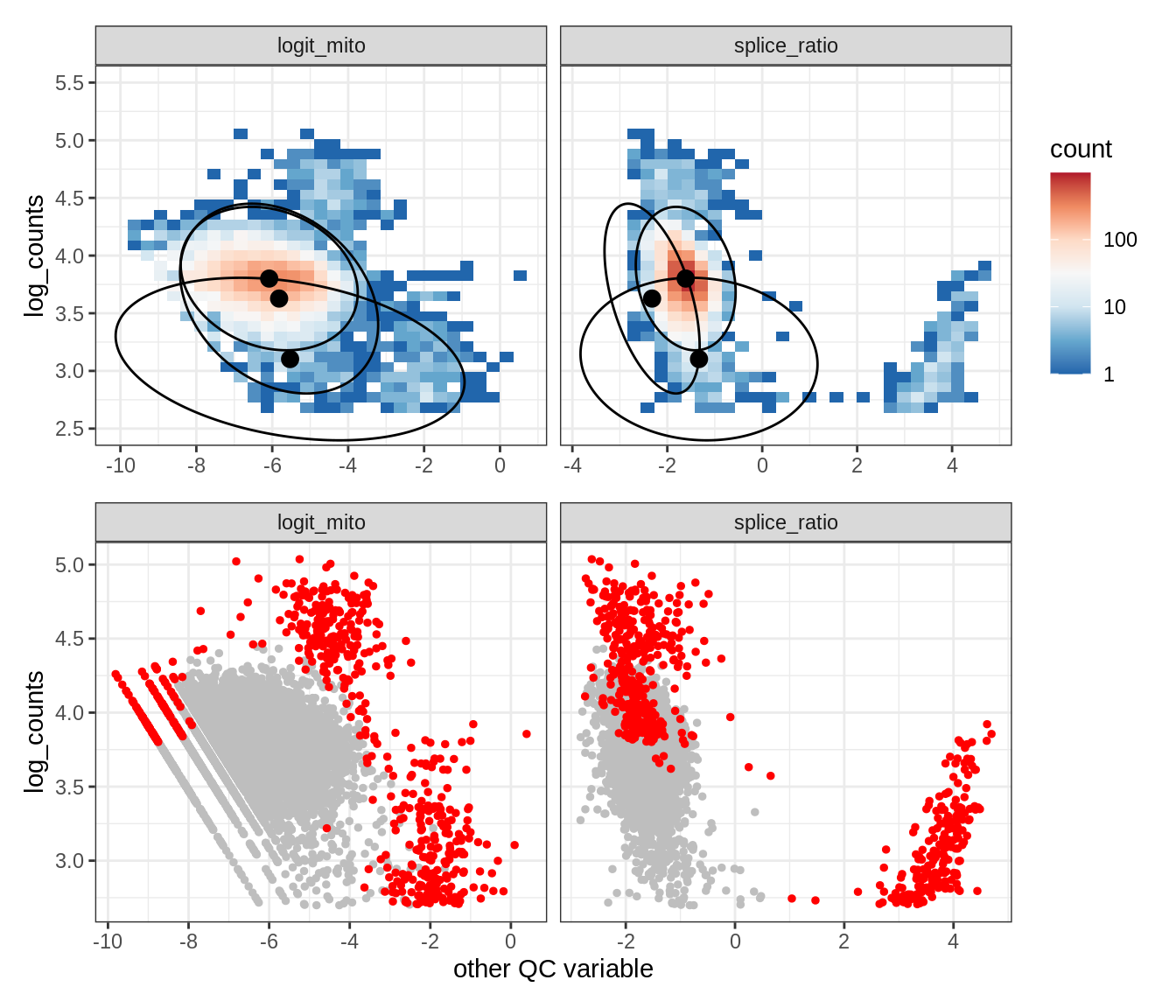
WM188
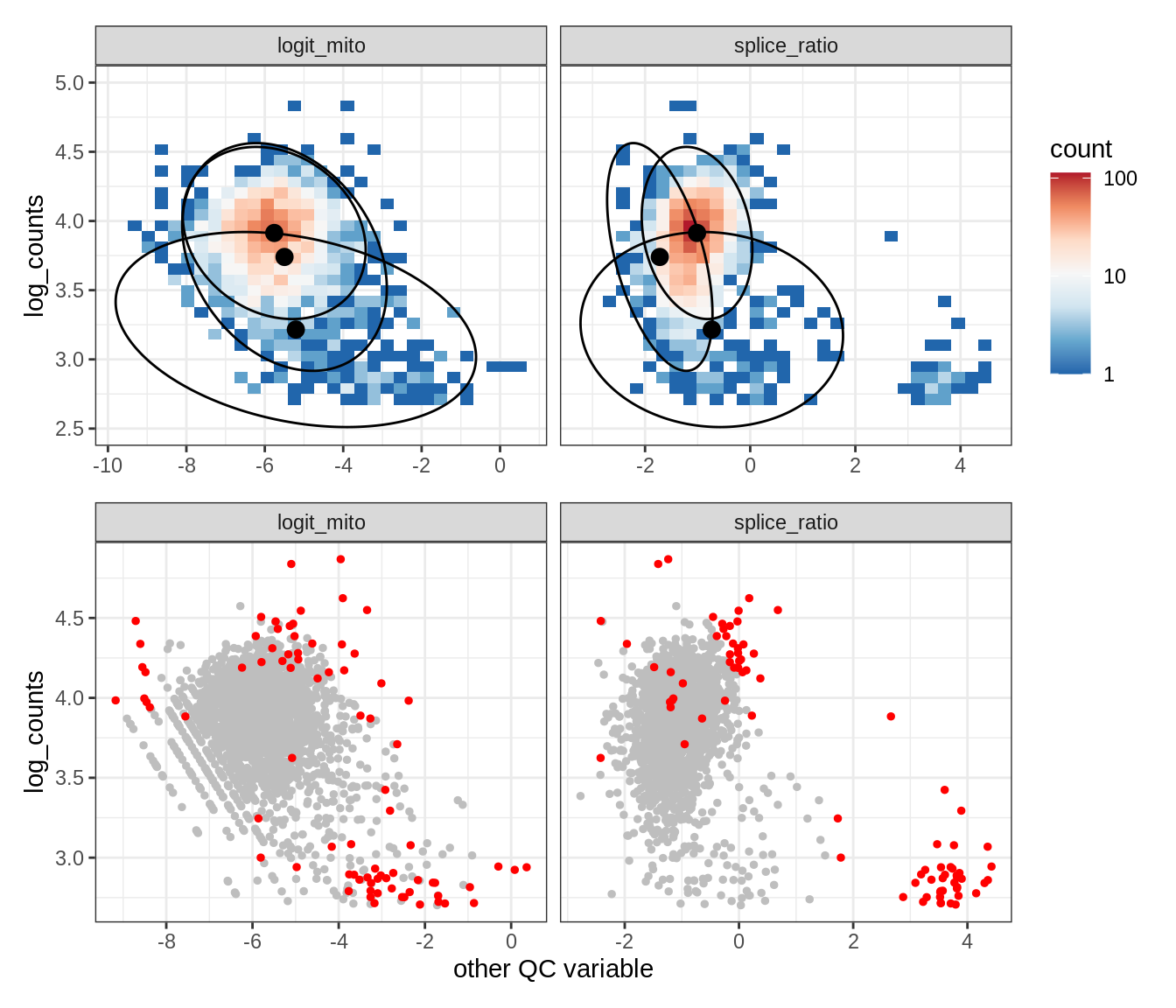
WM193
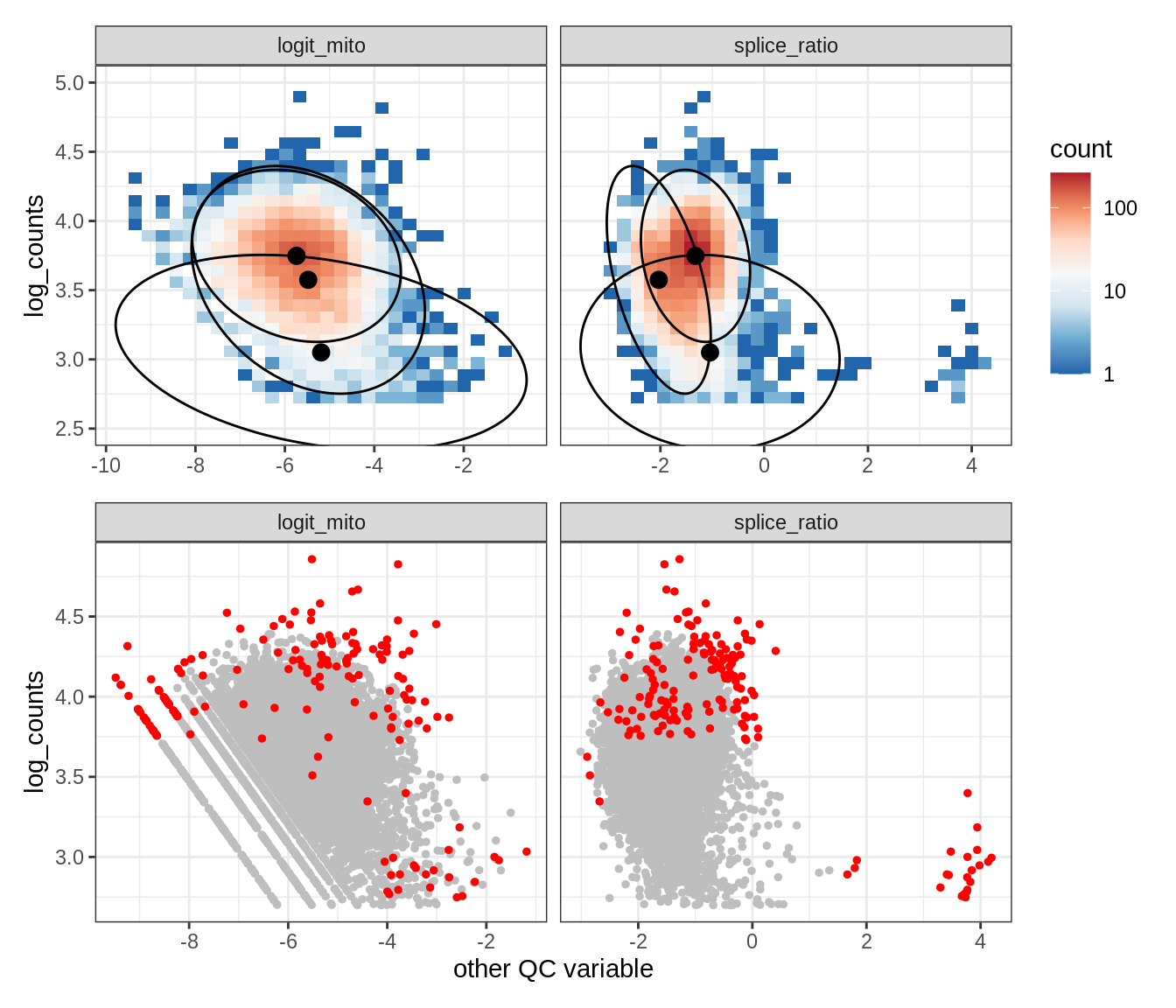
WM194
WM199
SG4
EU005
EU008
EU010
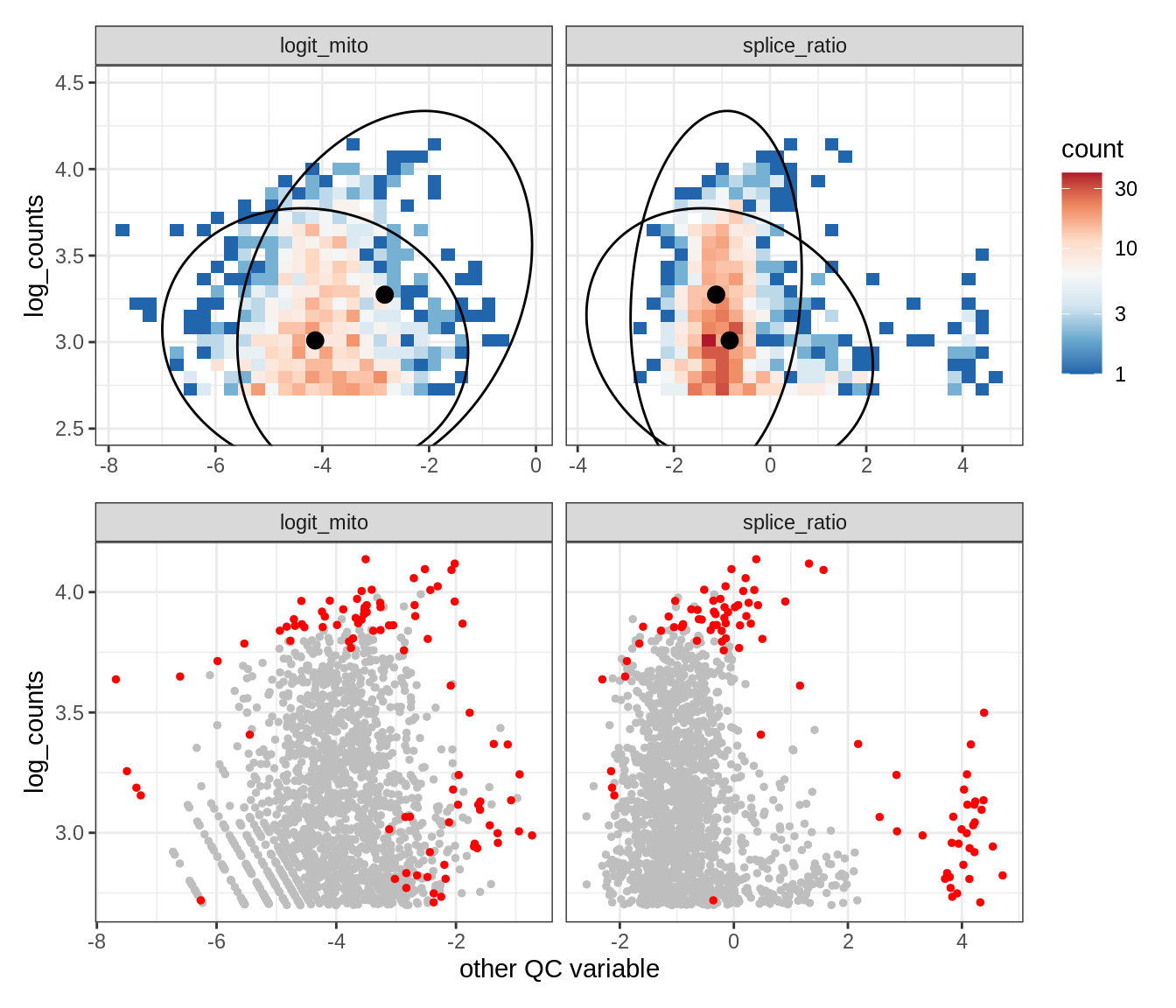
EU019
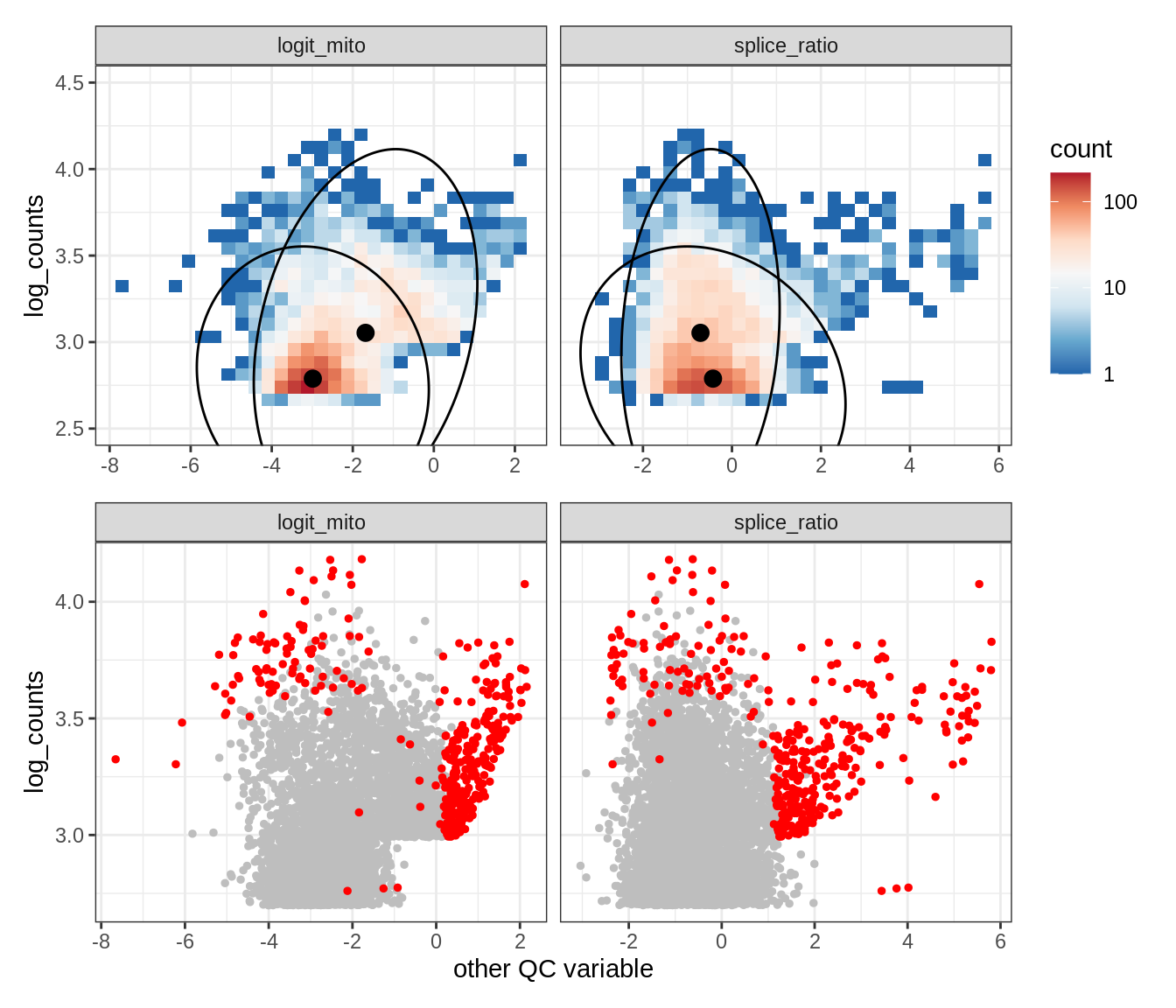
EU023
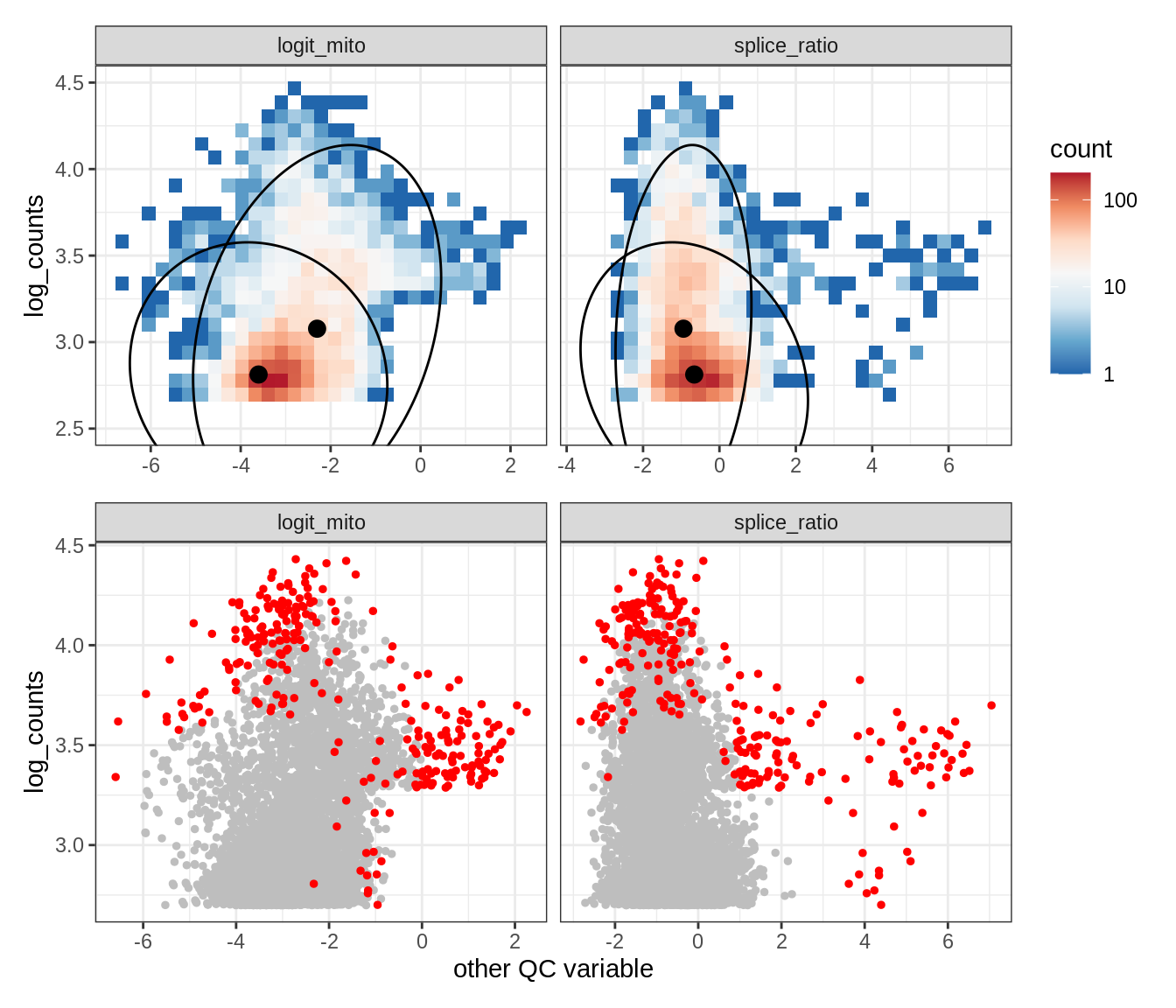
EU028
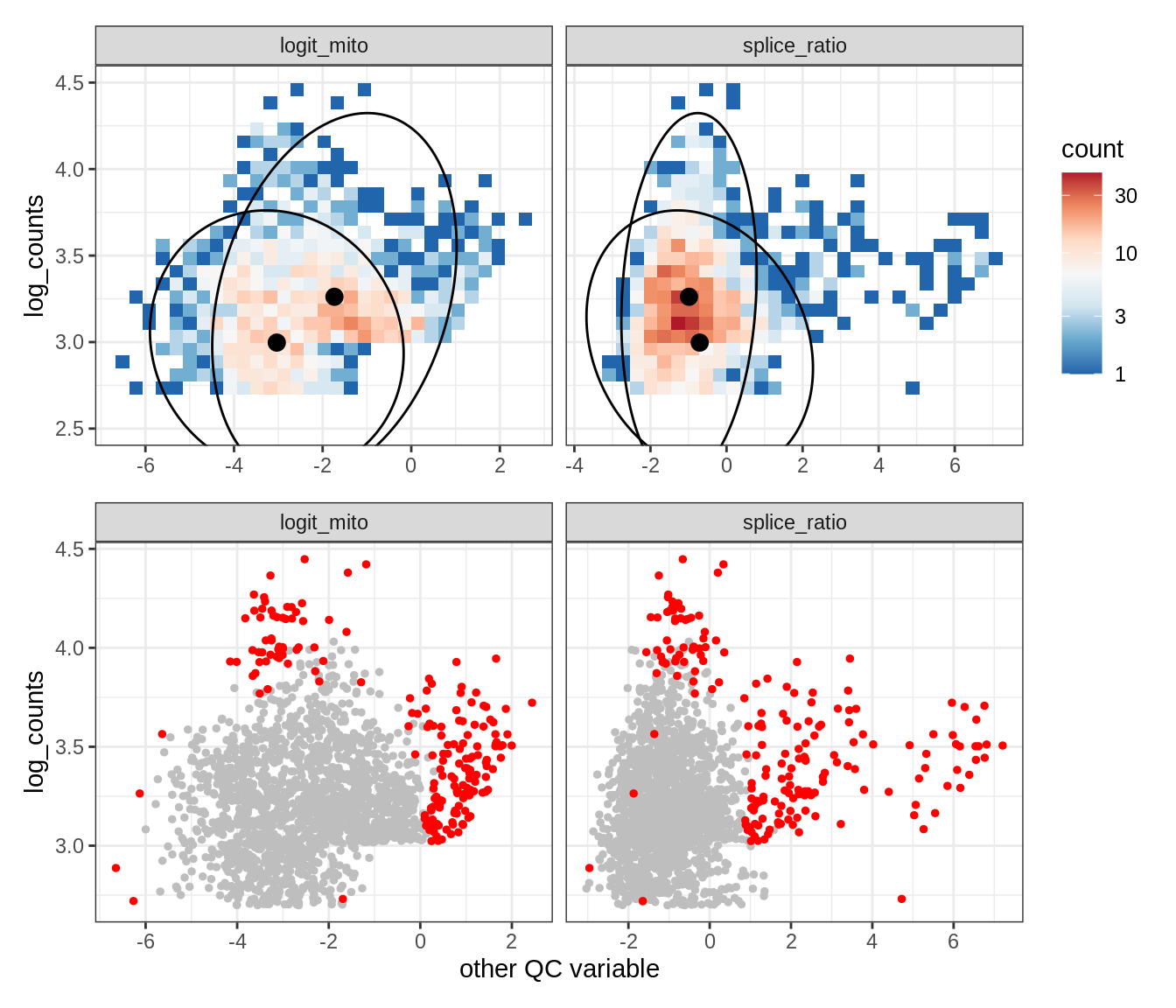
EU029
EU030
EU031
EU035
EU036
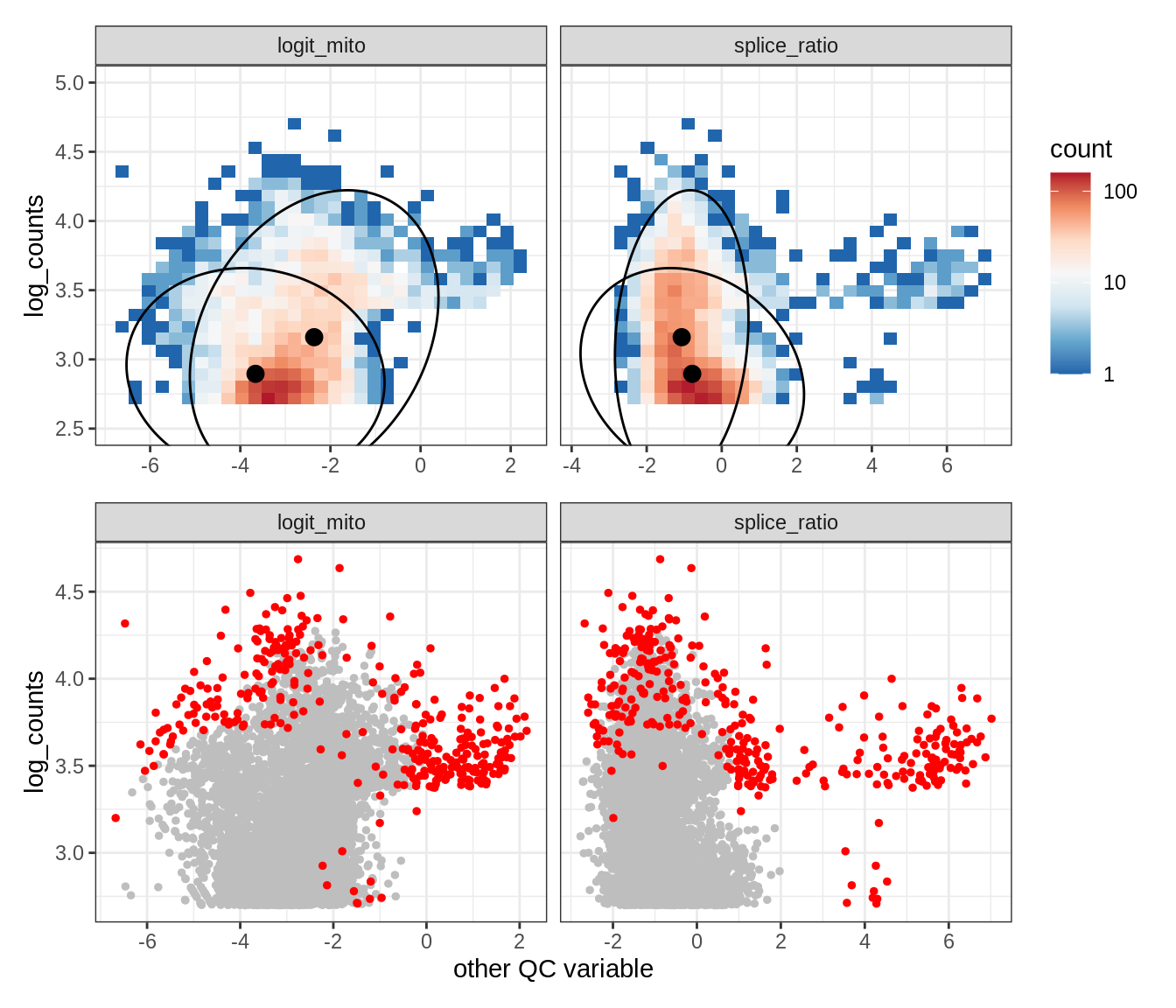
EU037
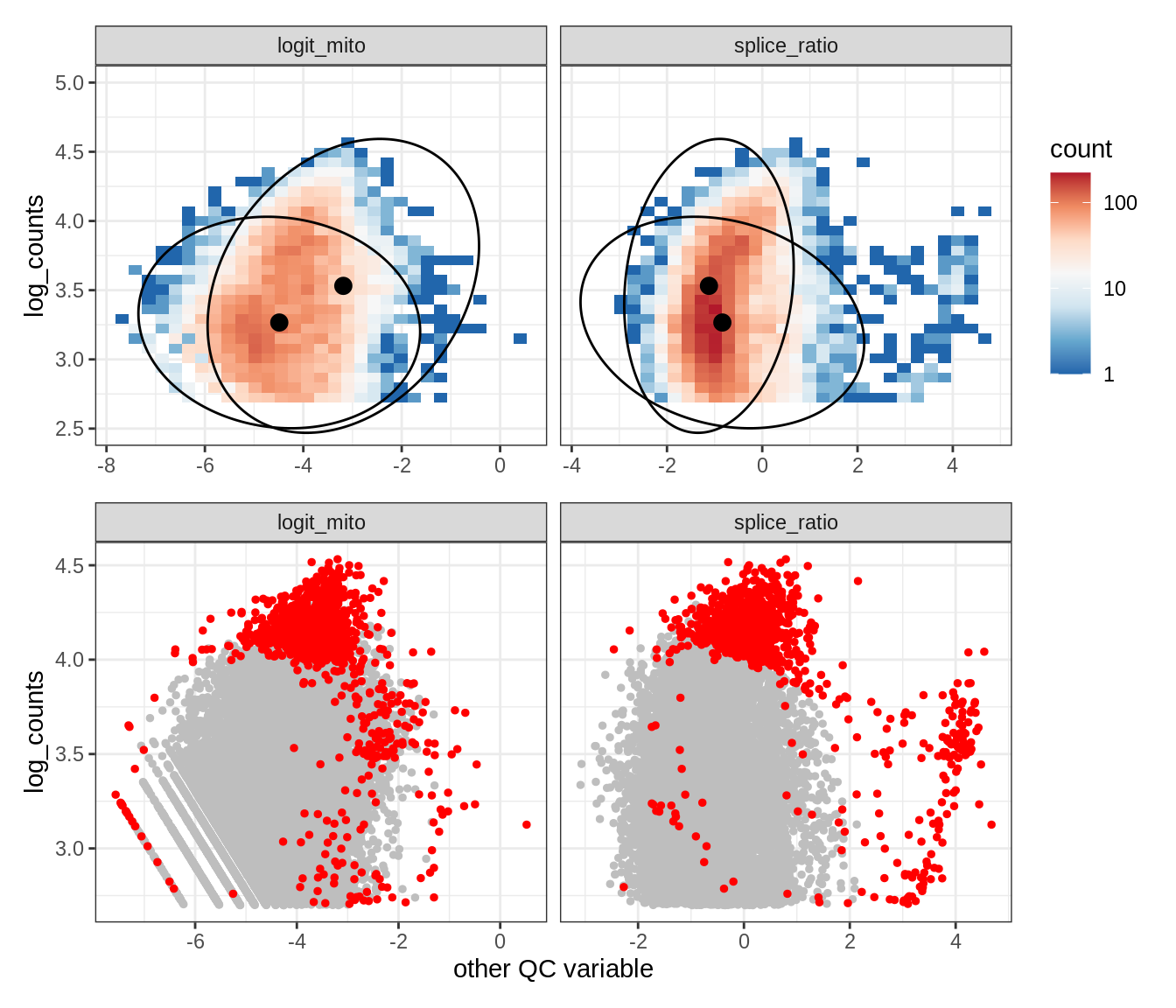
EU041
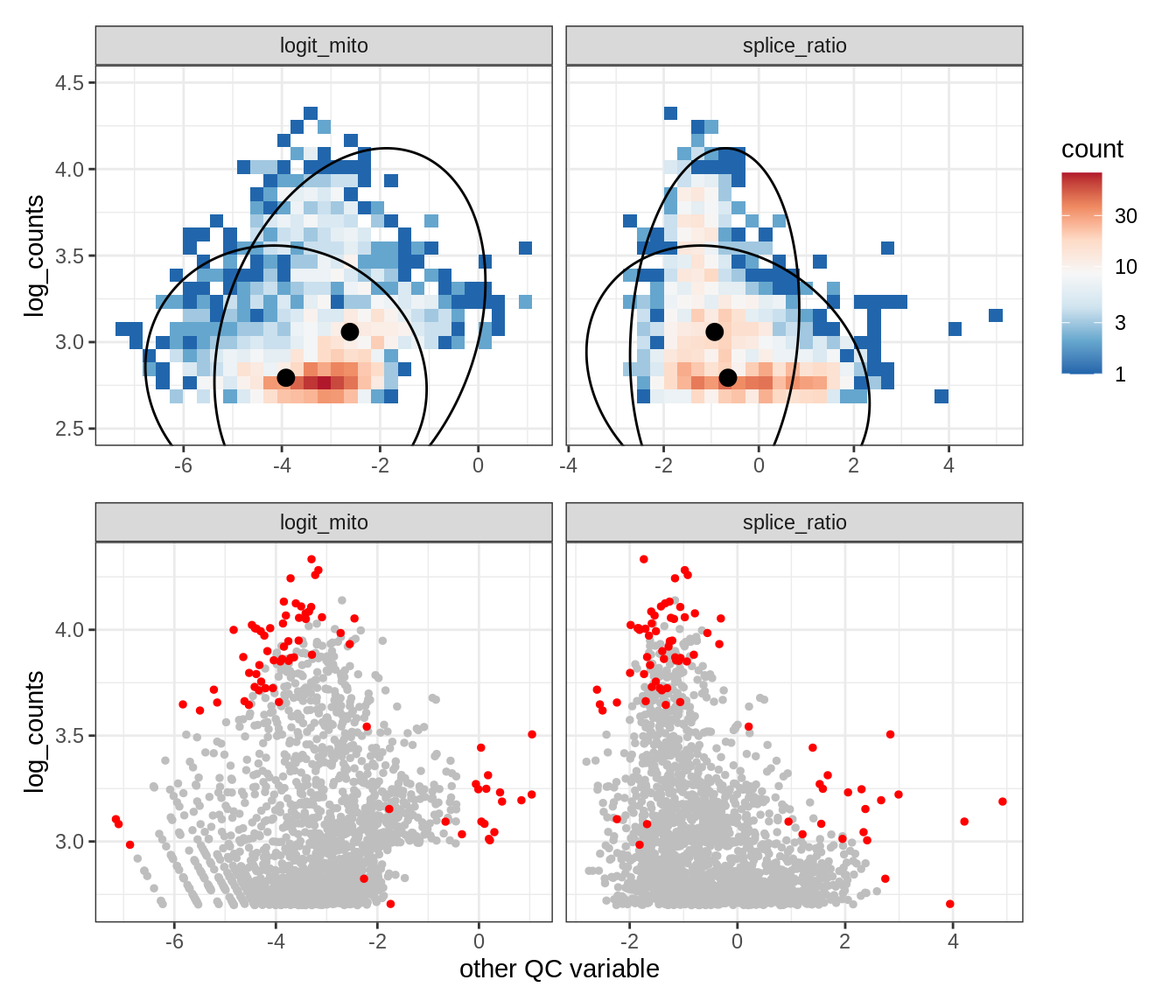
WM128
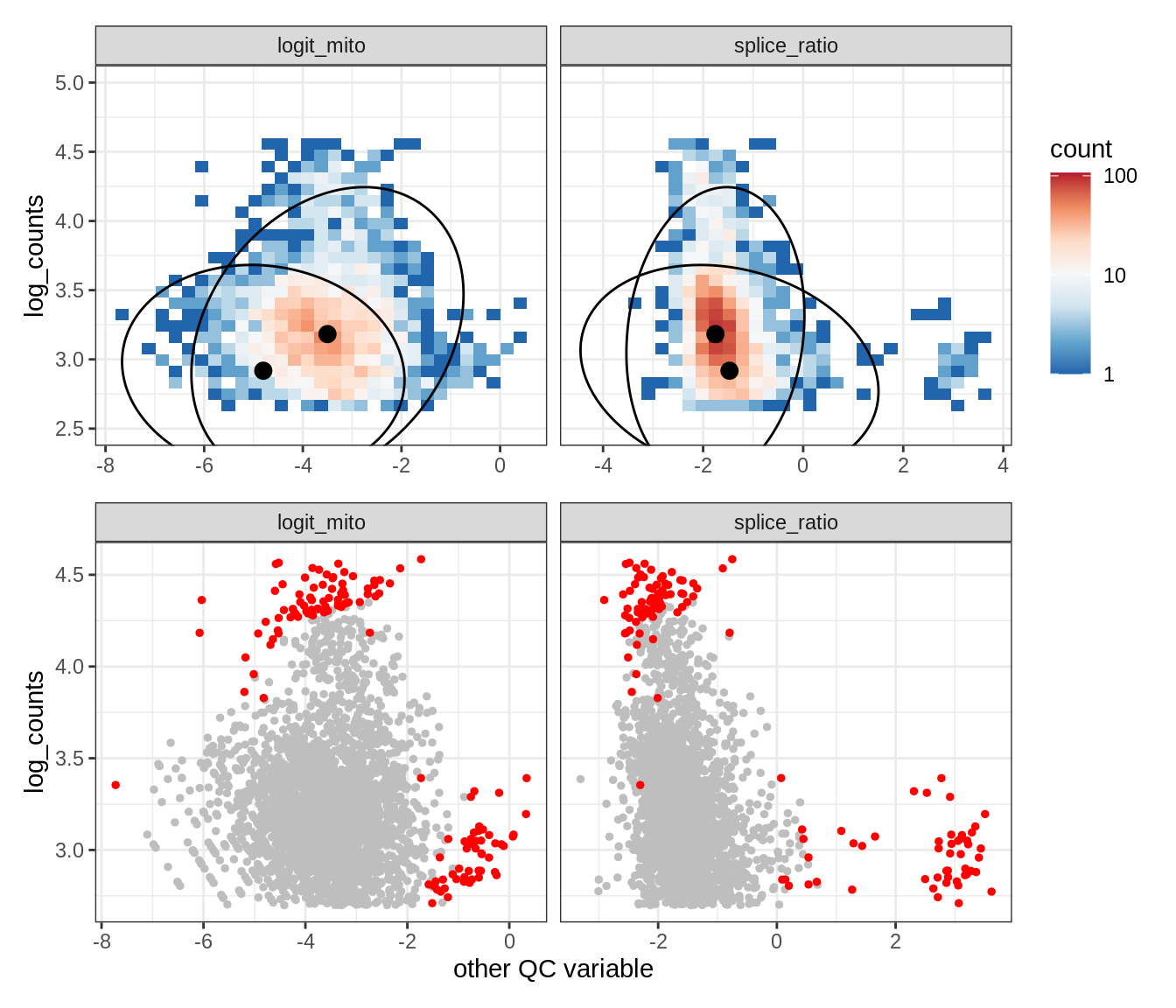
WM136
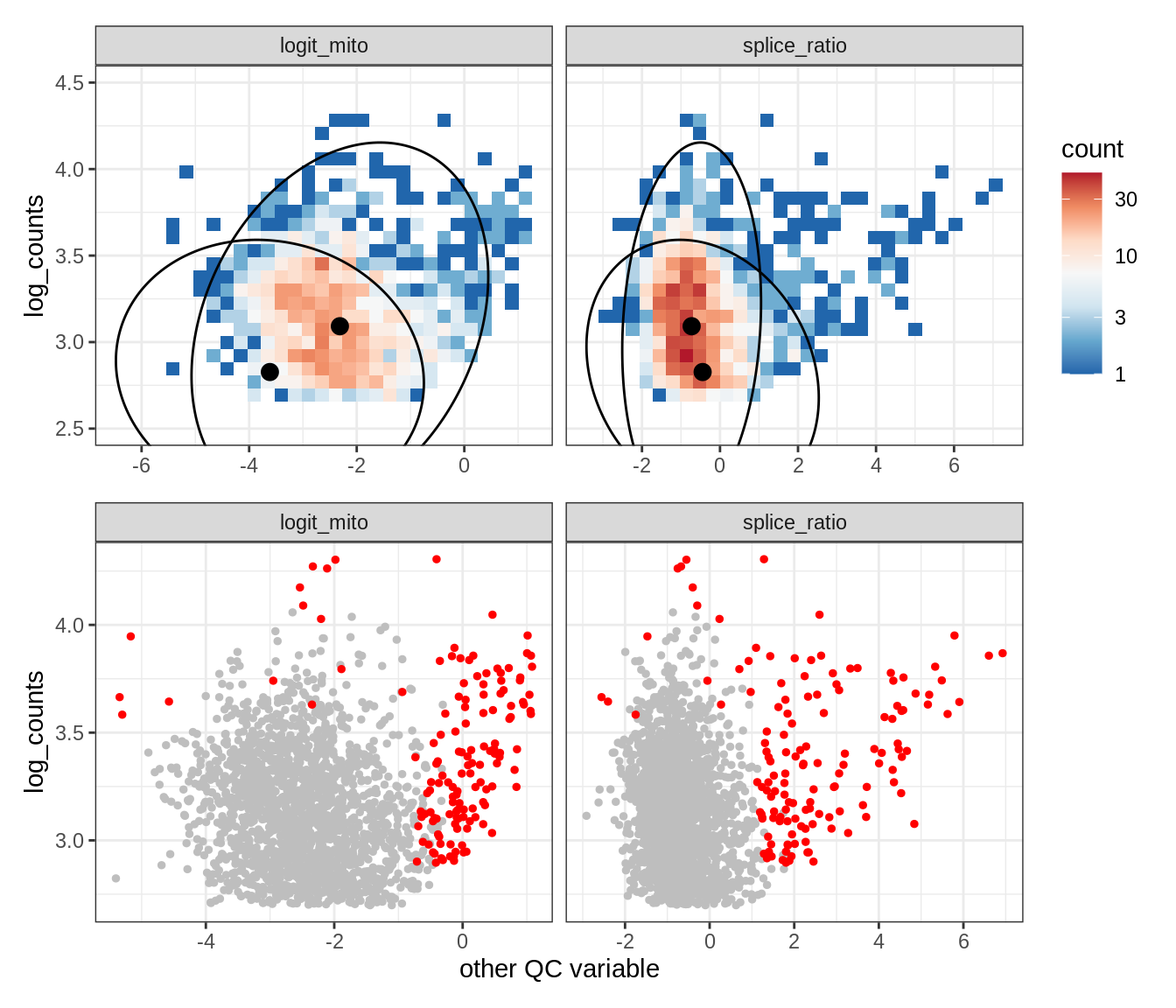
WM145
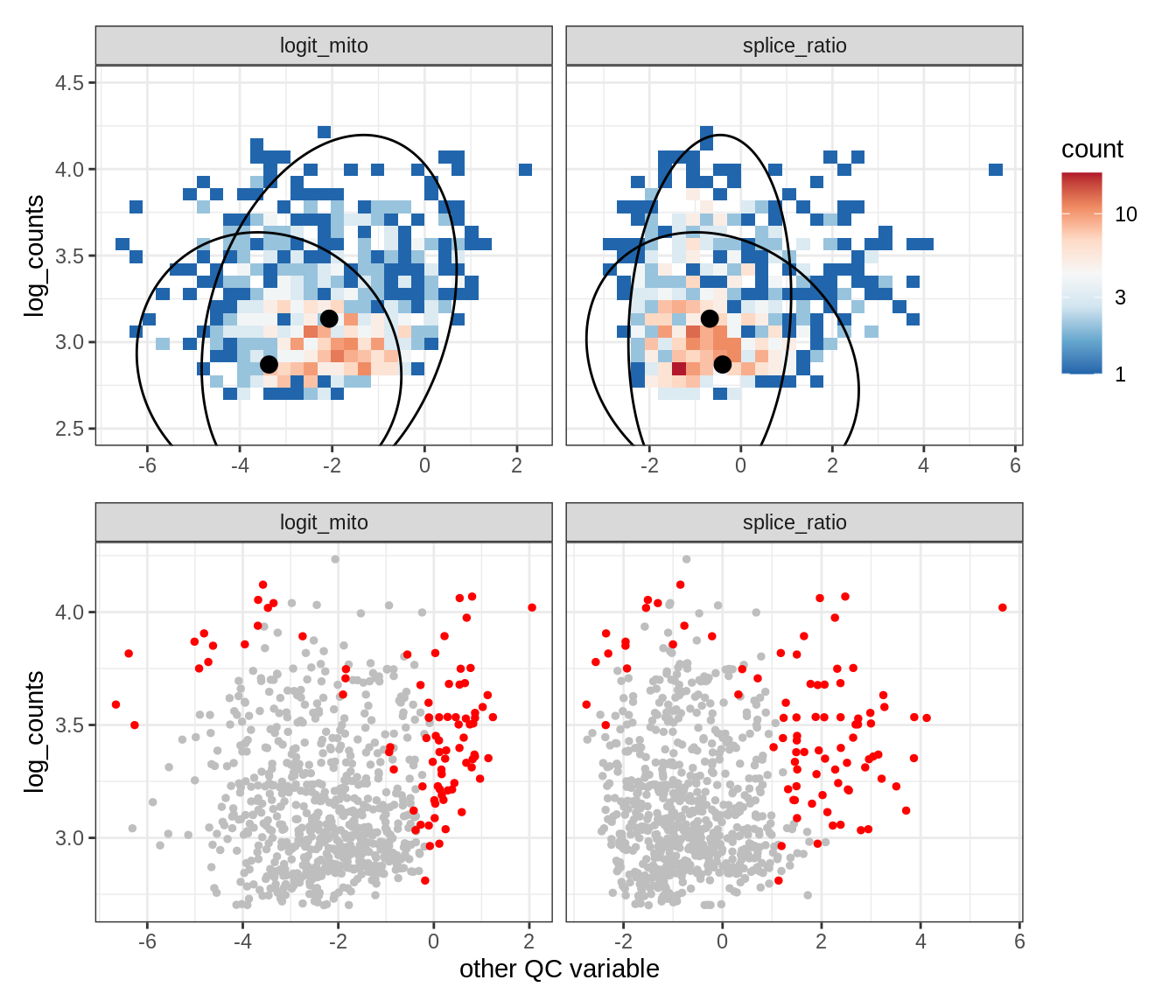
WM166
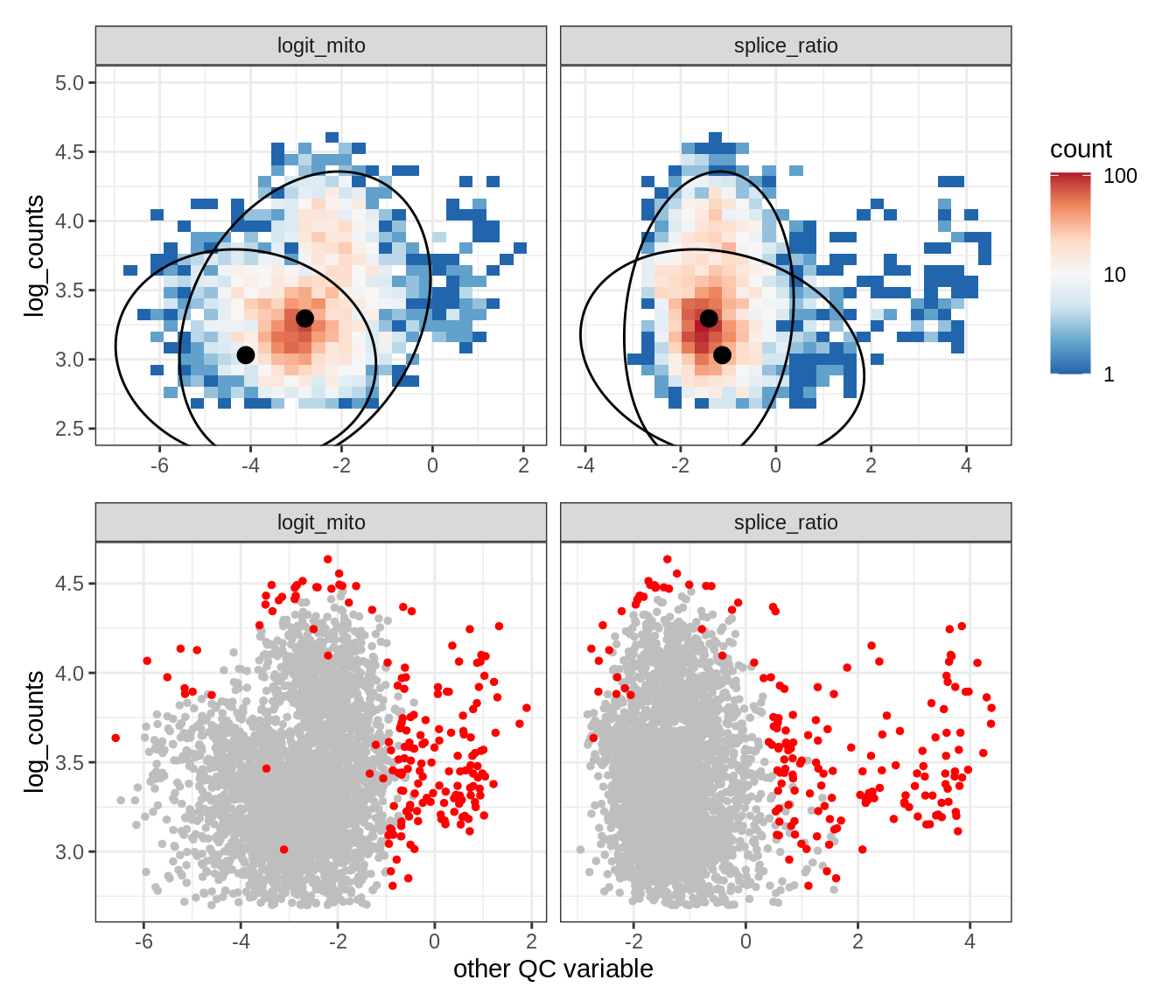
WM167
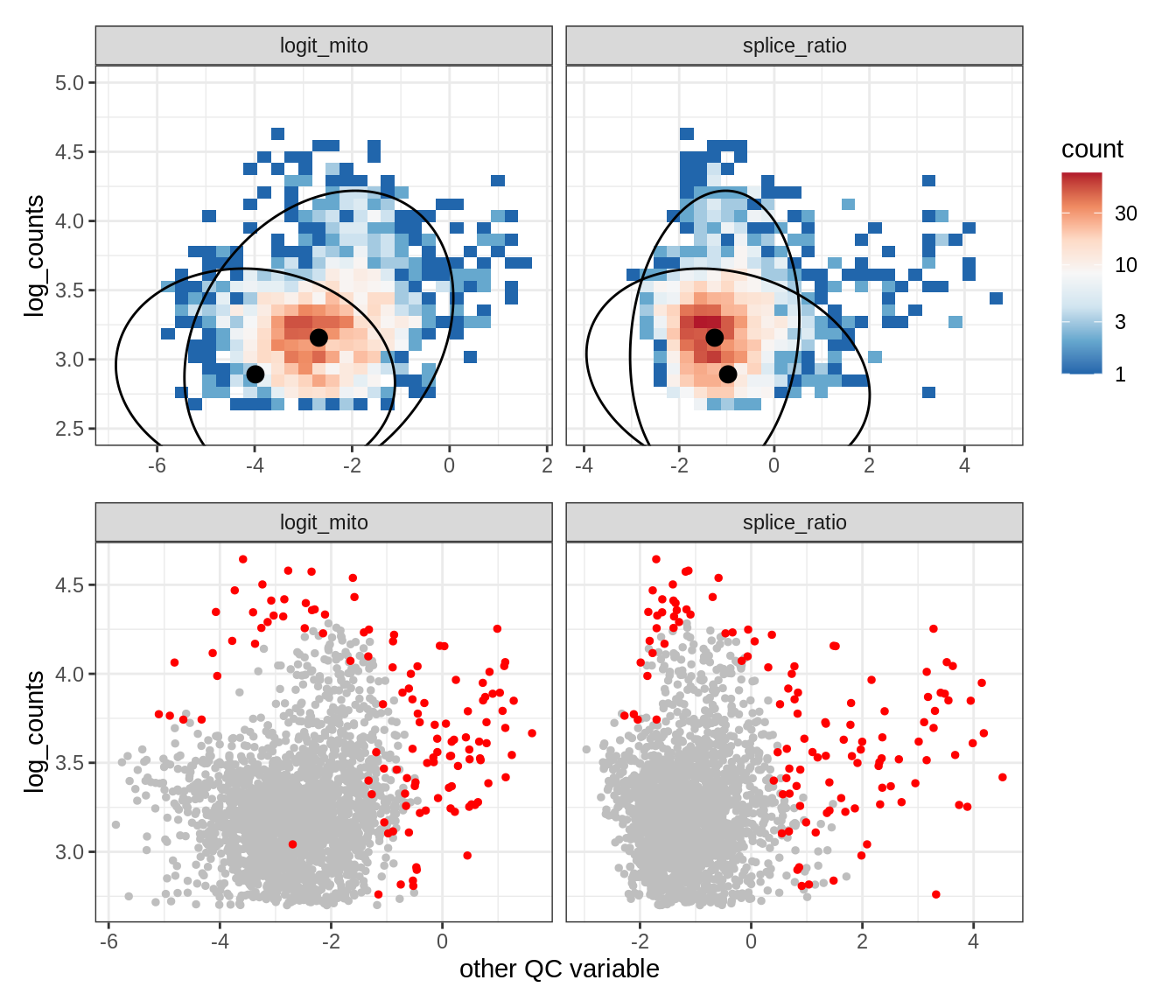
WM172
WM191
WM192
SG5
EU015
EU016
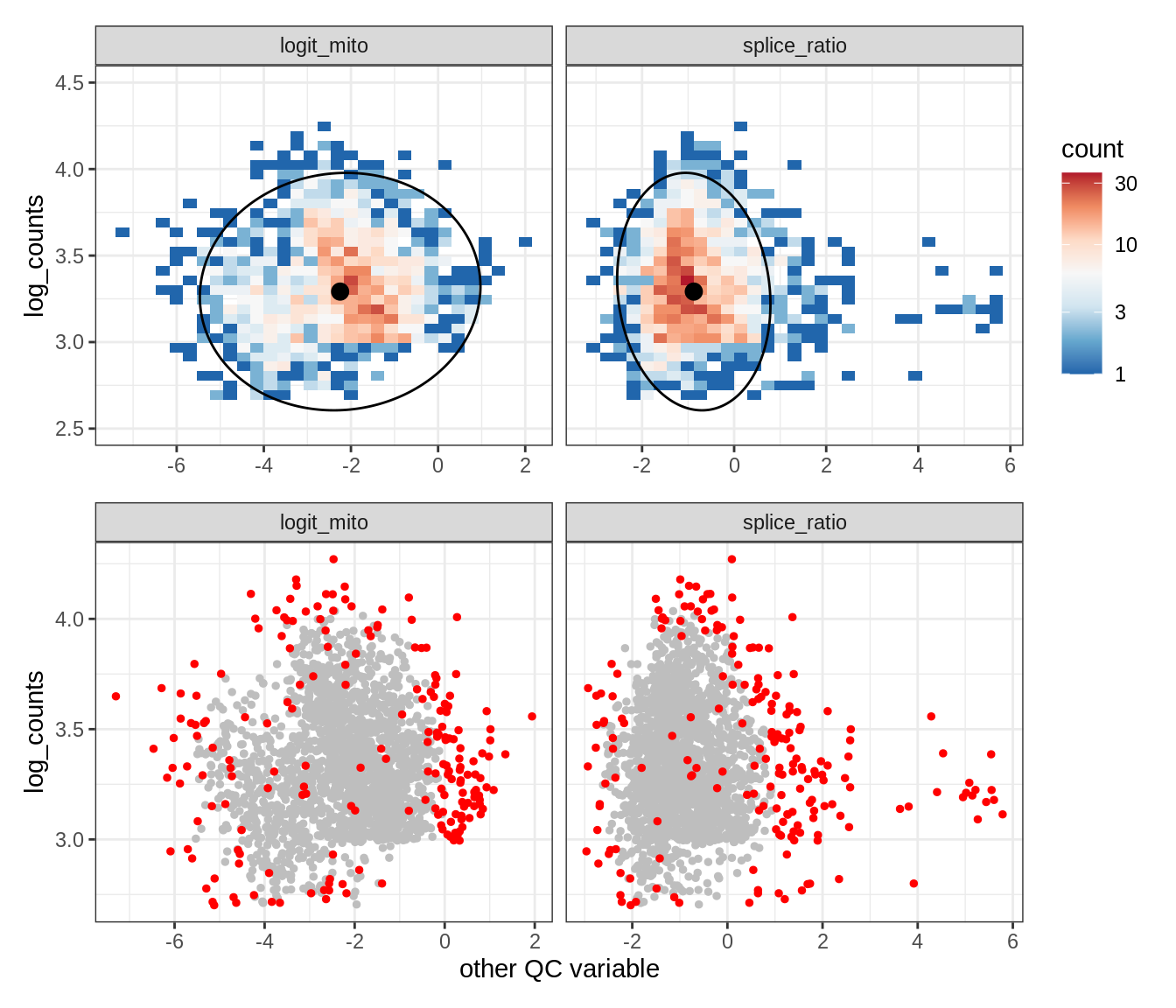
EU042
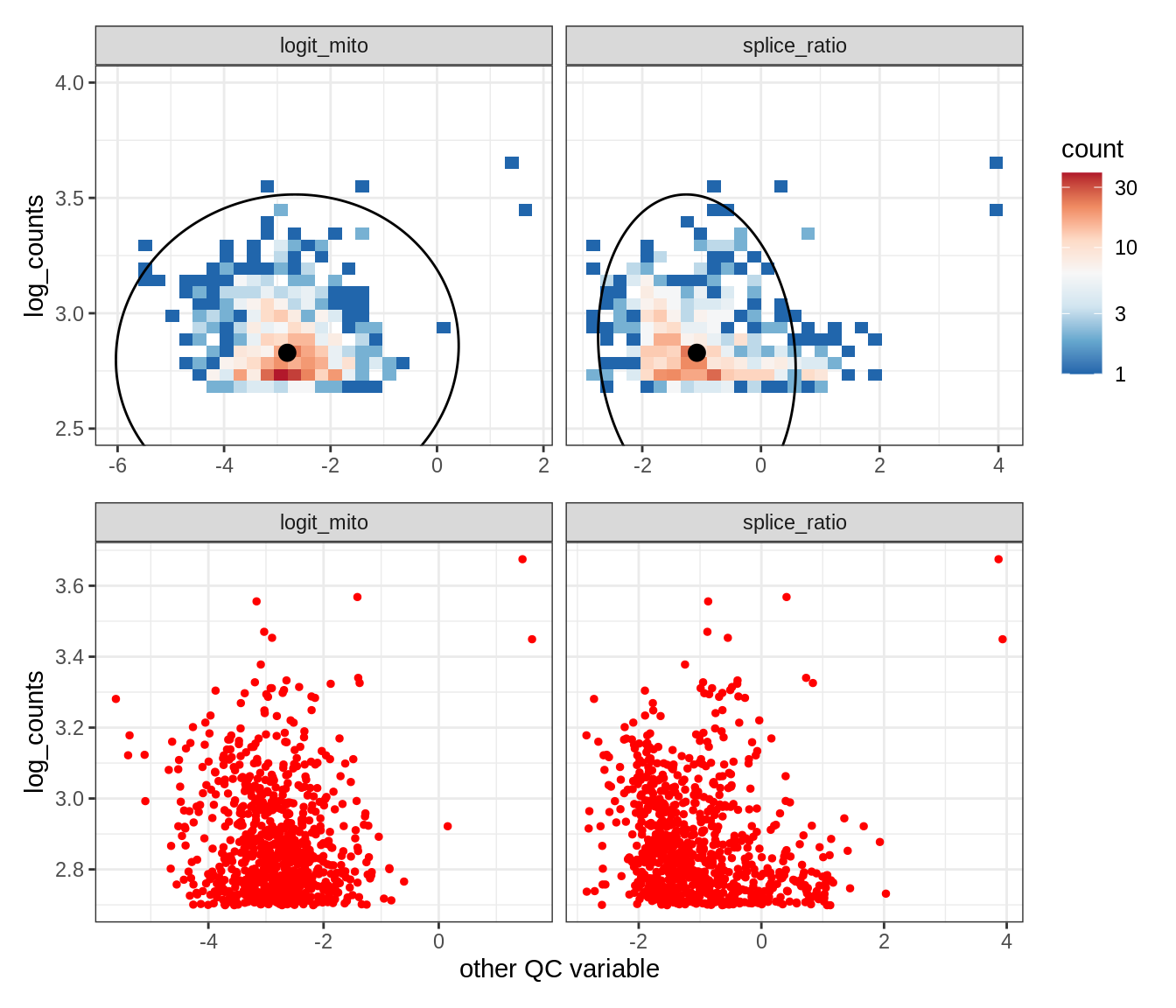
EU043
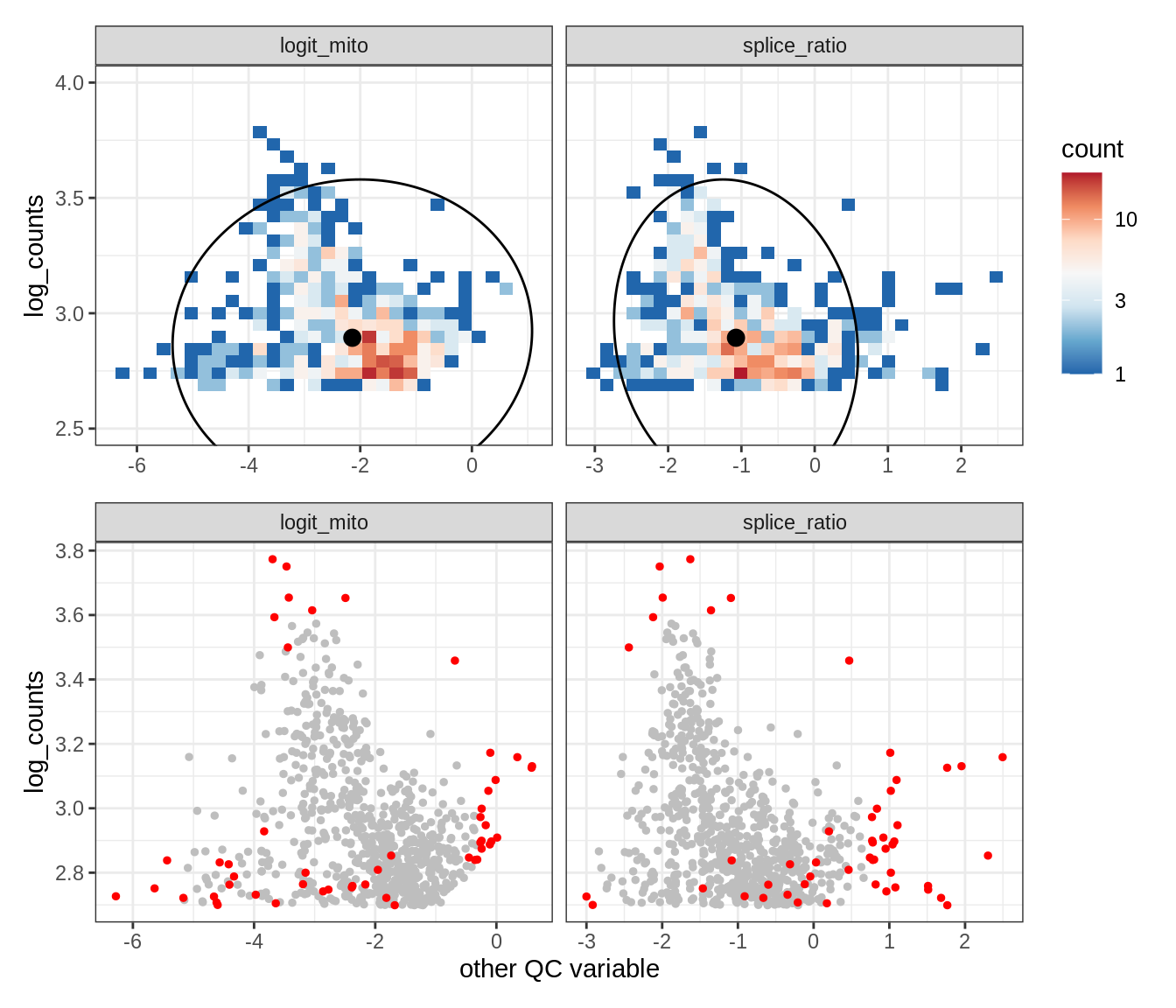
EU044
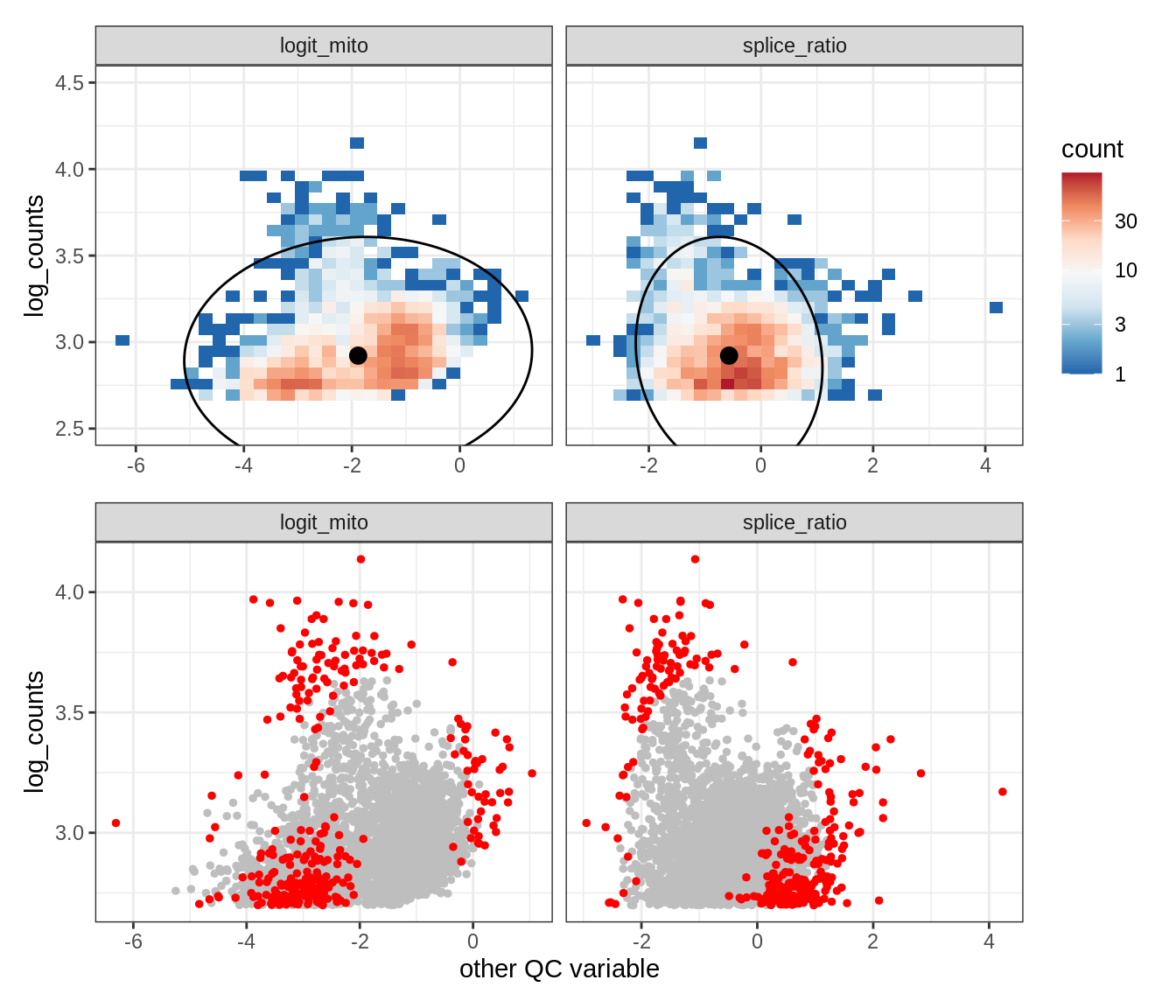
WM106
WM129
WM138
WM140
WM141
WM142
WM146
WM159
WM161
WM181
WM197
WM198
SG6
EU018
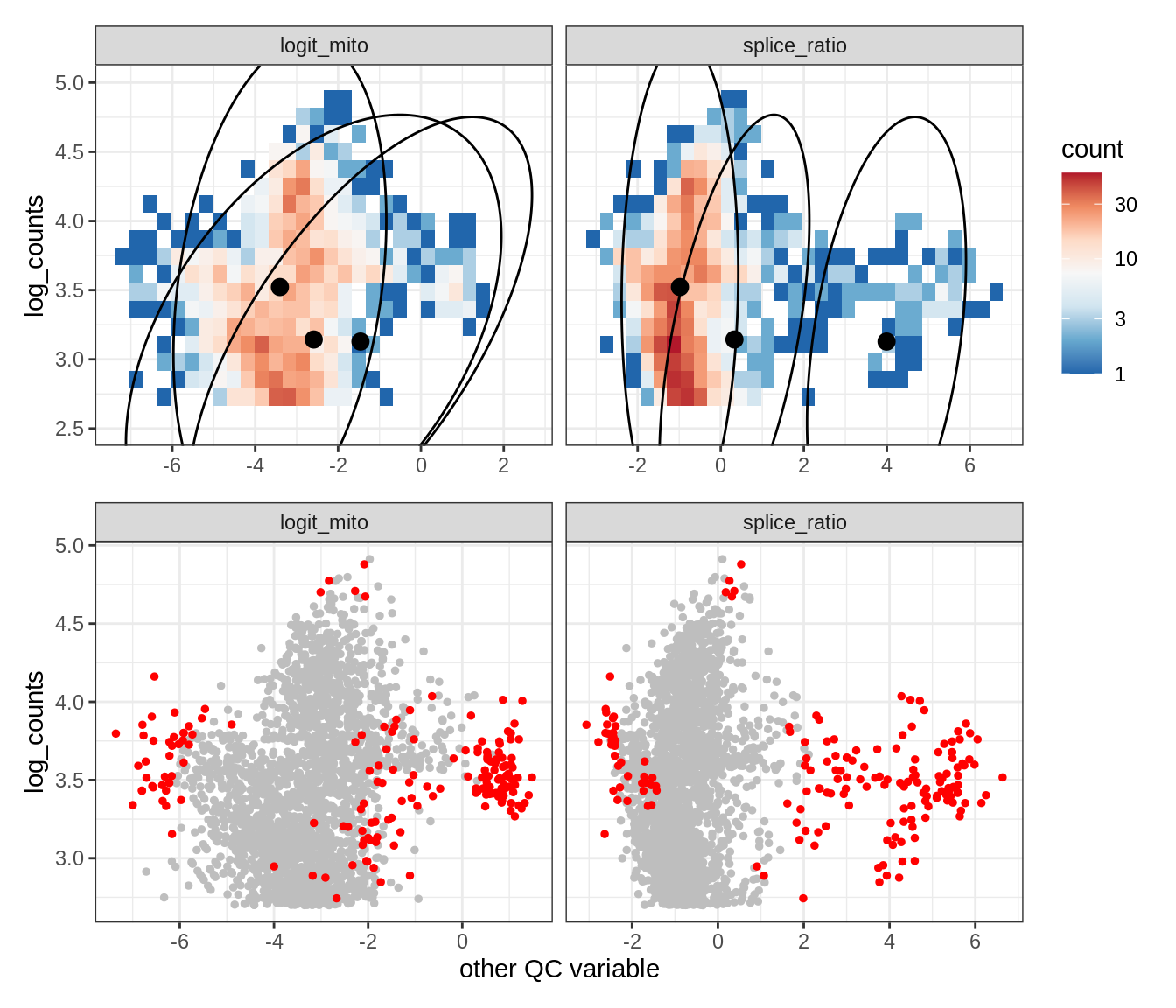
EU024
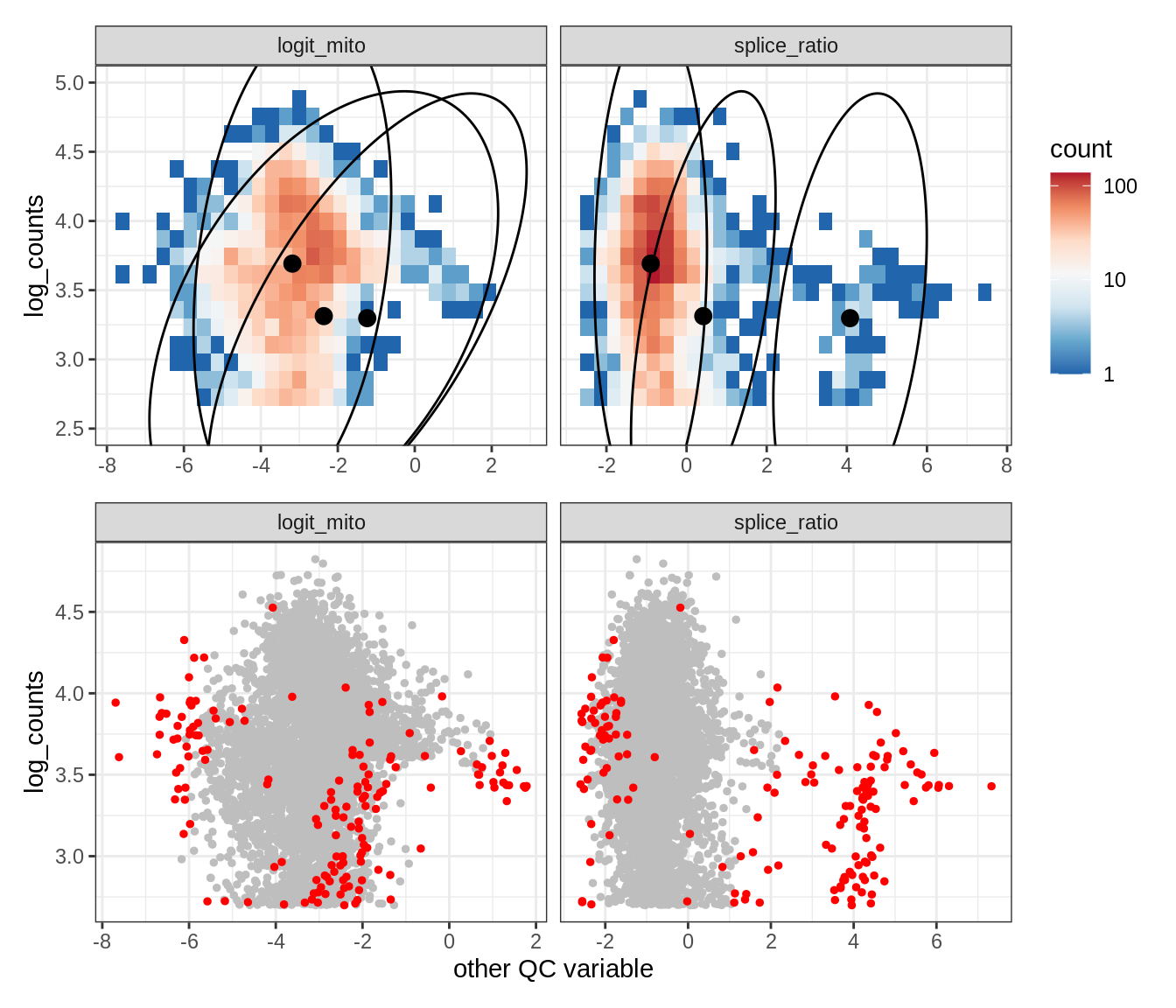
EU033
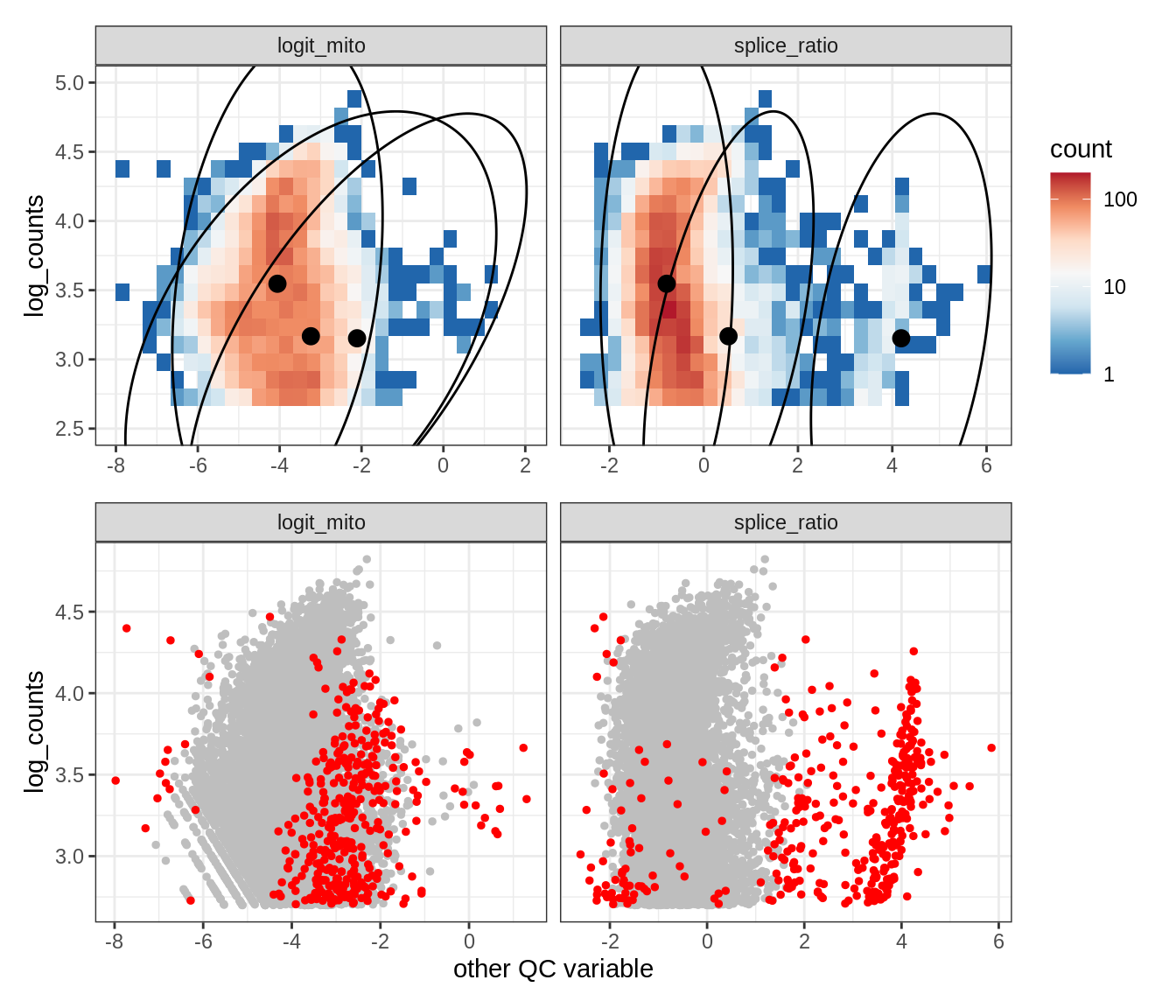
EU038
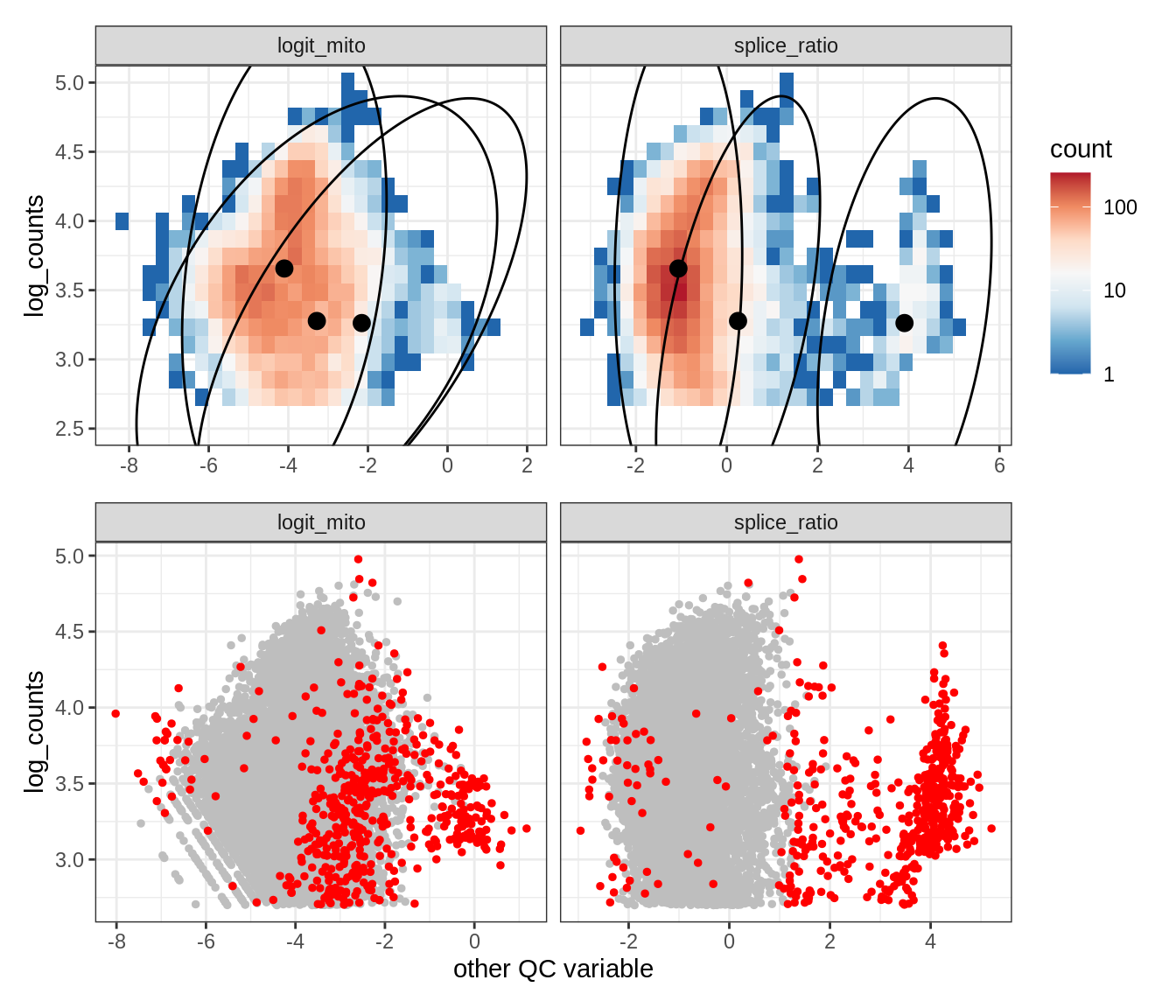
EU039
EU040
EU045
EU047
EU055
EU068
WM110
WM125
WM127
WM148
WM150
WM173
SG7
EU017
EU022
EU025
EU032
EU046
EU048
EU050
EU058
EU064
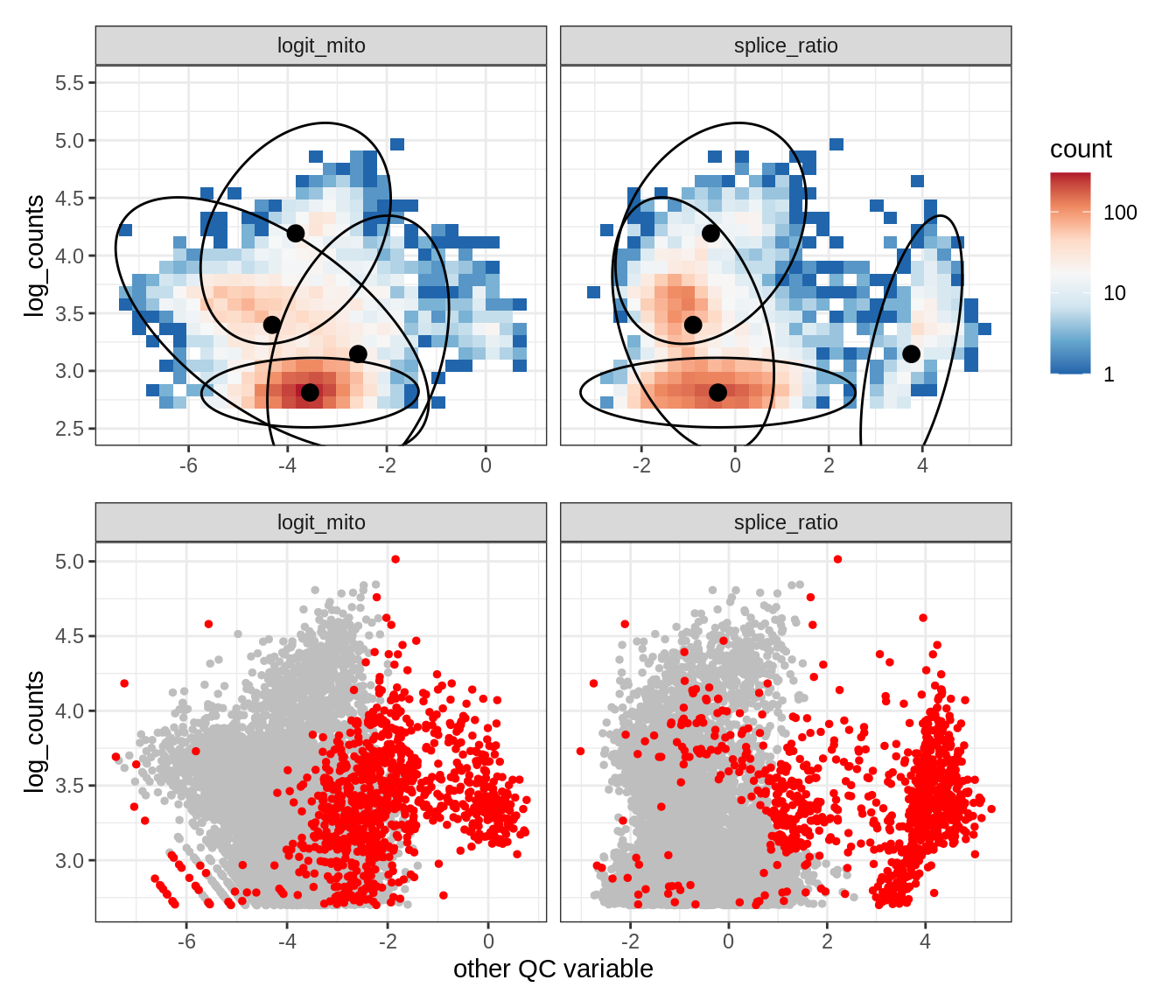
EU066
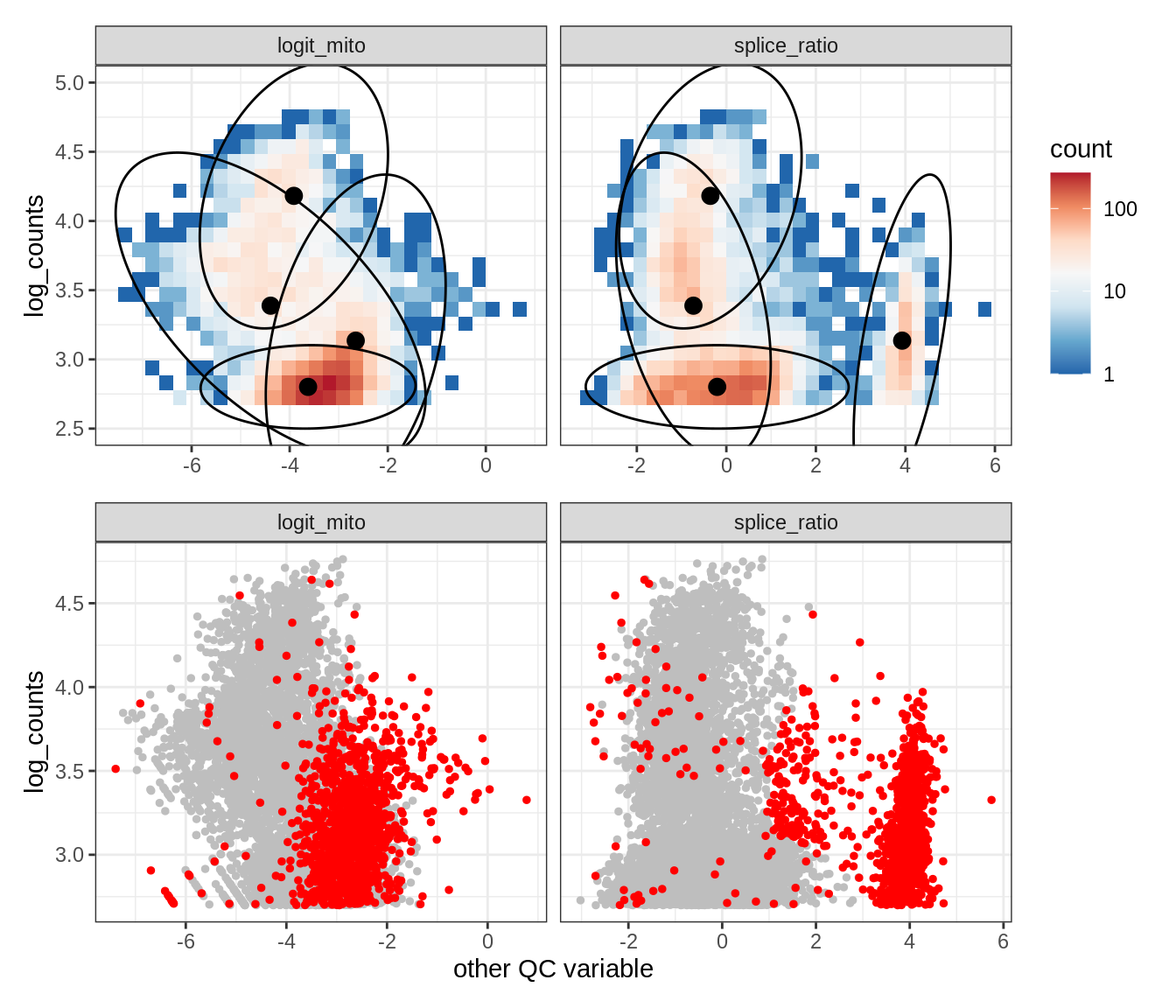
EU067
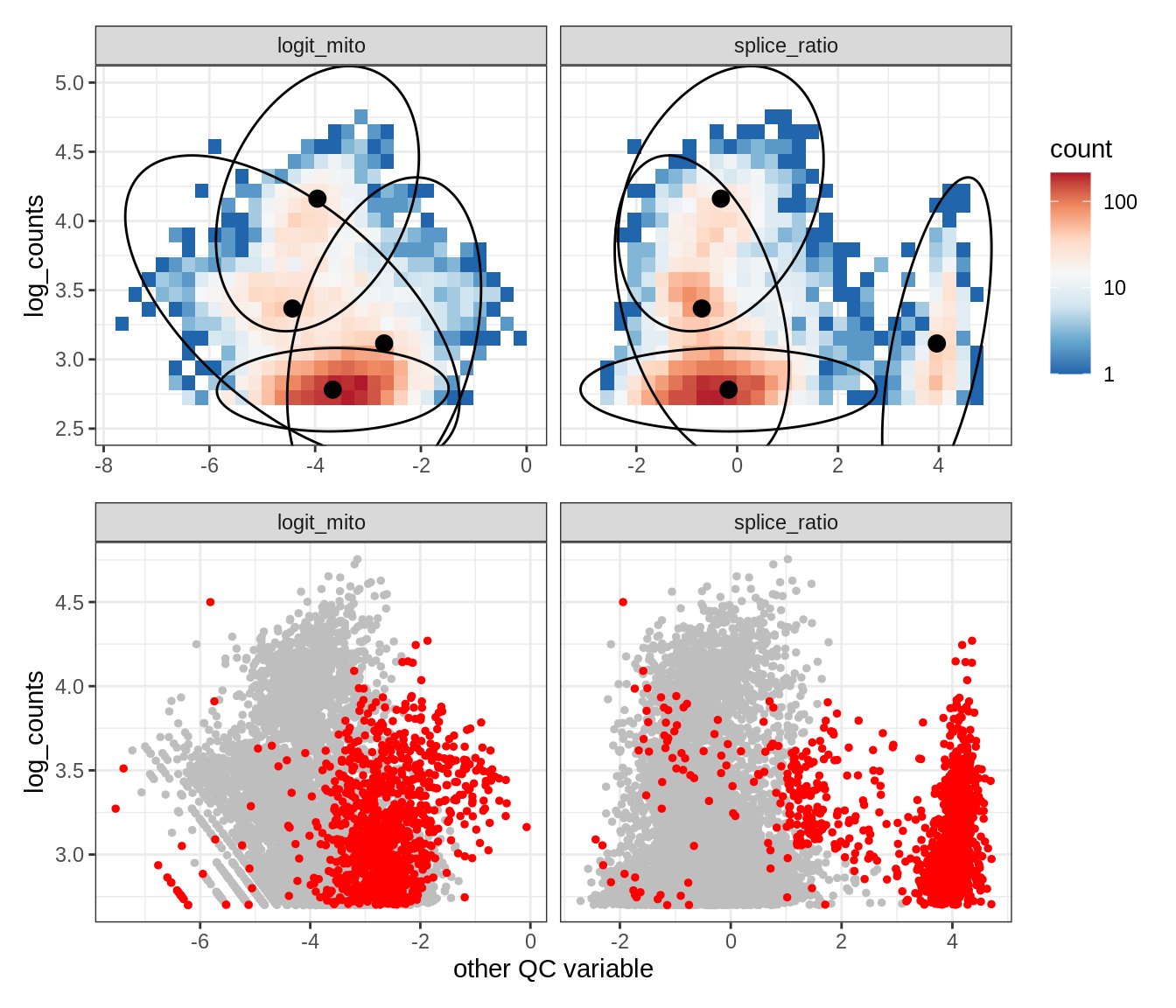
EU071
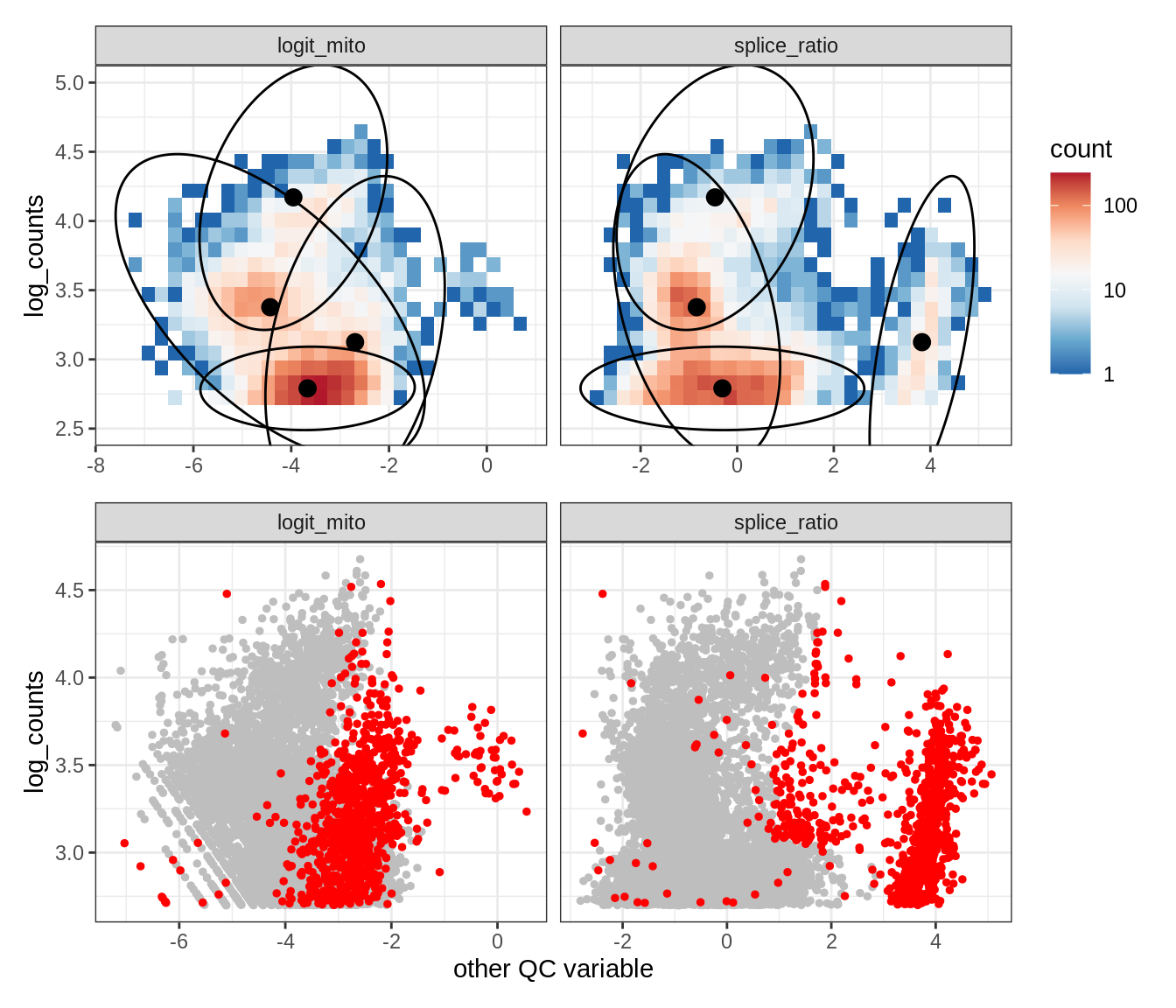
EU072
EU077
WM174
SG8
EU007
EU011
EU034
EU069
EU070
EU073
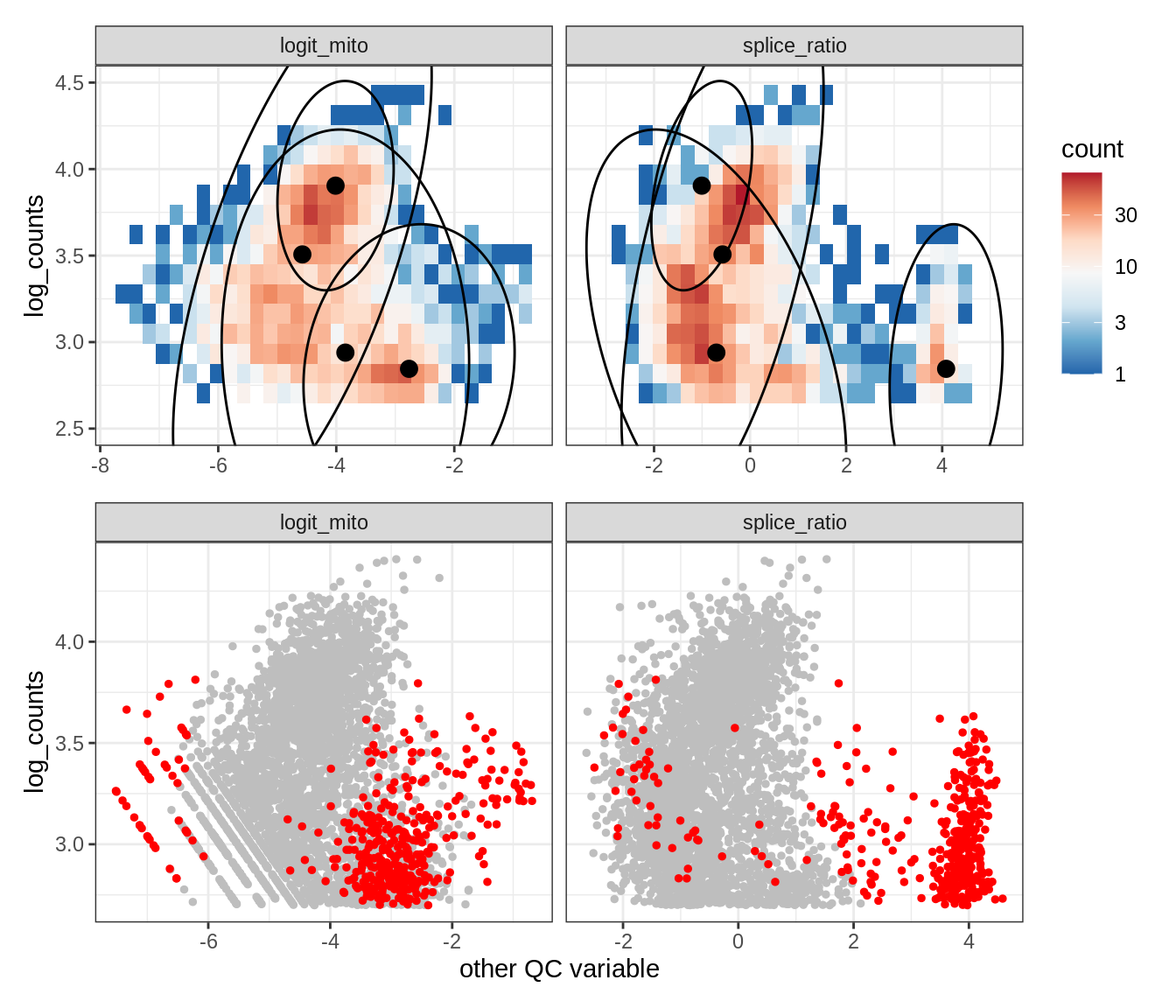
EU075
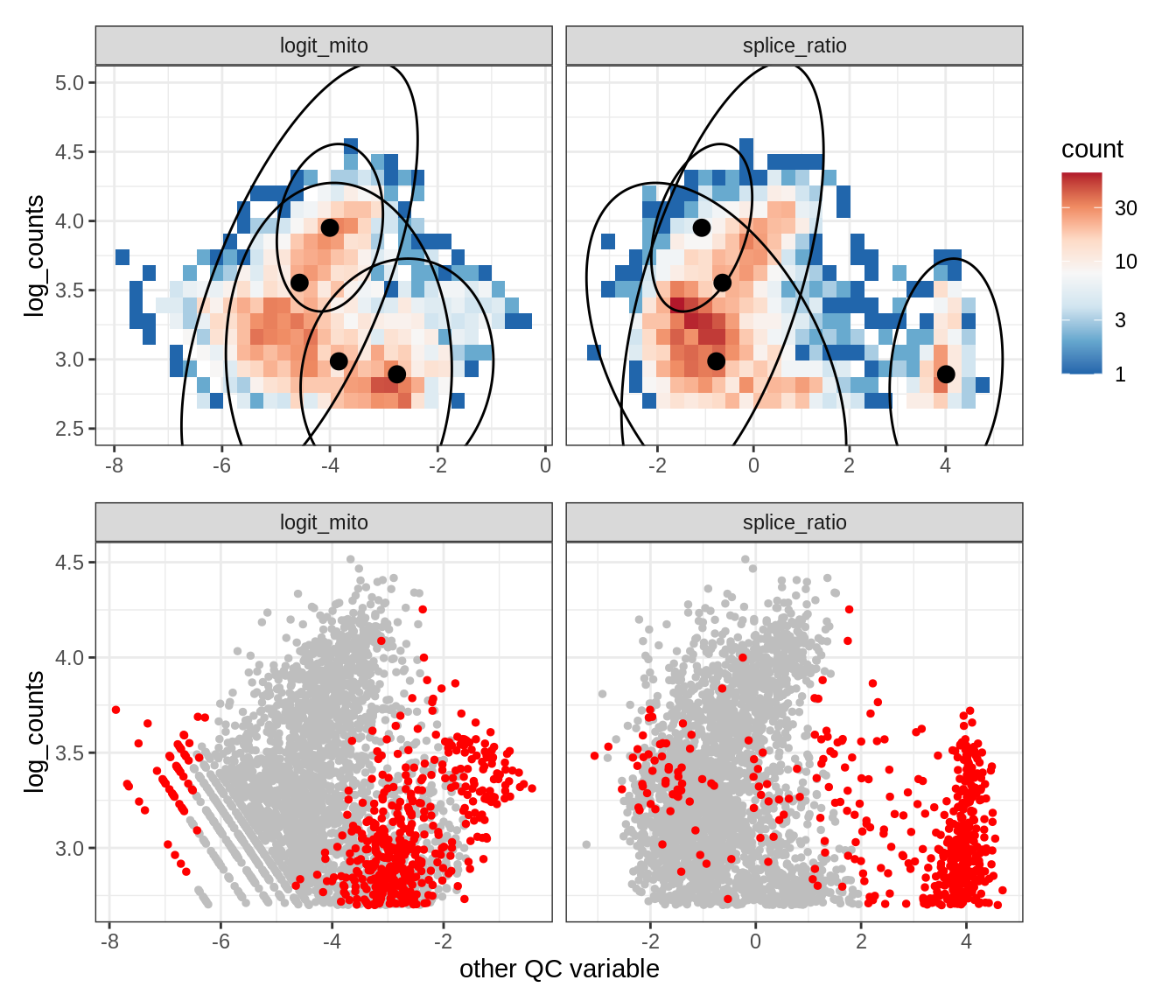
EU076
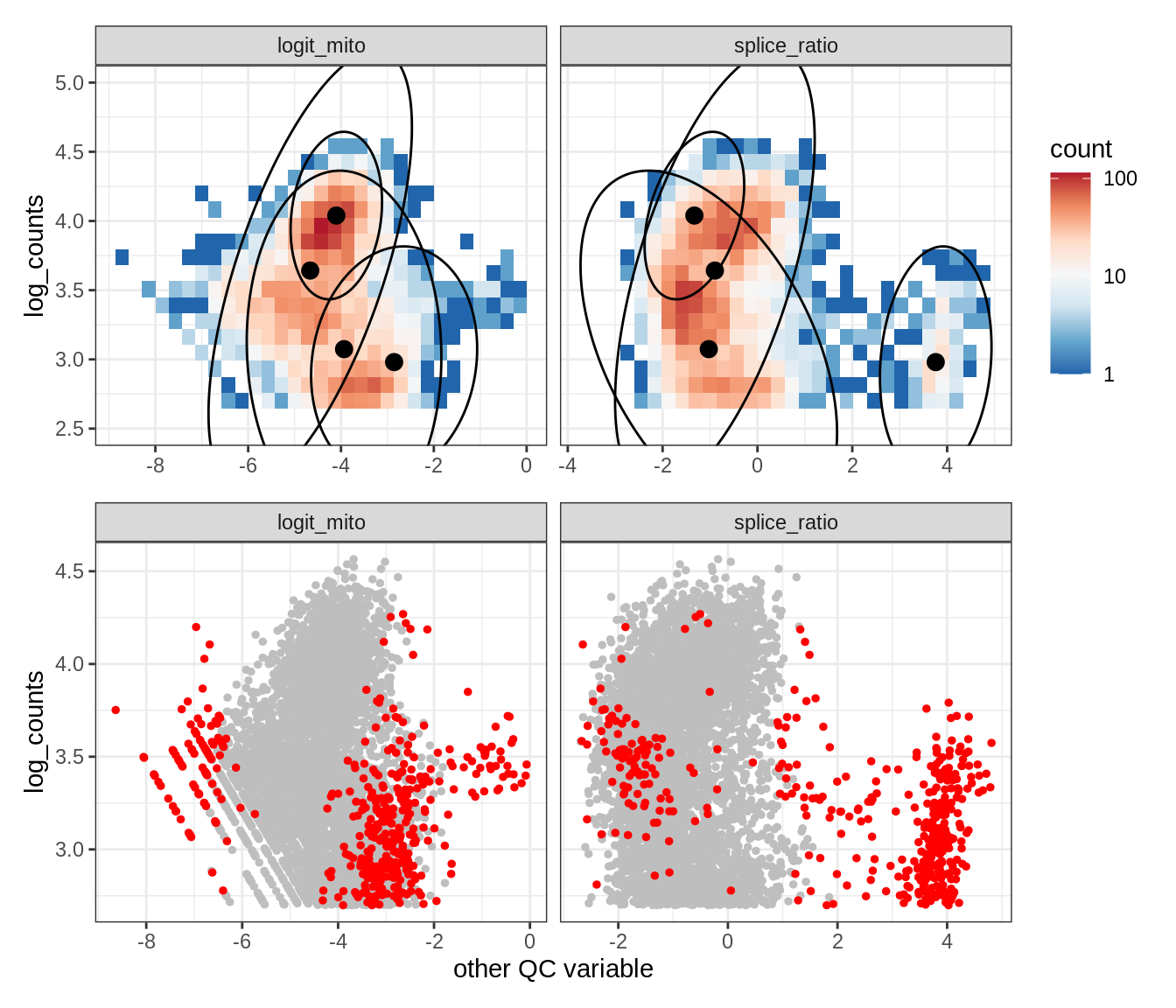
EU078
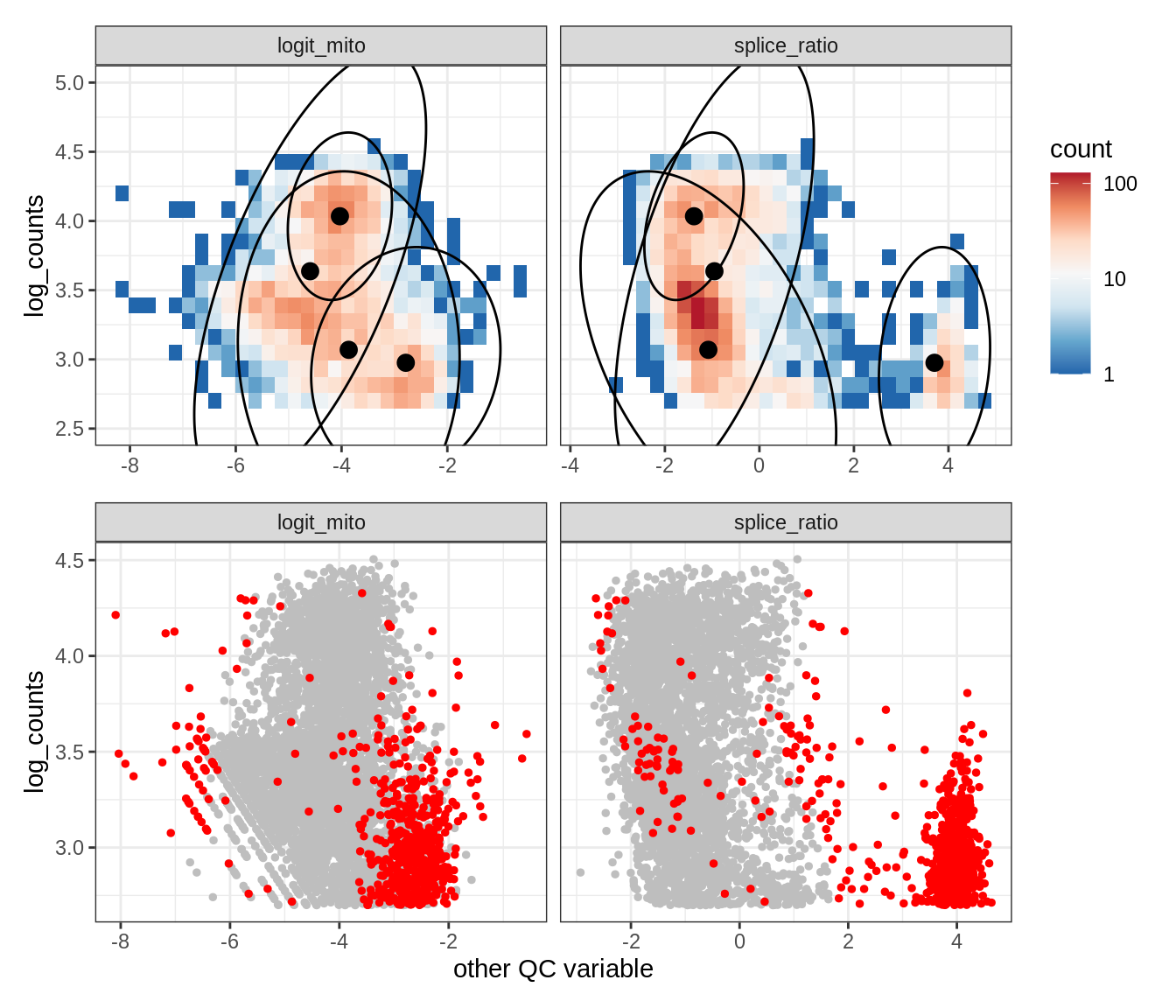
WM101
SG9
WM126
WM149
WM176
WM178
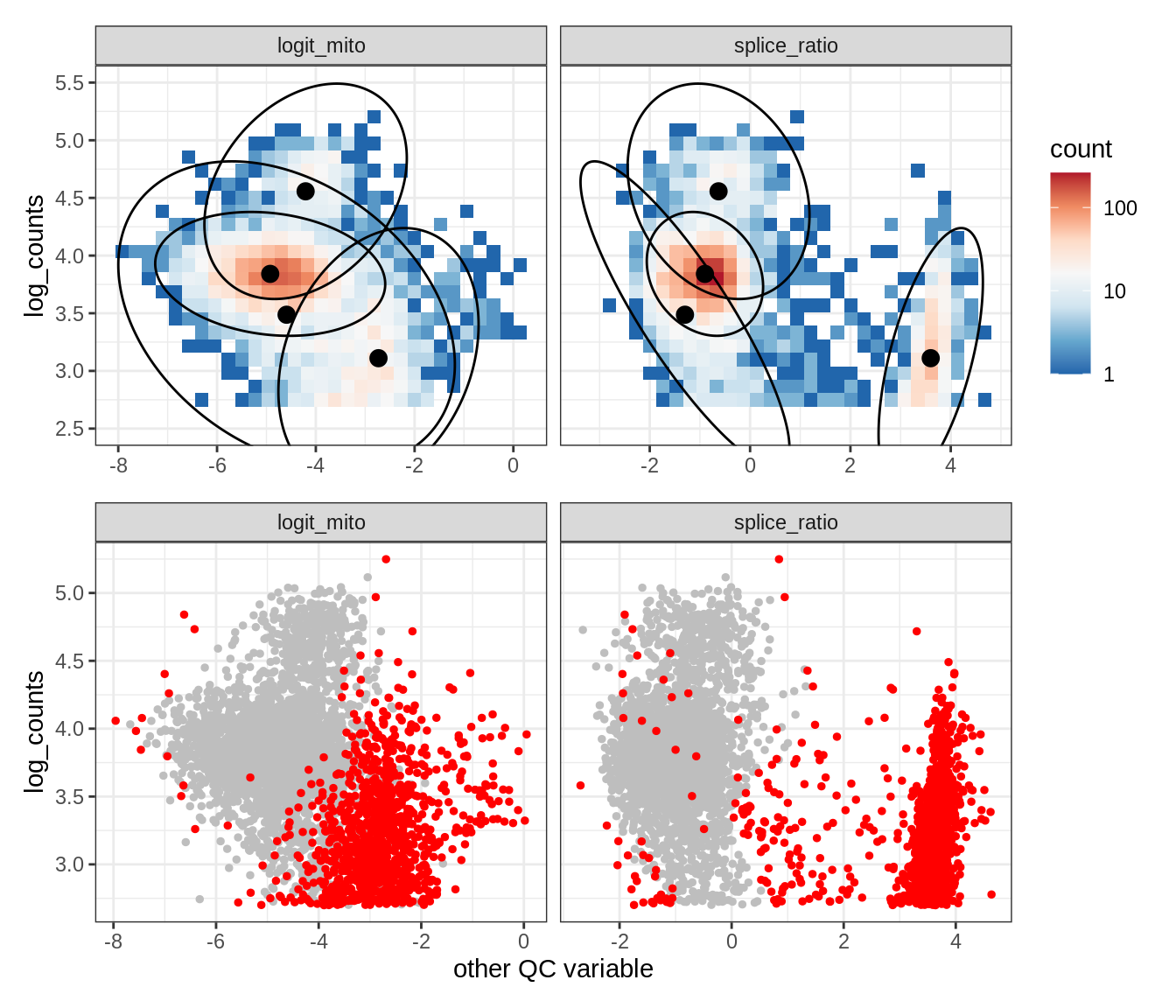
WM180
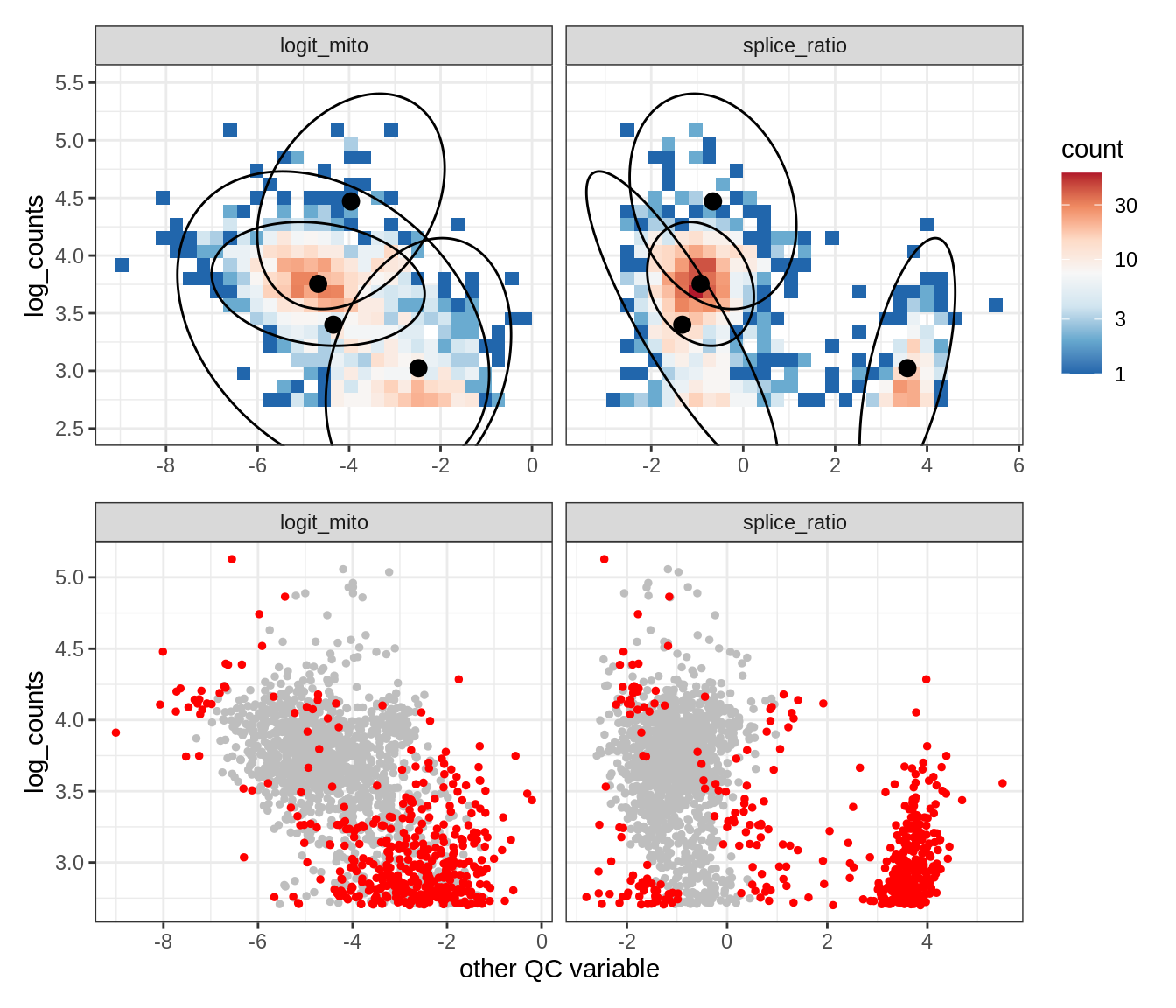
WM189
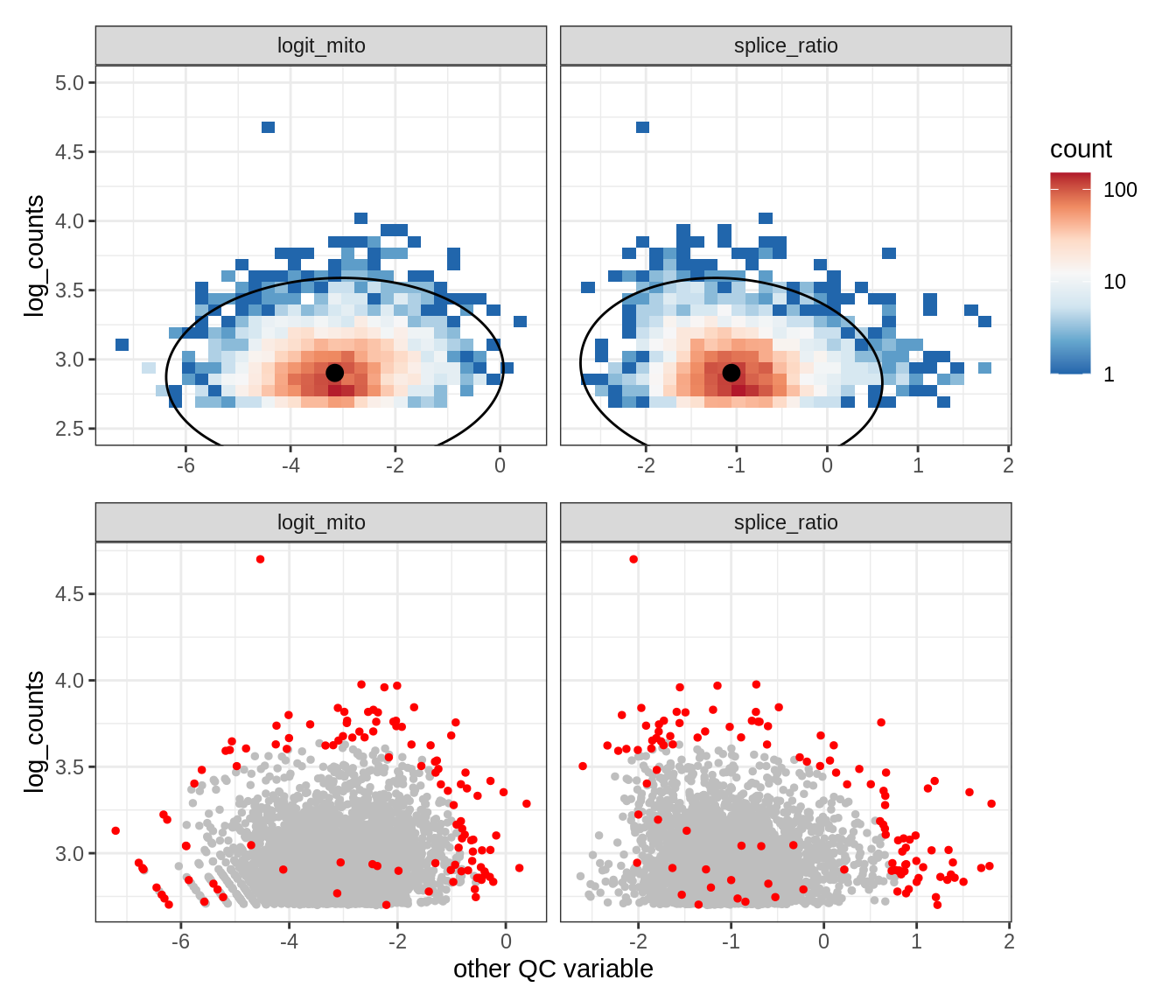
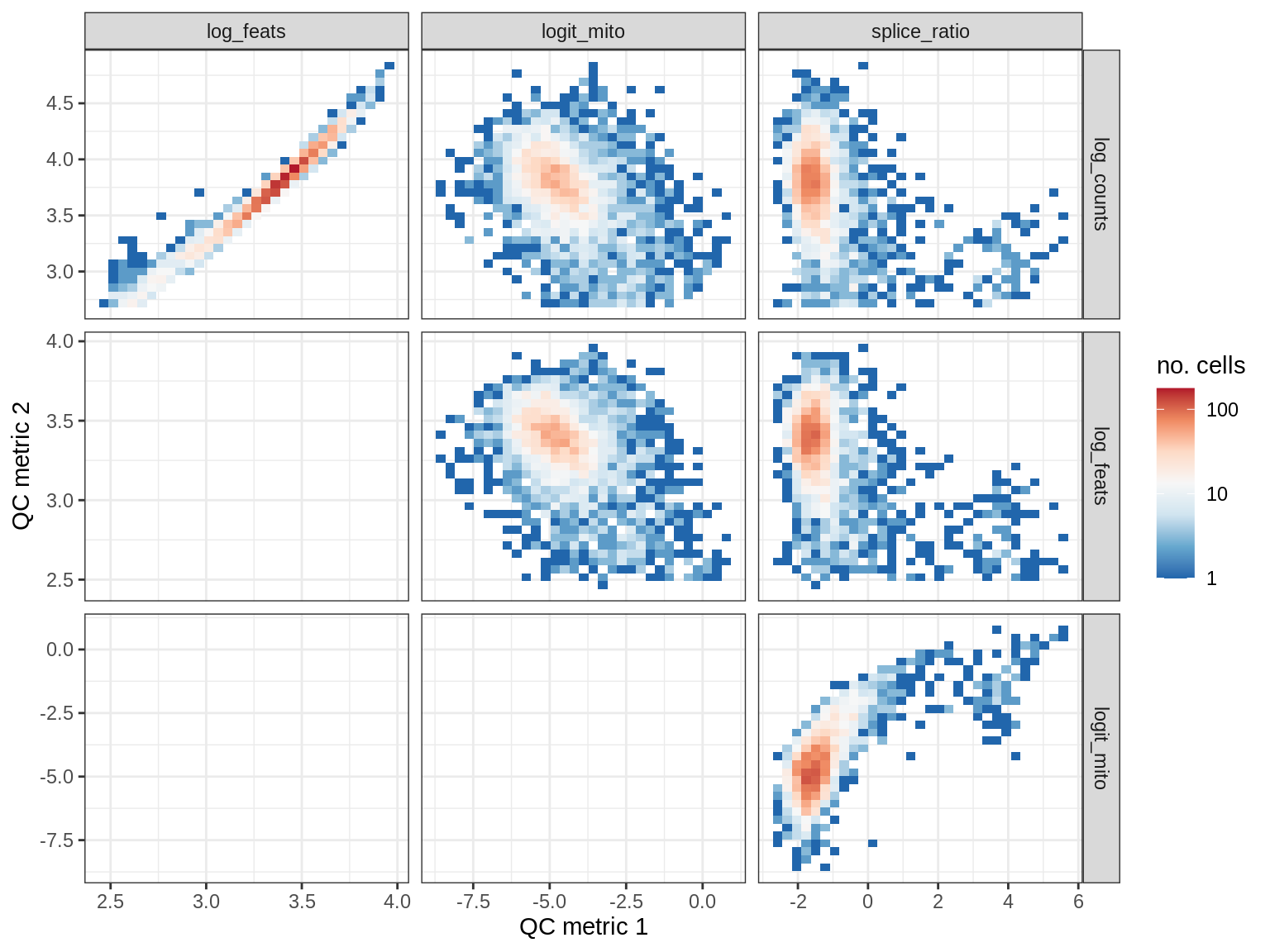
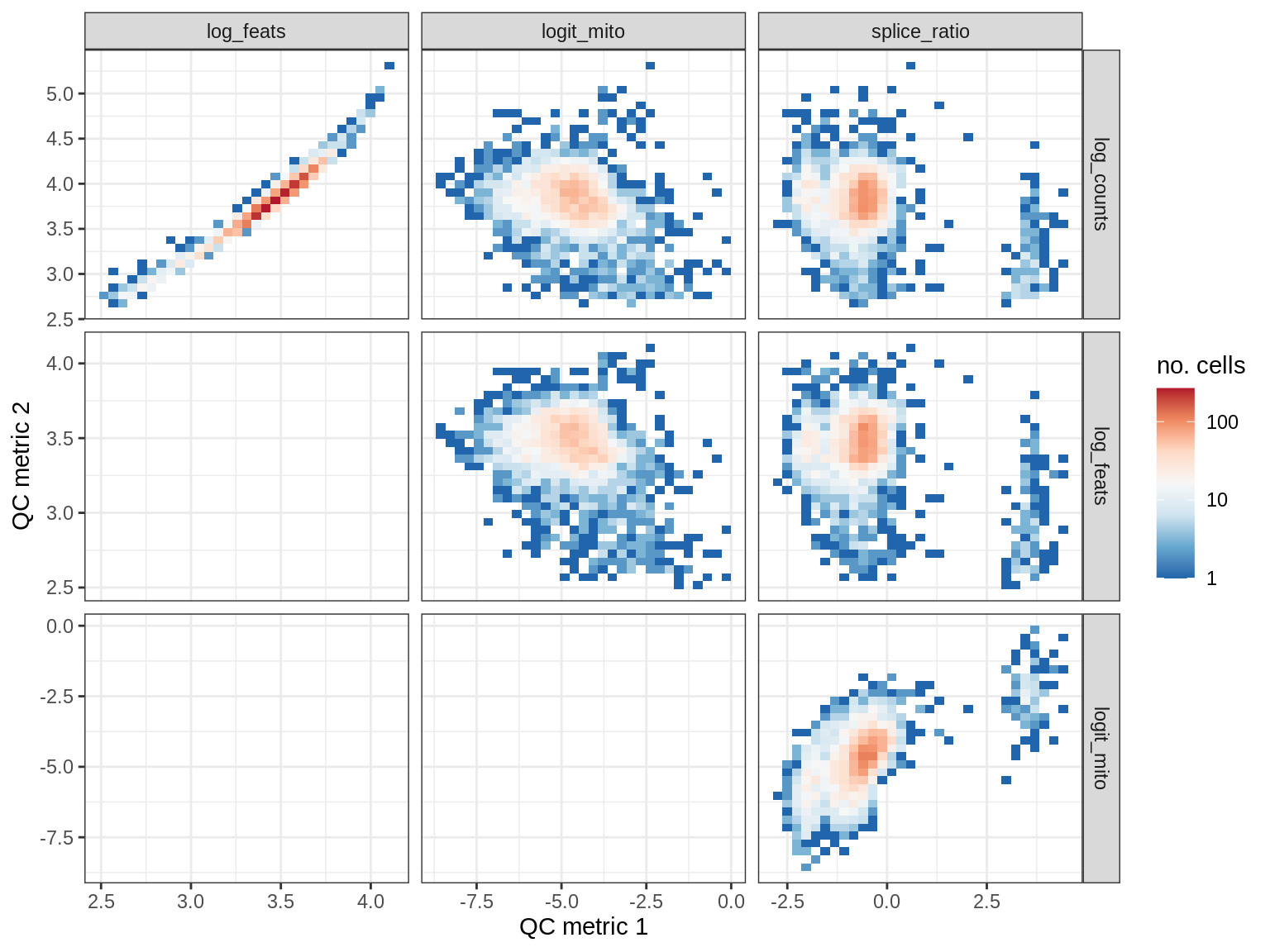
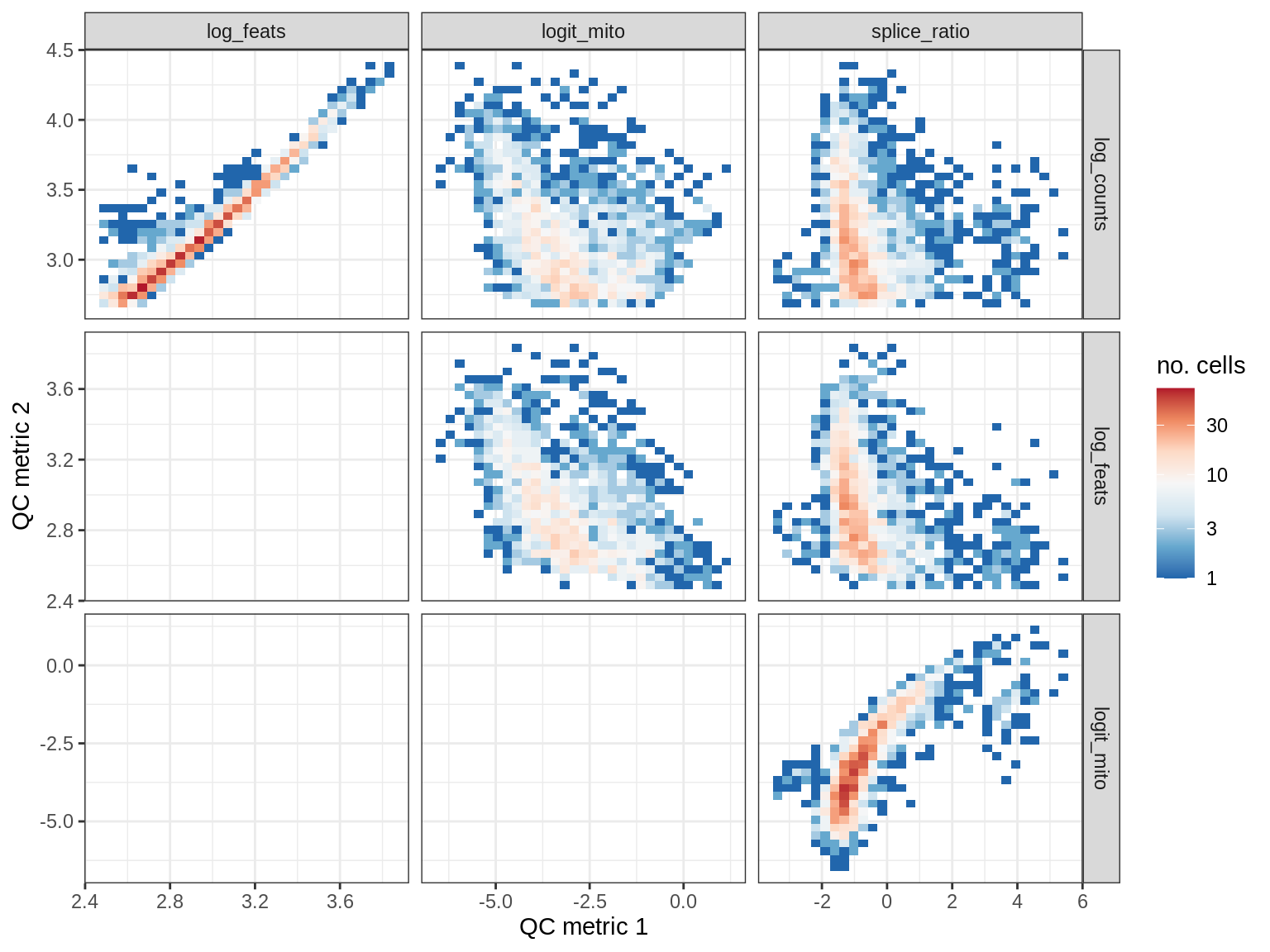
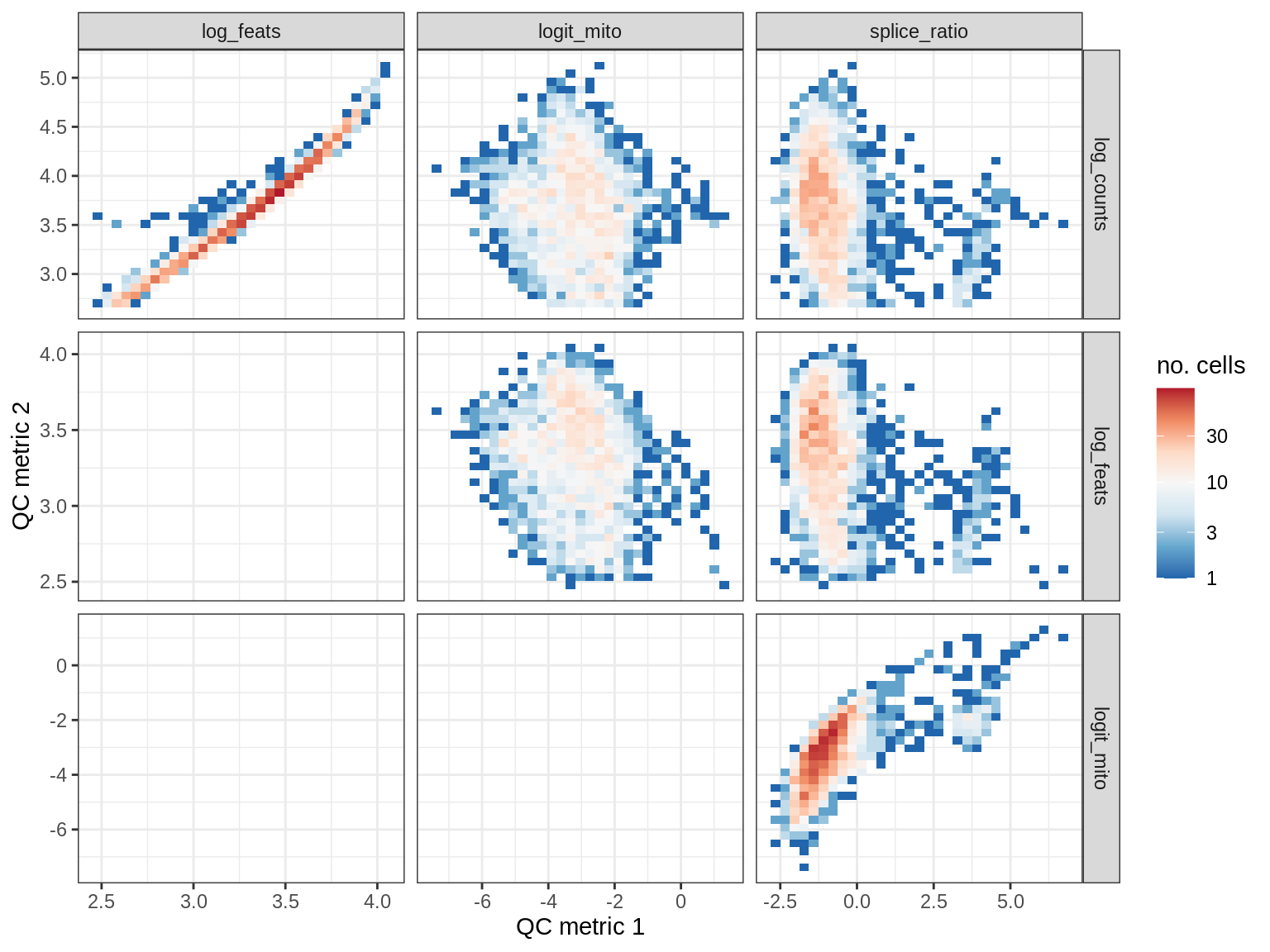
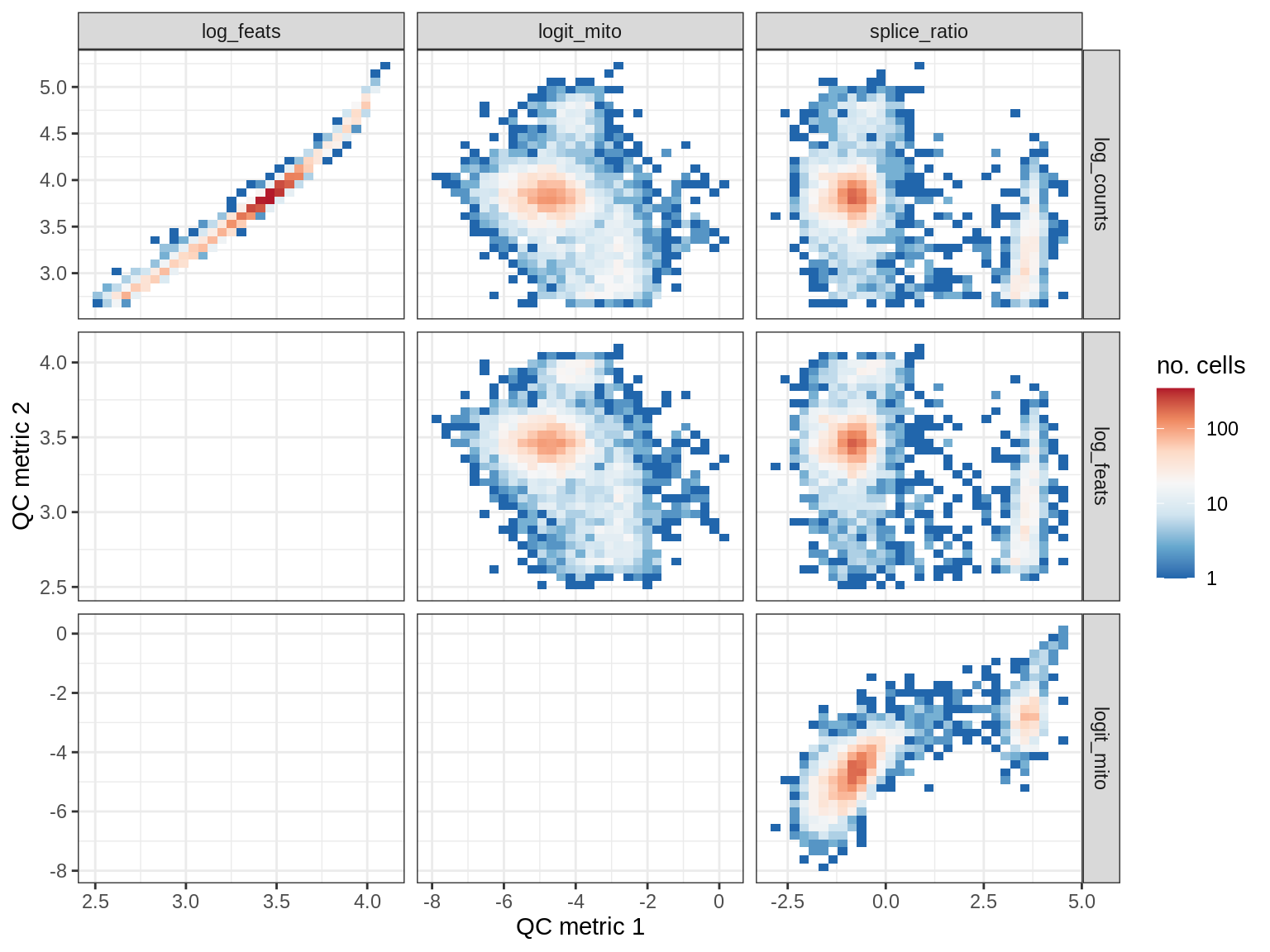
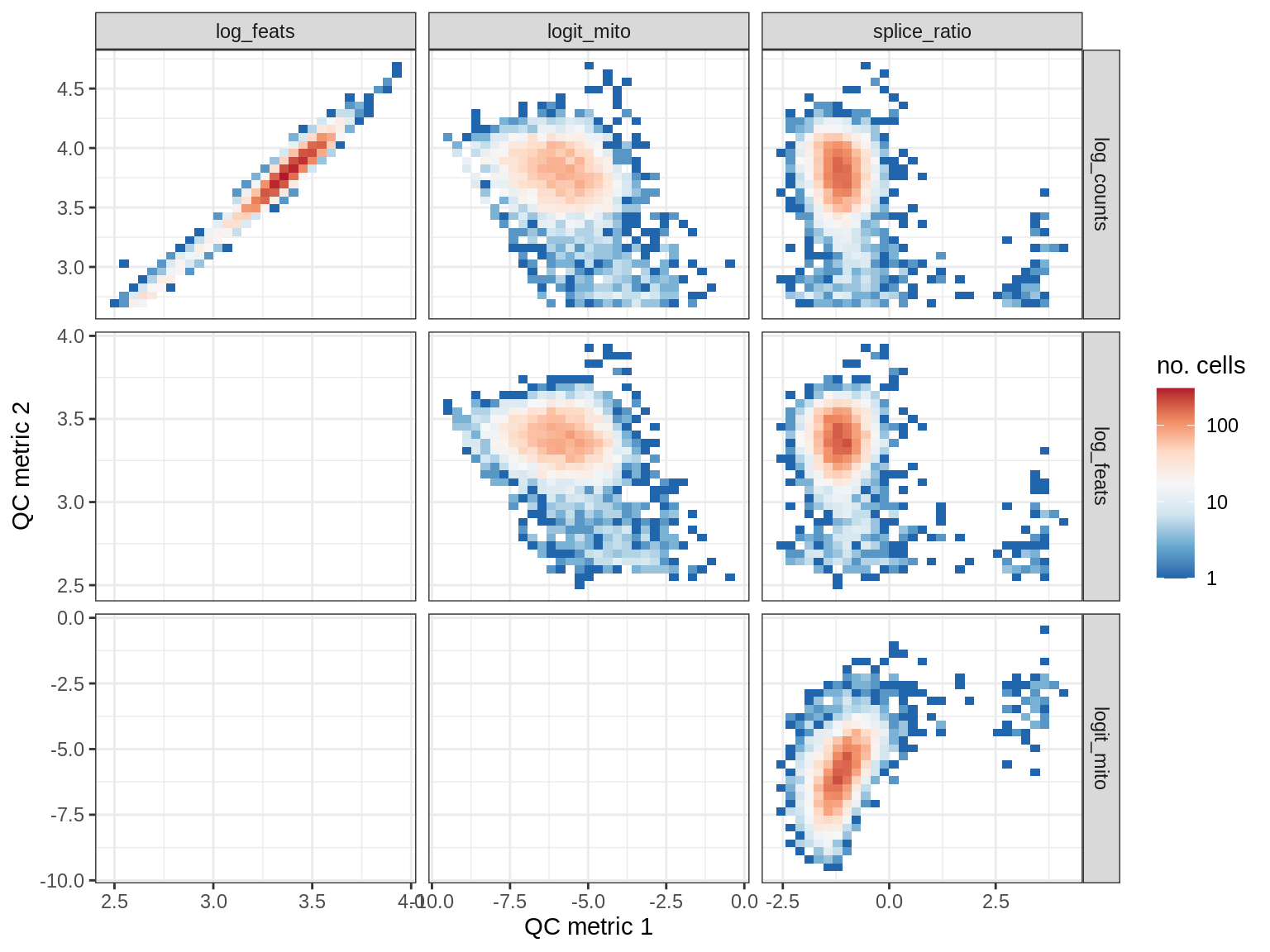
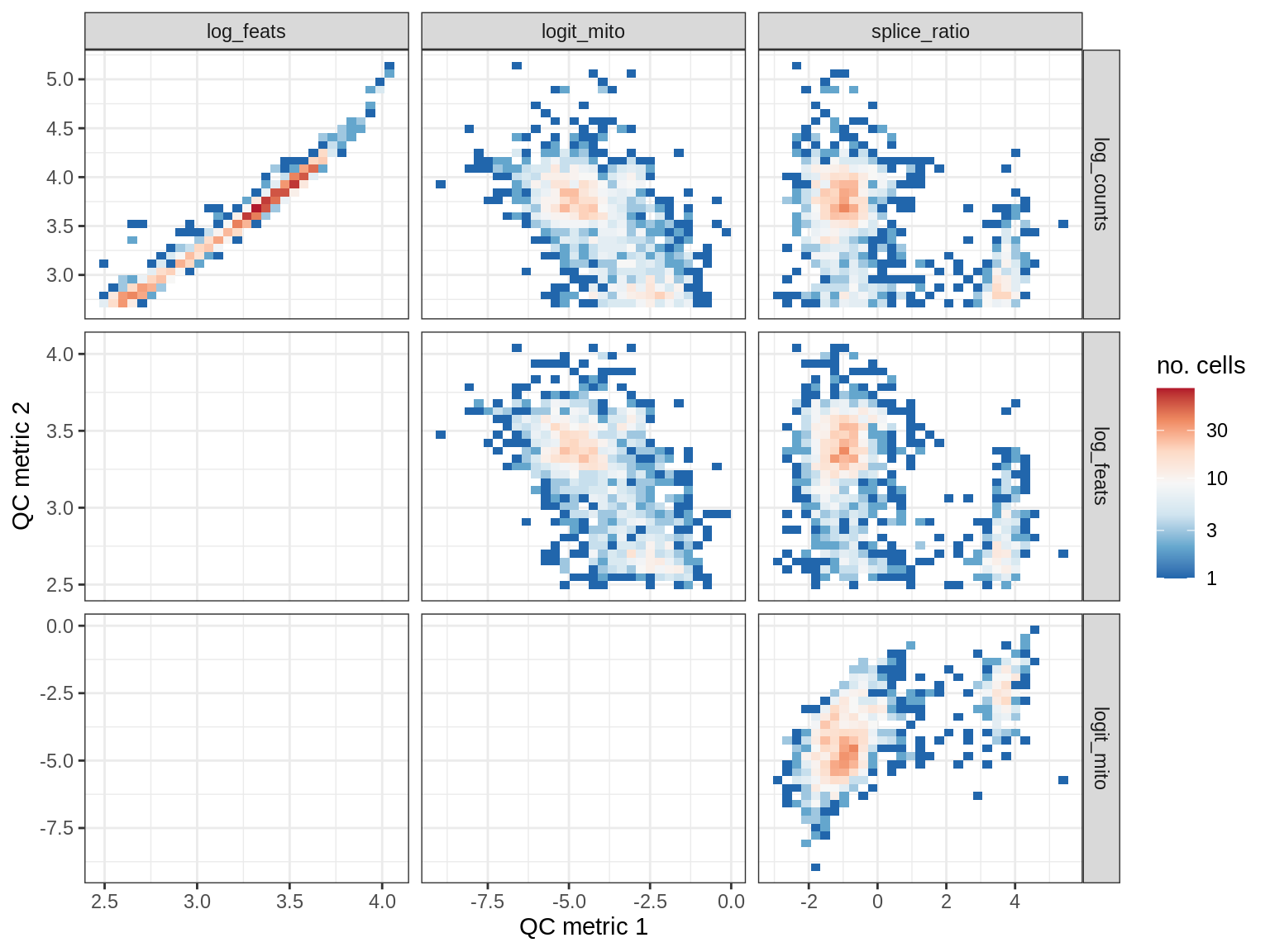
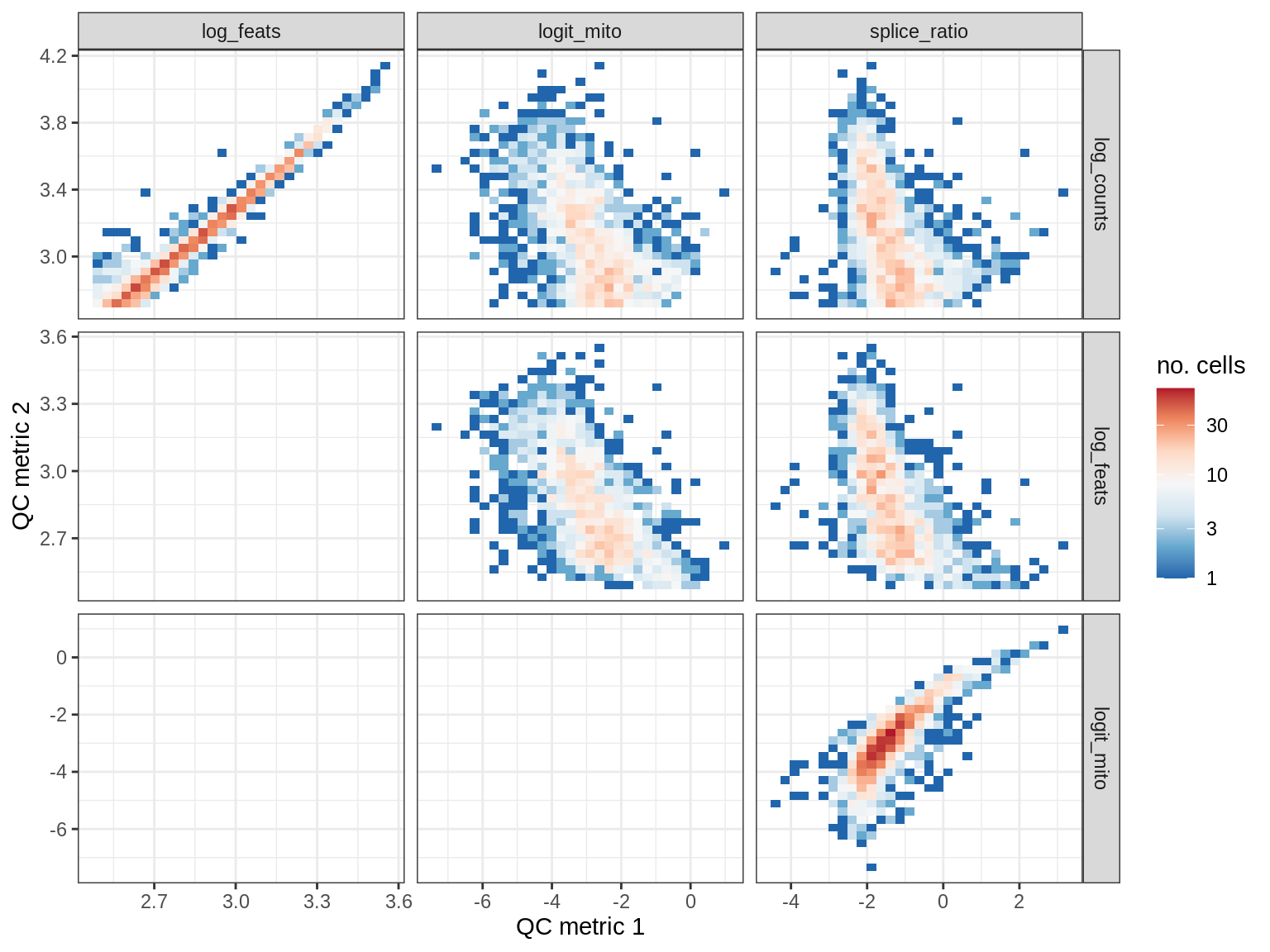
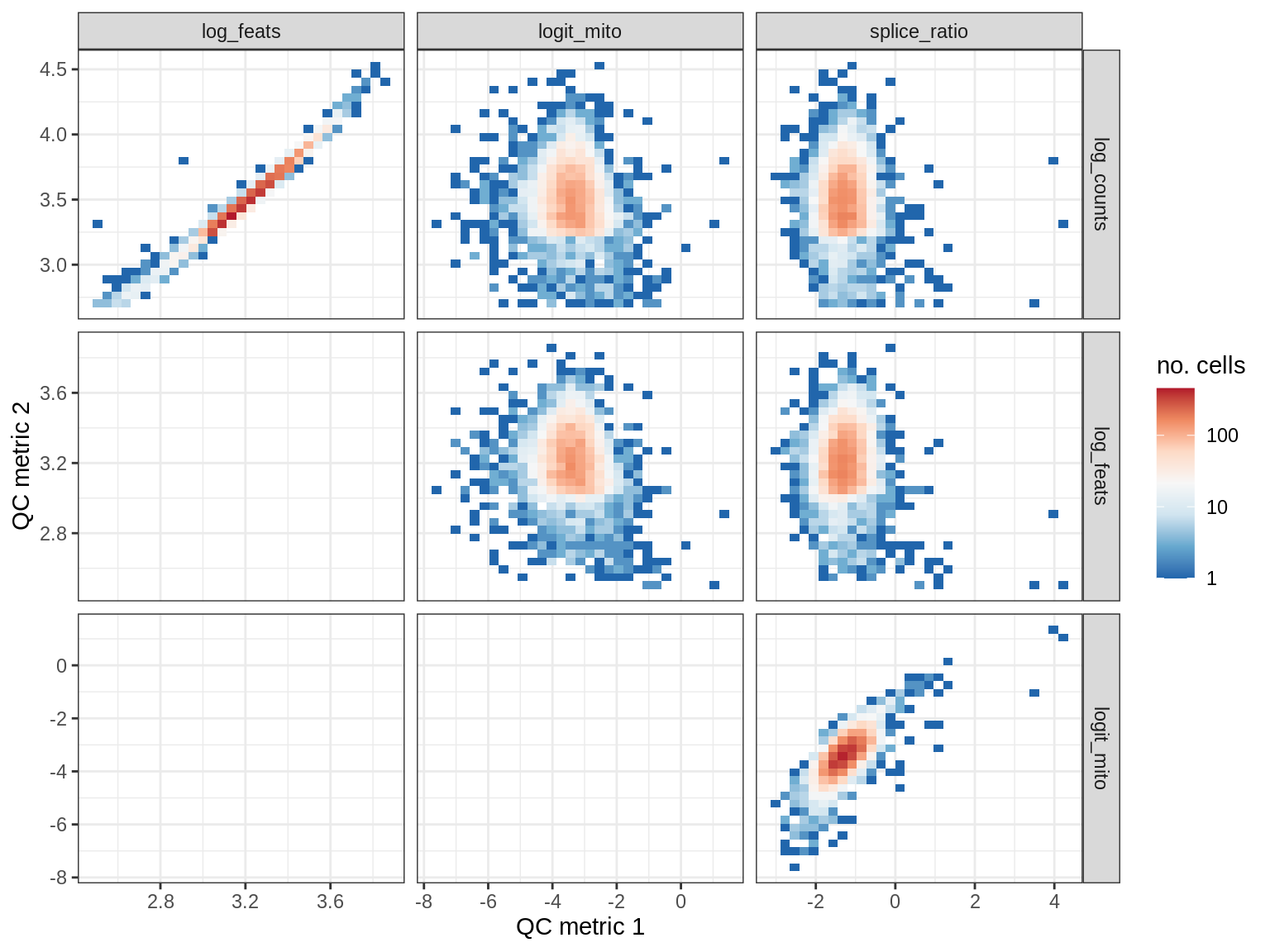
QC metrics scatterplots
samples = qc_dt$sample_id %>% unique %>% sort
# qc_names = c('log_counts', 'logit_mito', 'splice_ratio')
qc_names = c('log_counts', 'log_feats', 'logit_mito', 'splice_ratio')
for (s in samples) {
cat('### ', s, '\n')
print(plot_qc_metric_scatter(qc_dt[ sample_id == s ], qc_names))
cat('\n\n')
}EU005
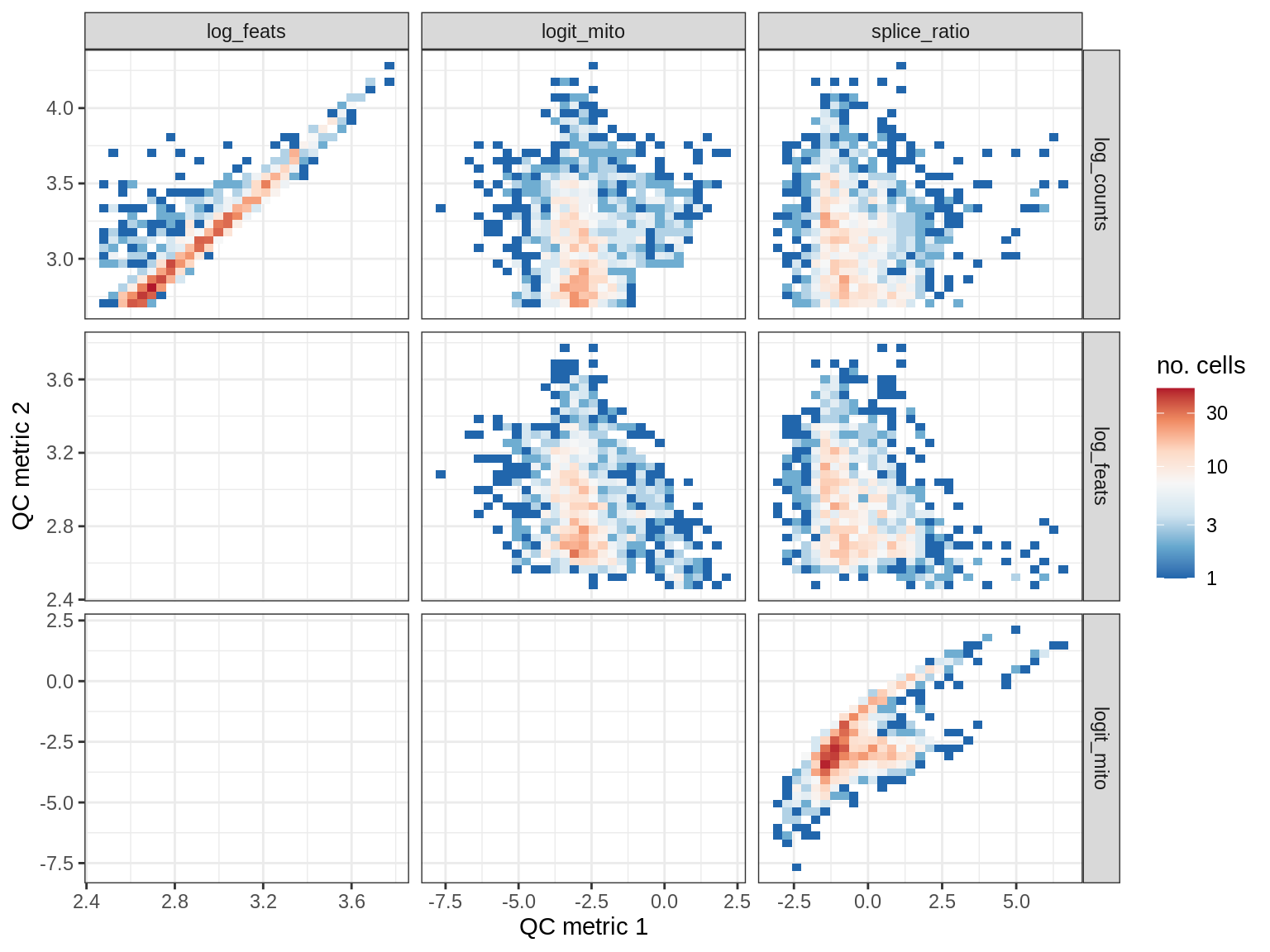
EU006
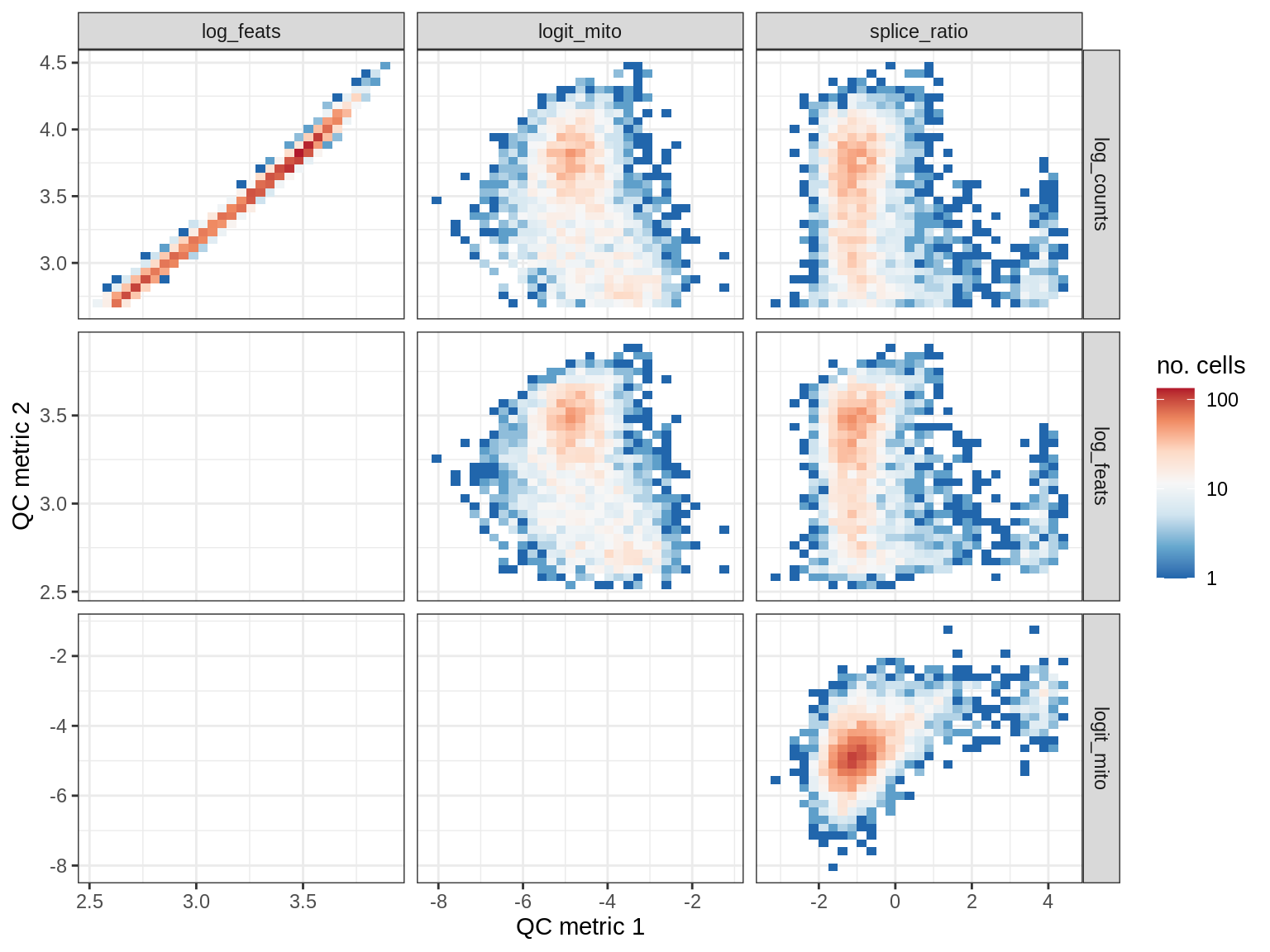
EU007
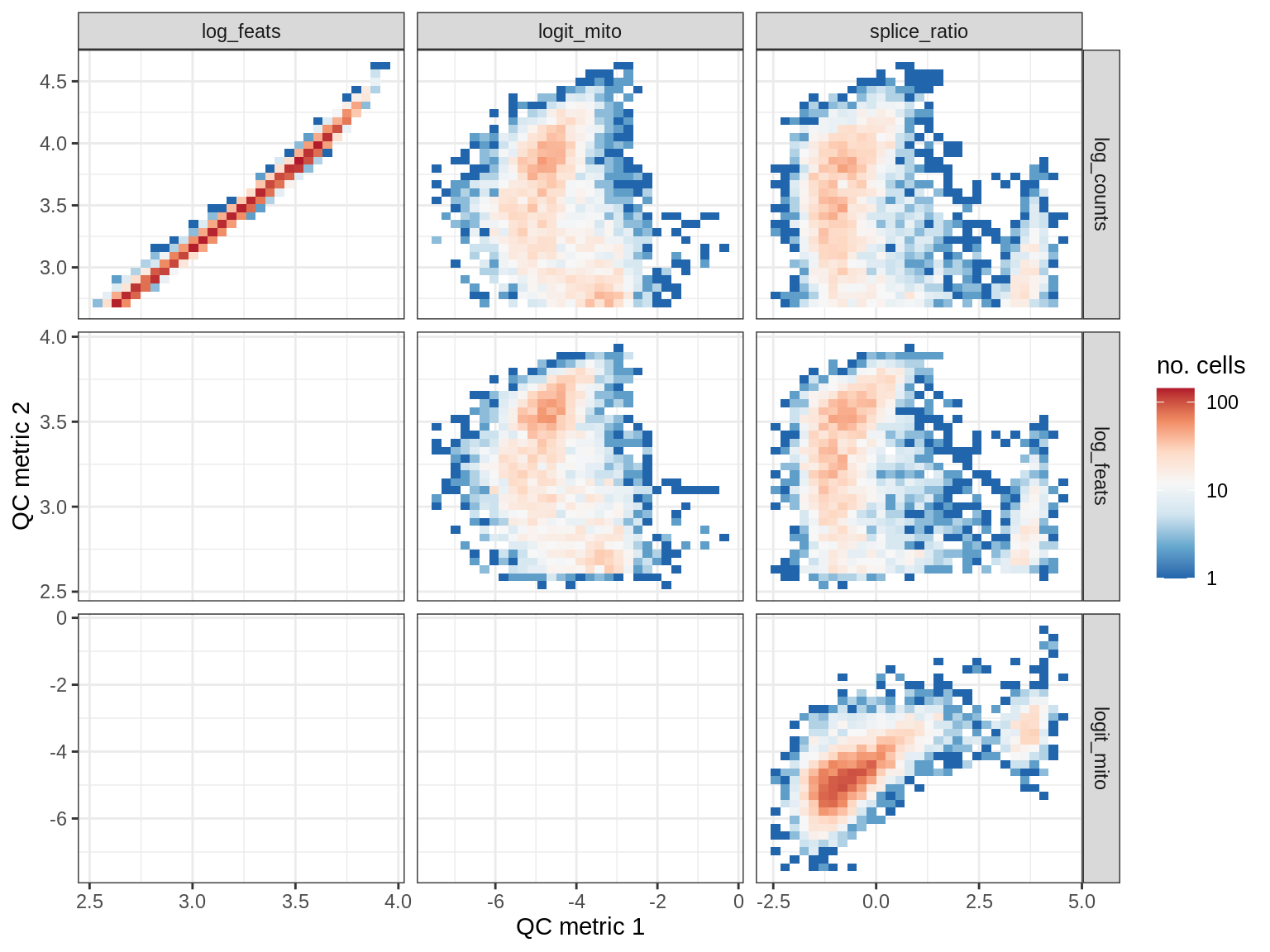
EU008
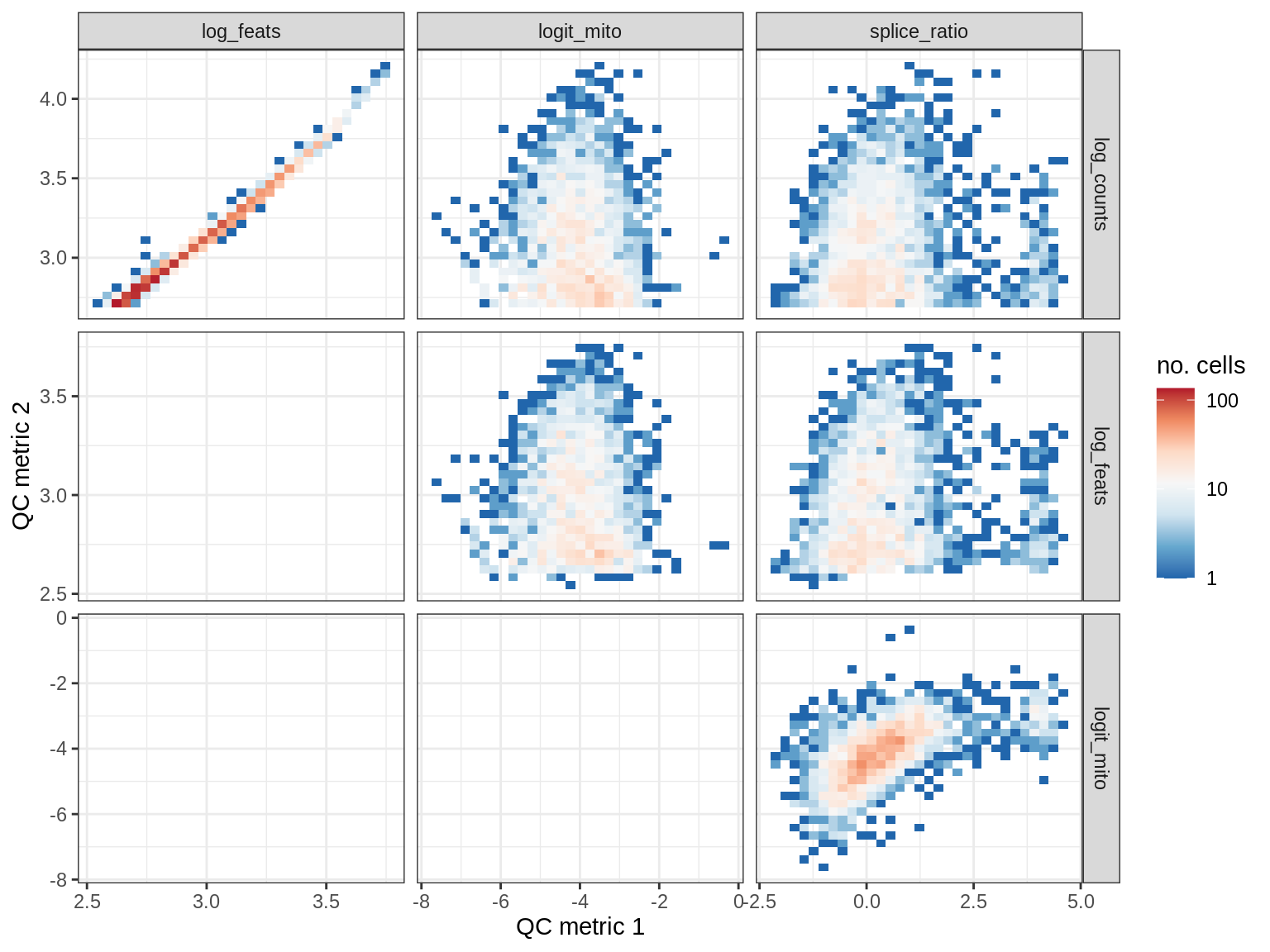
EU009
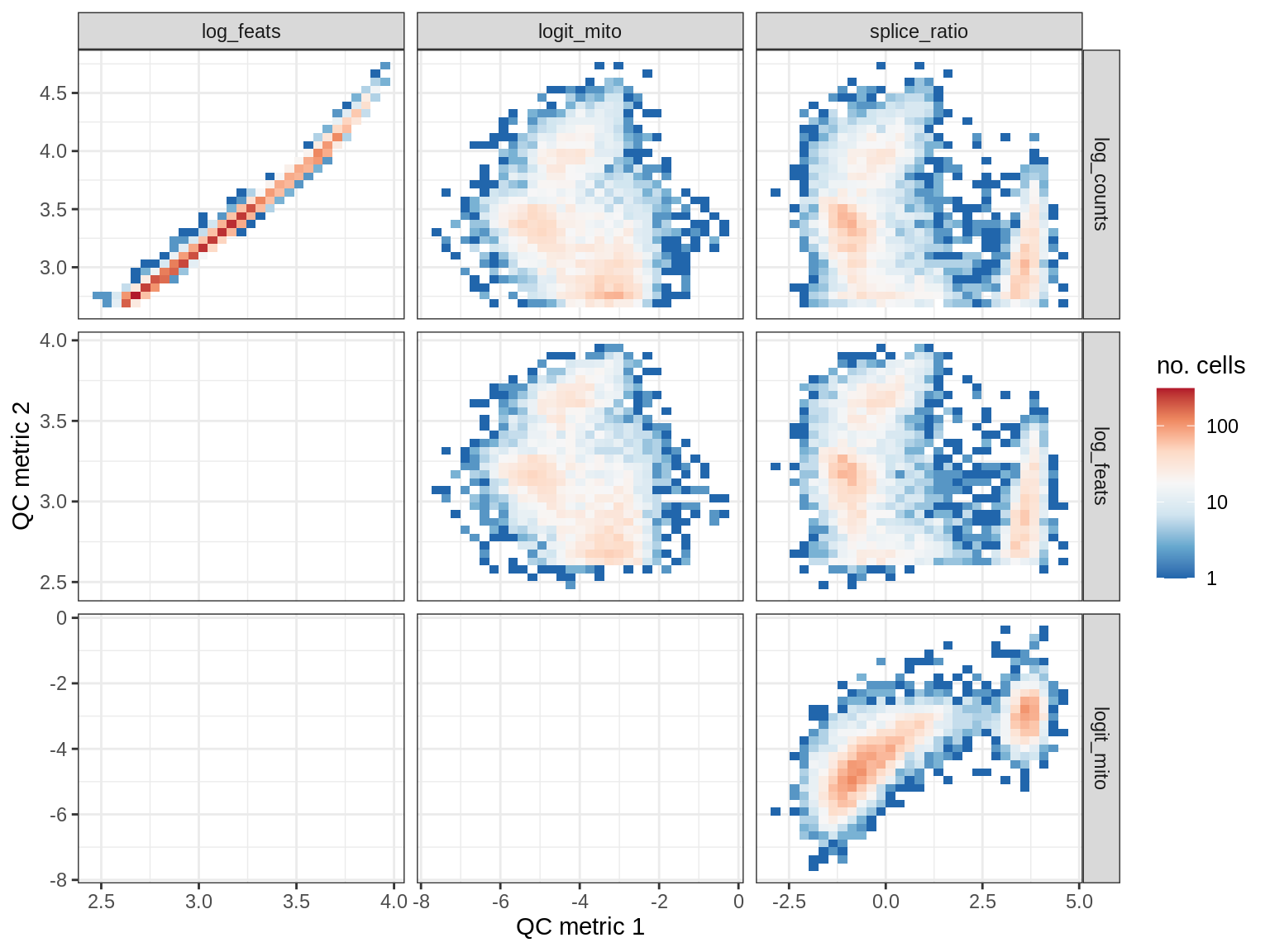
EU010
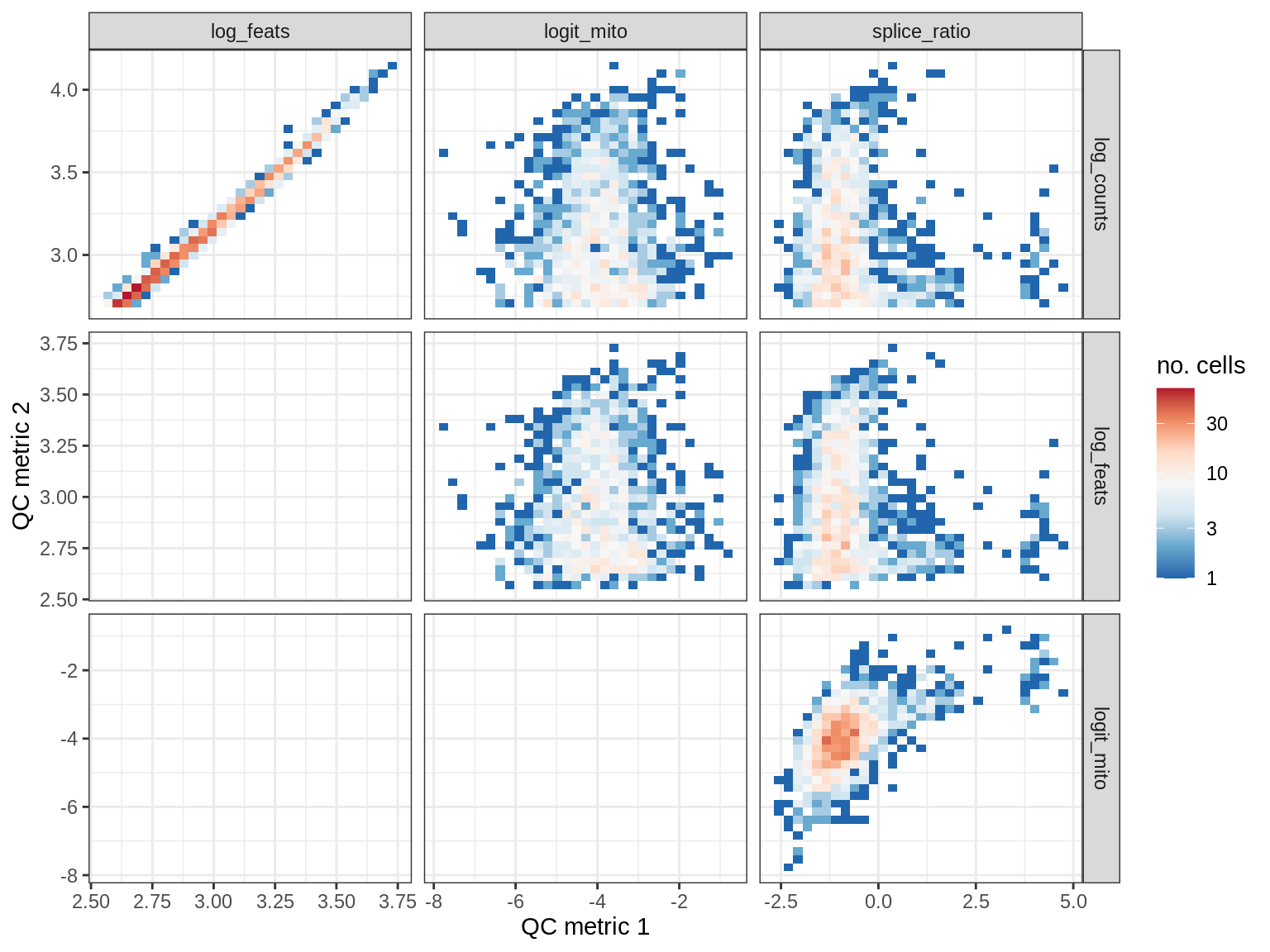
EU011
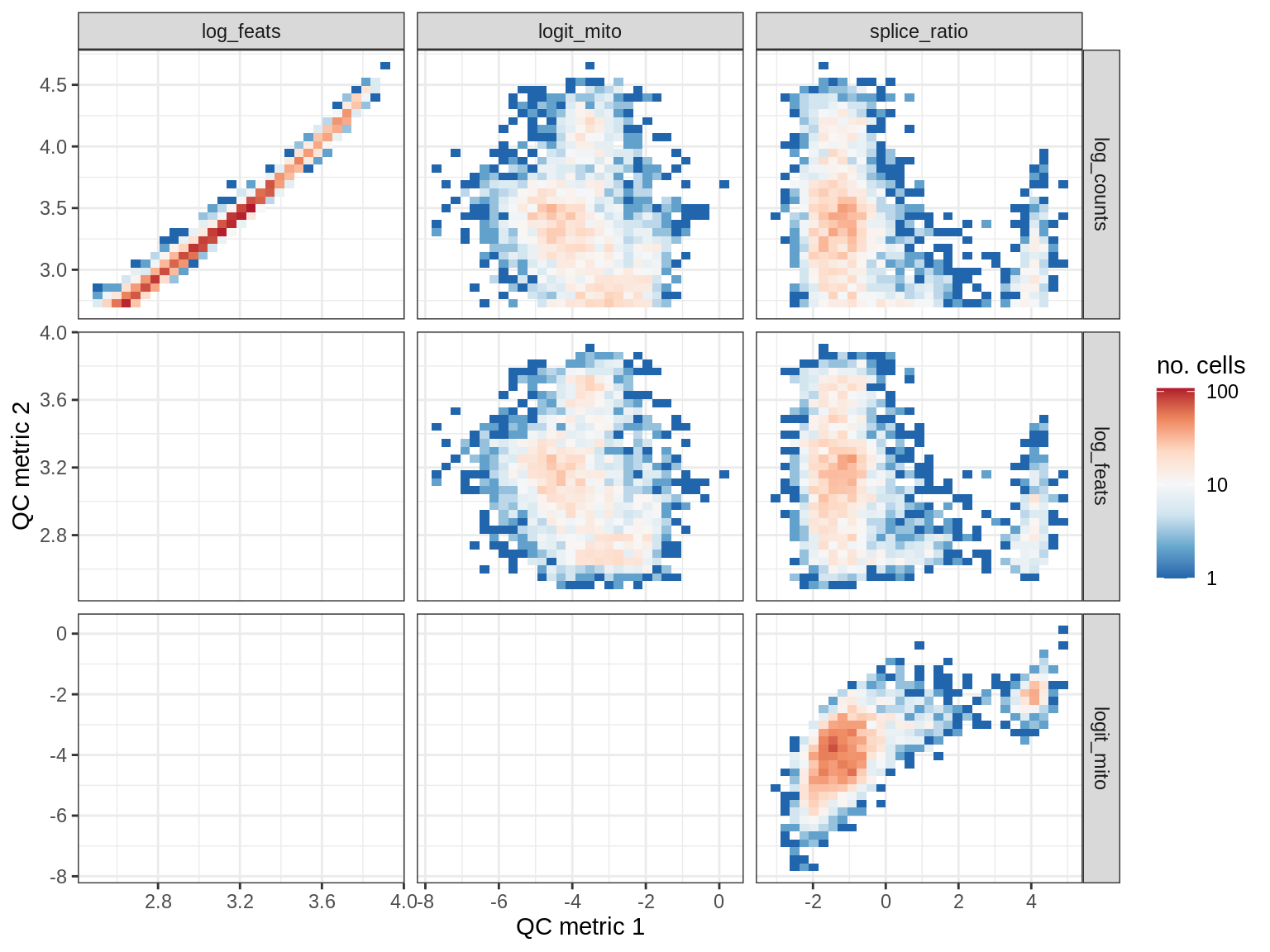
EU012
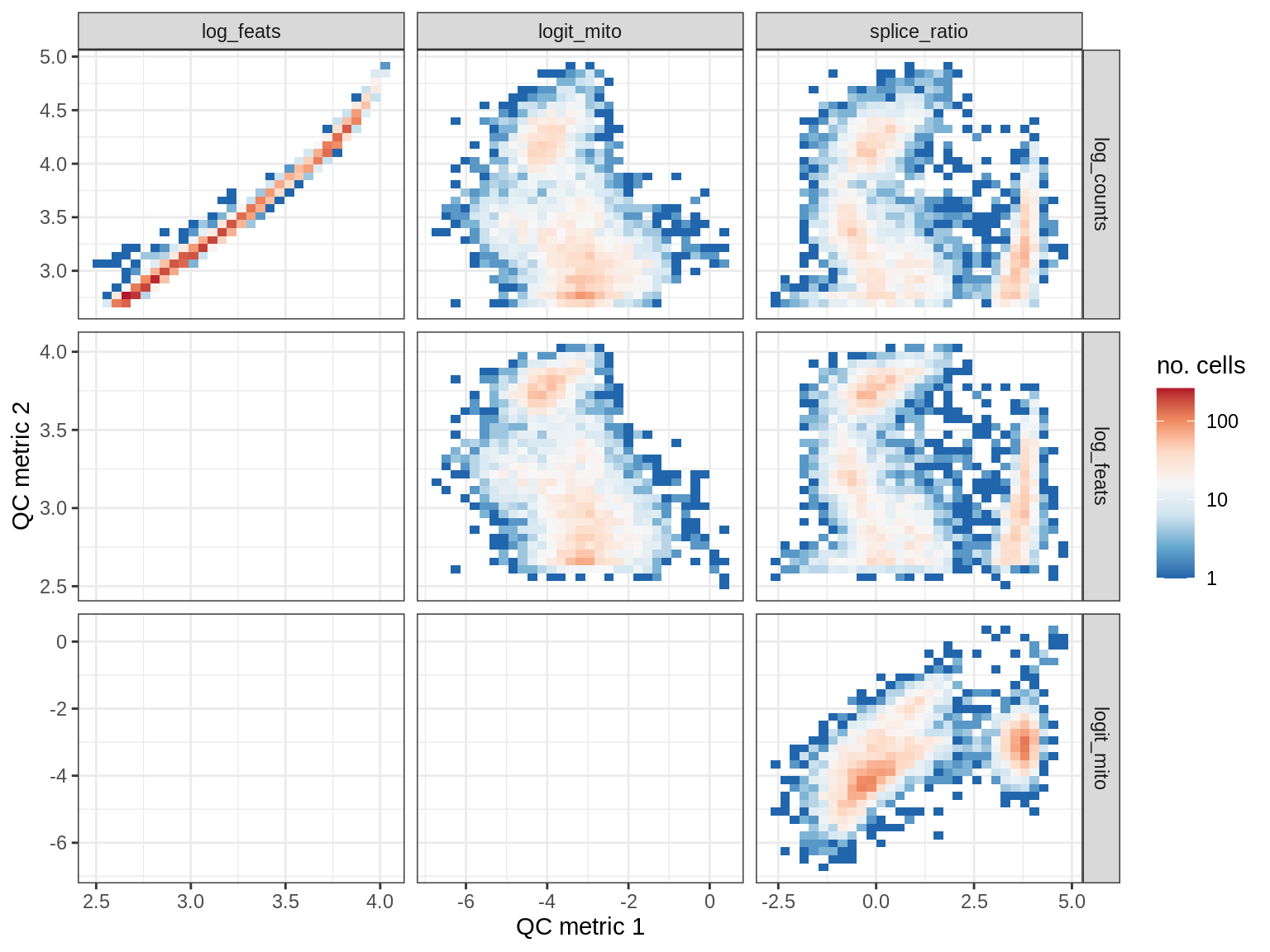
EU013
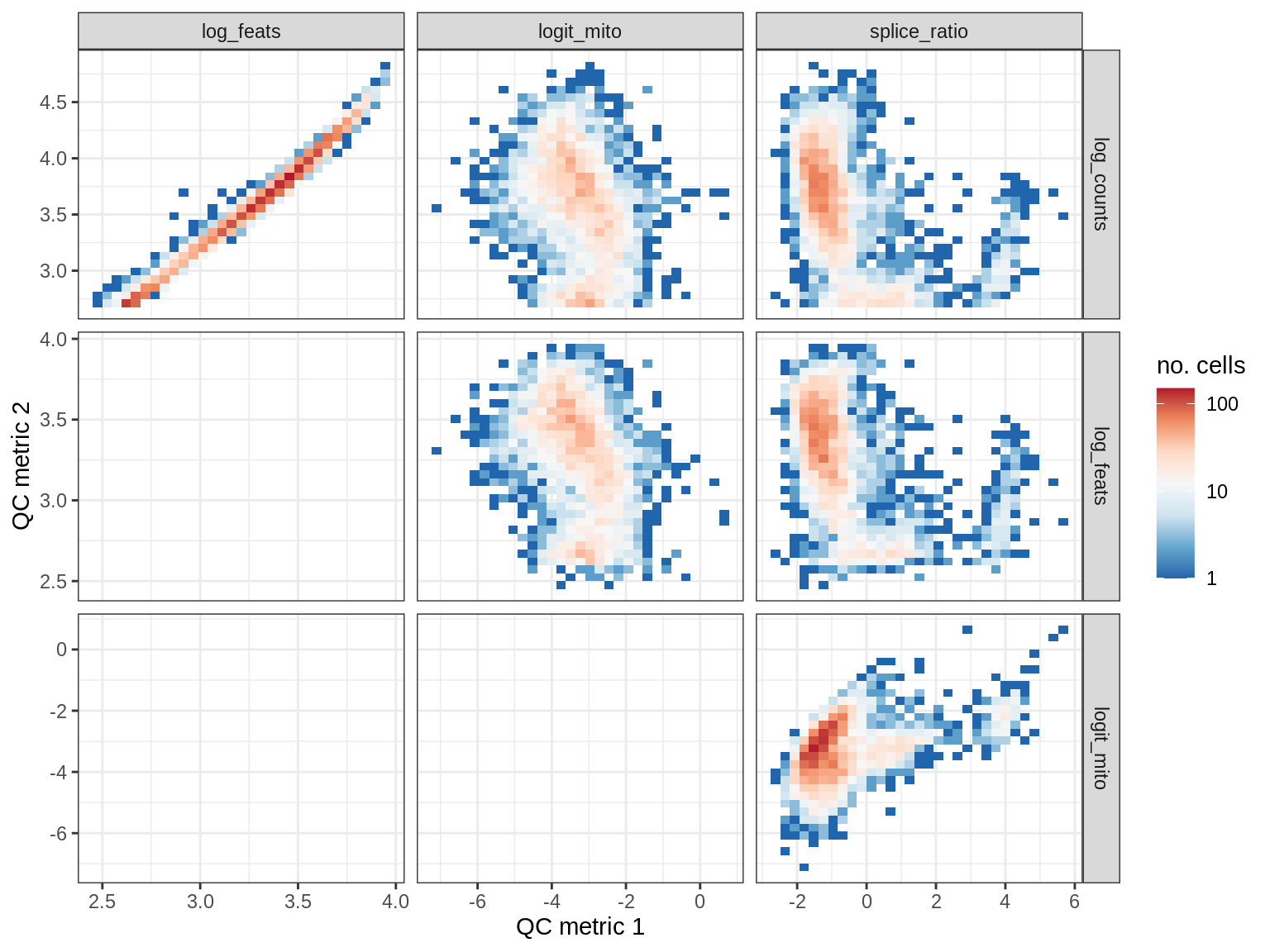
EU014
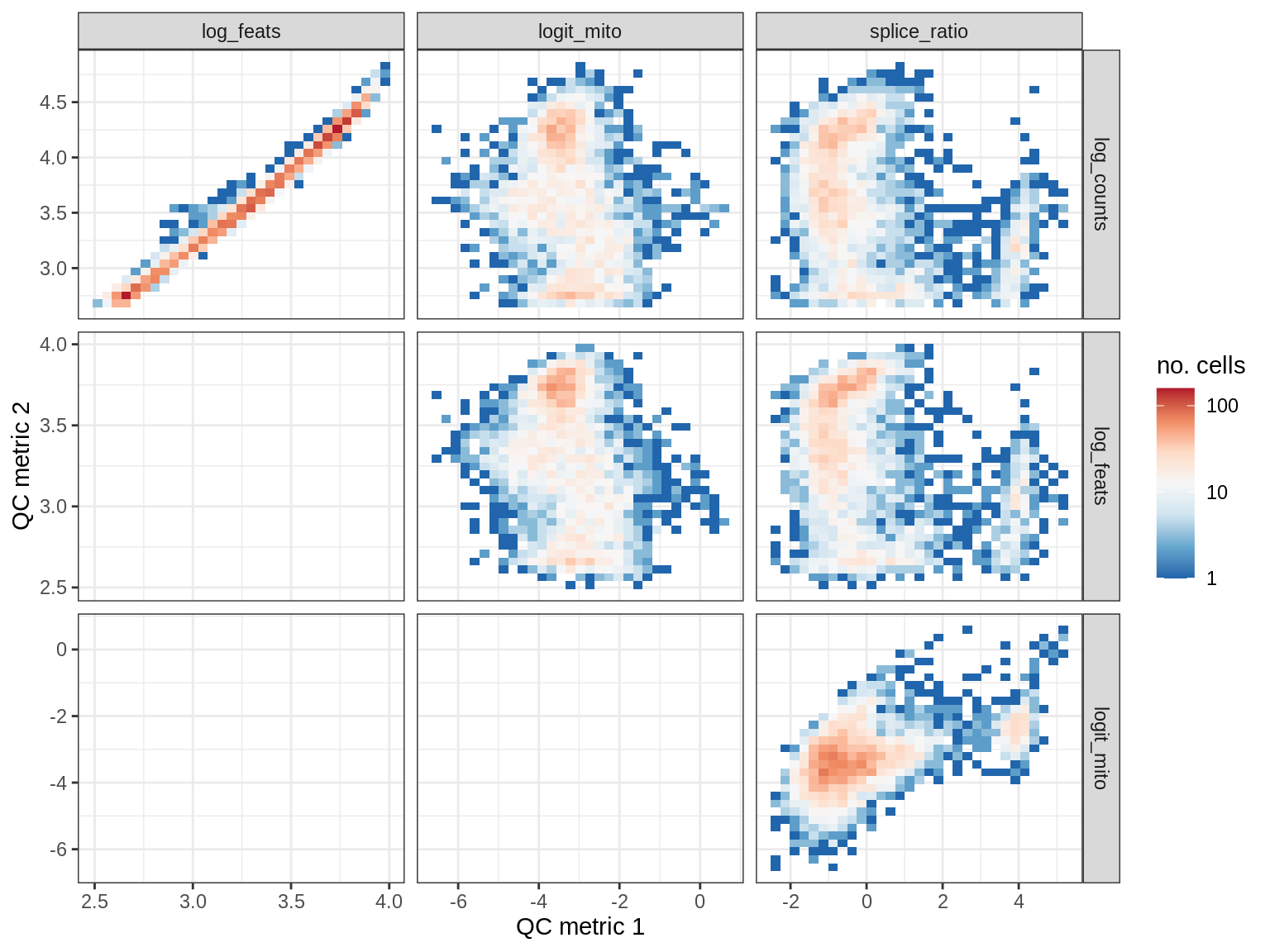
EU015
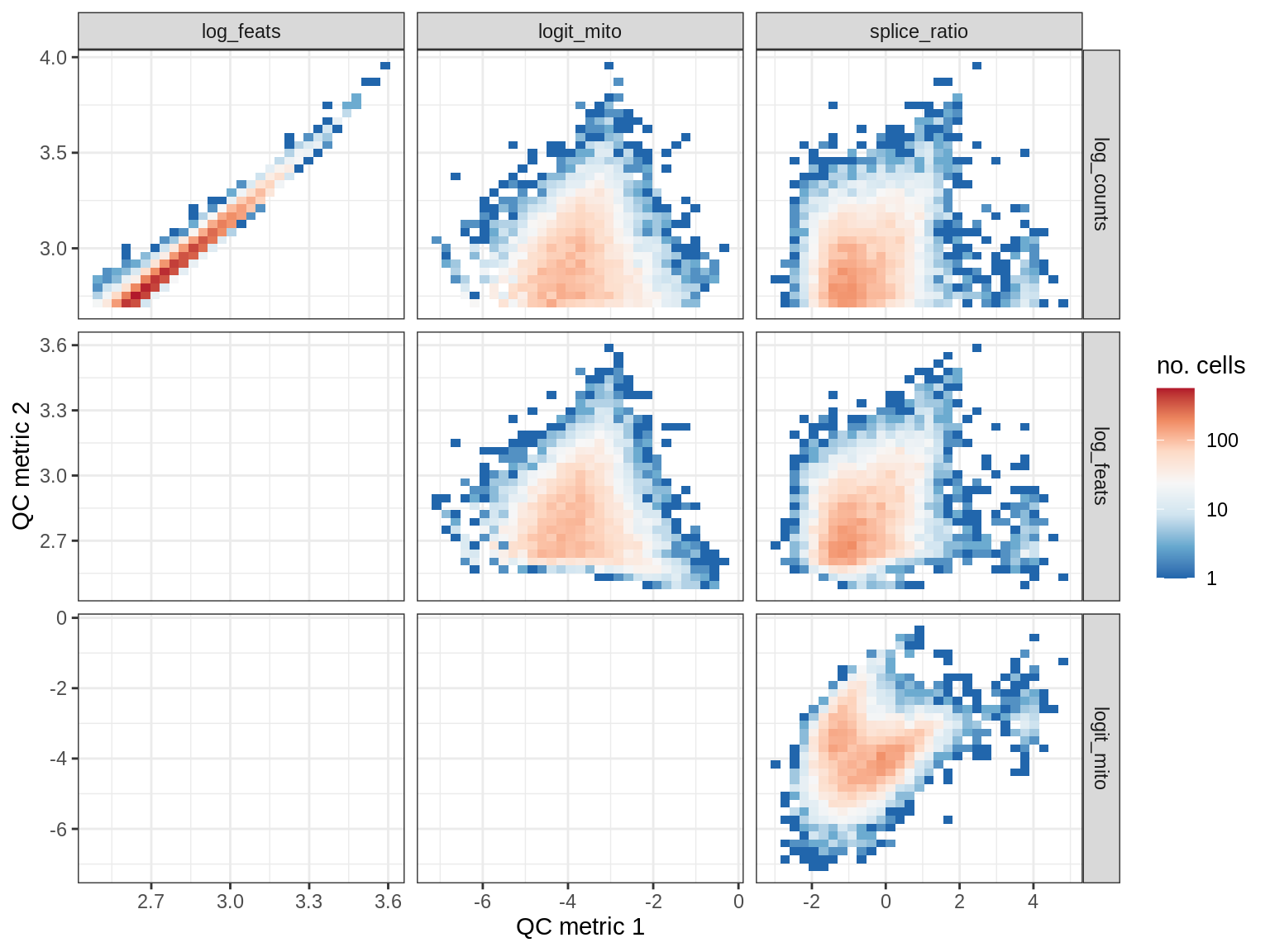
EU016
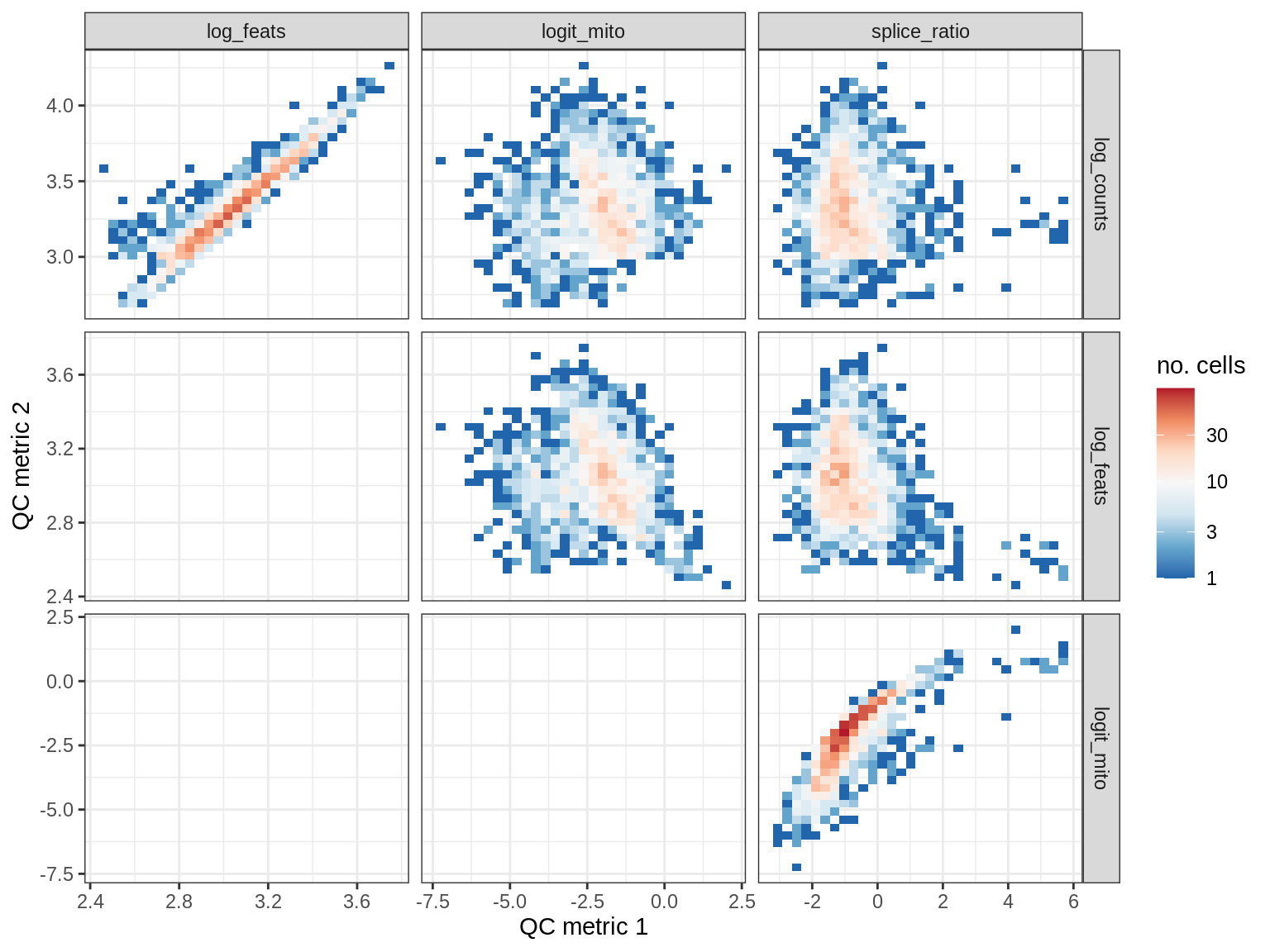
EU017
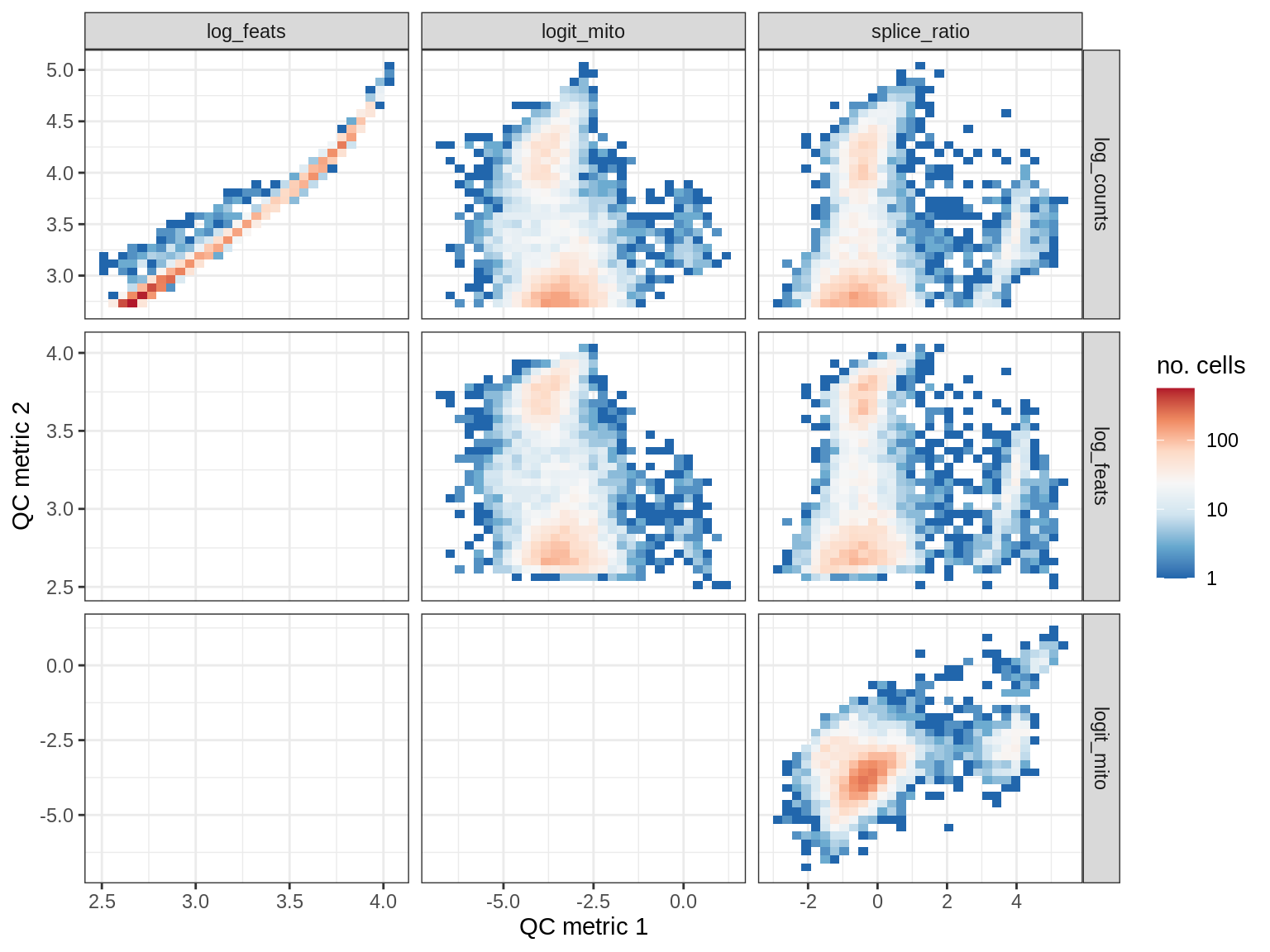
EU018
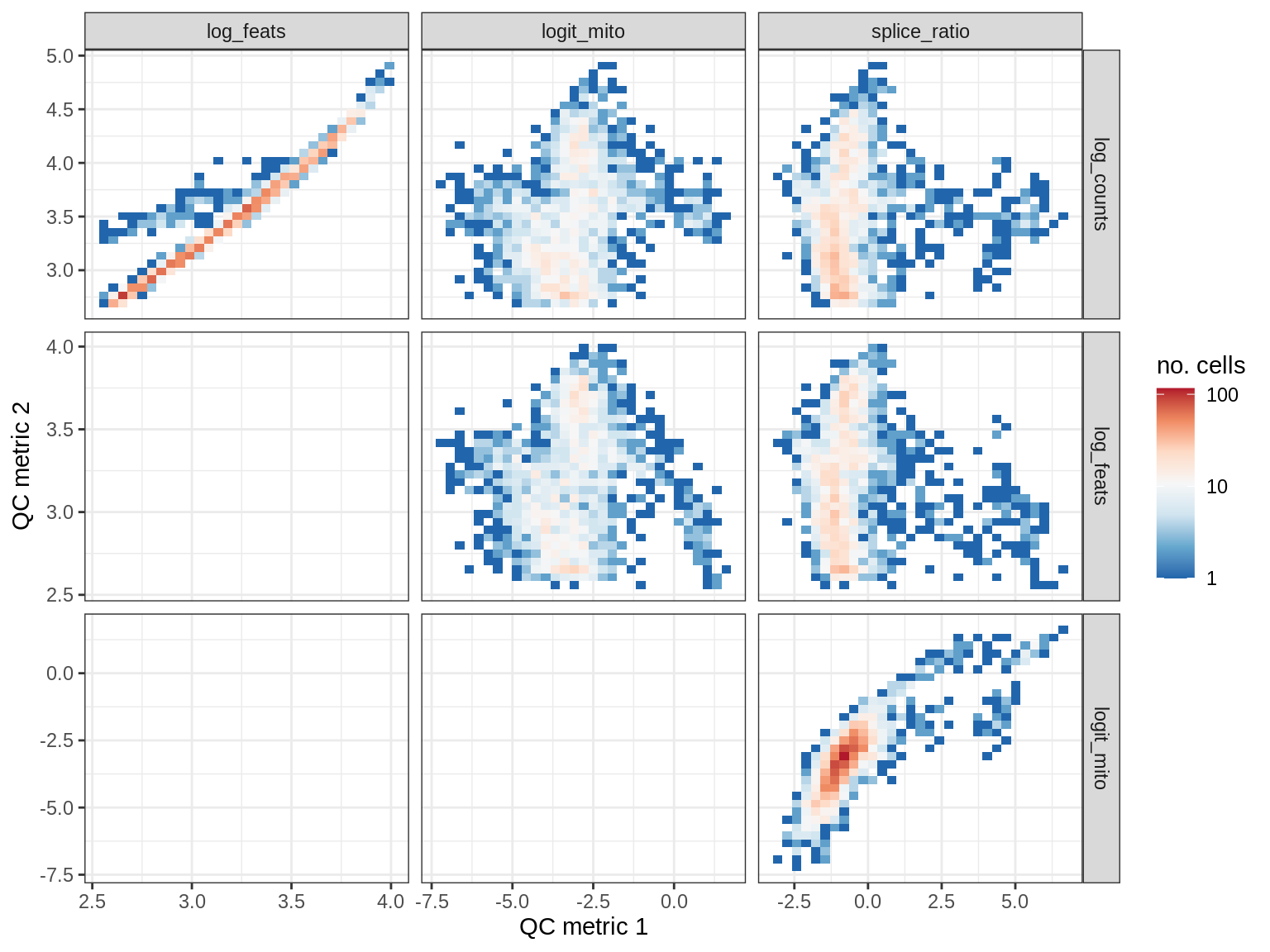
EU019
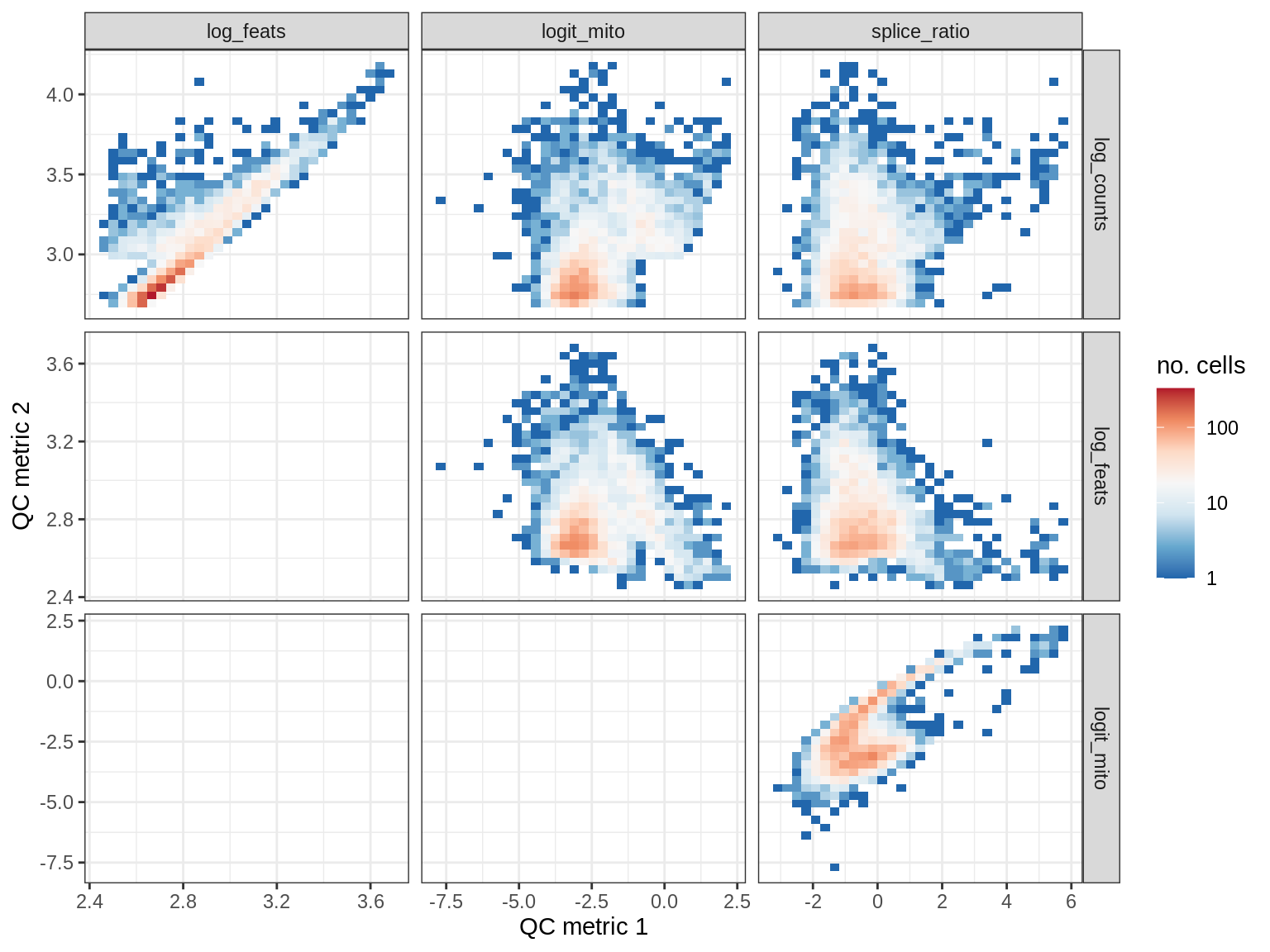
EU020
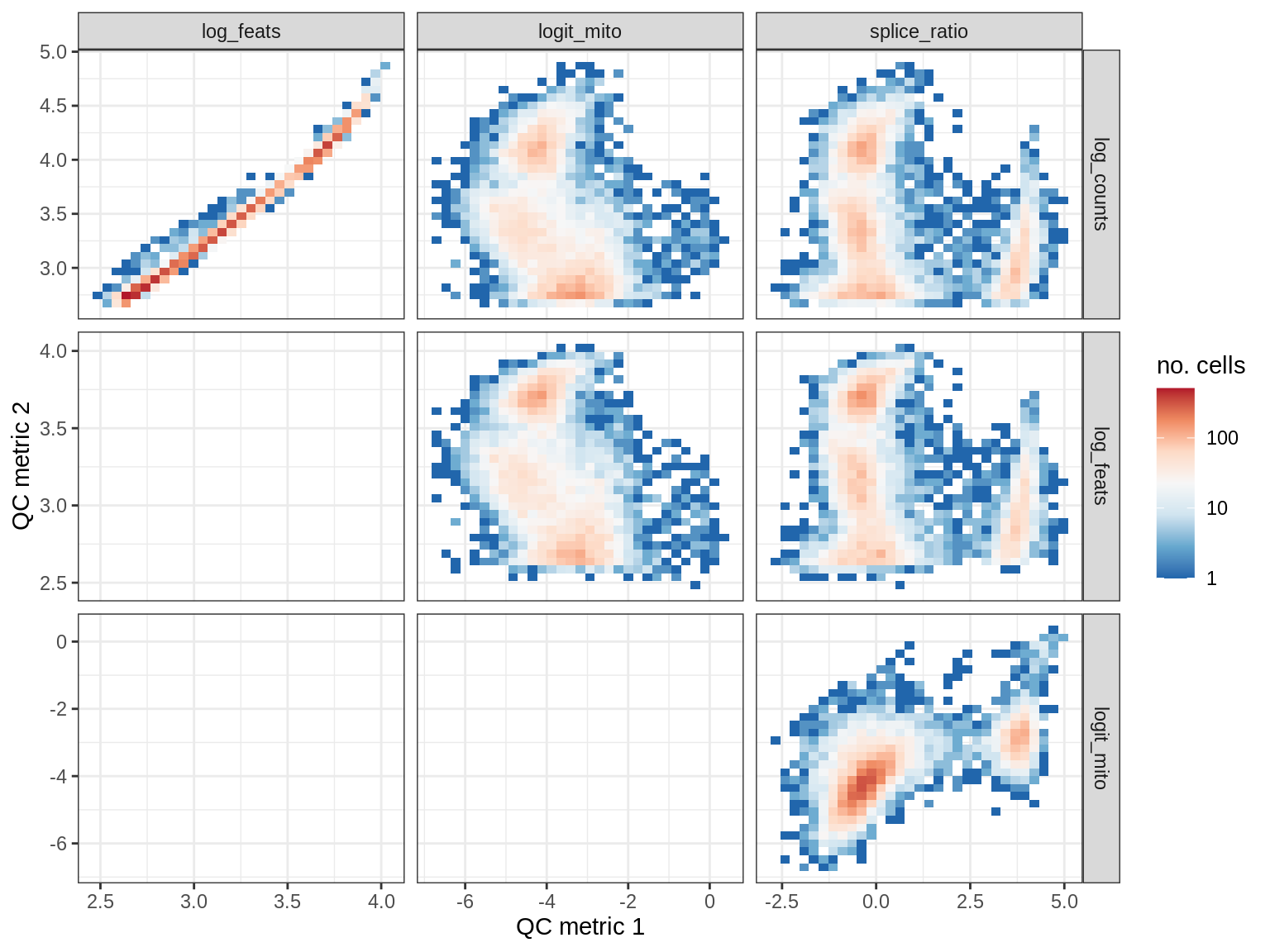
EU021
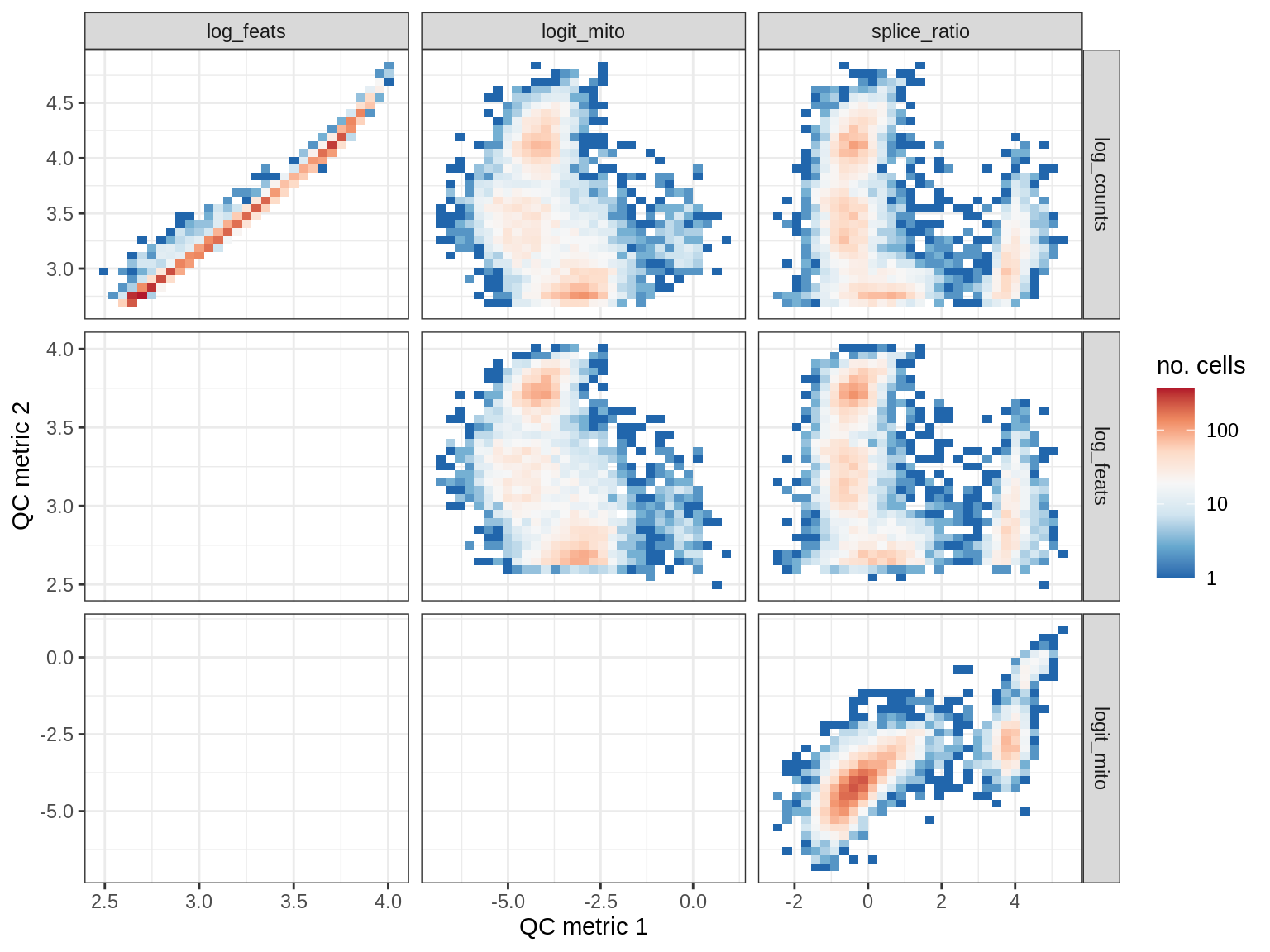
EU022
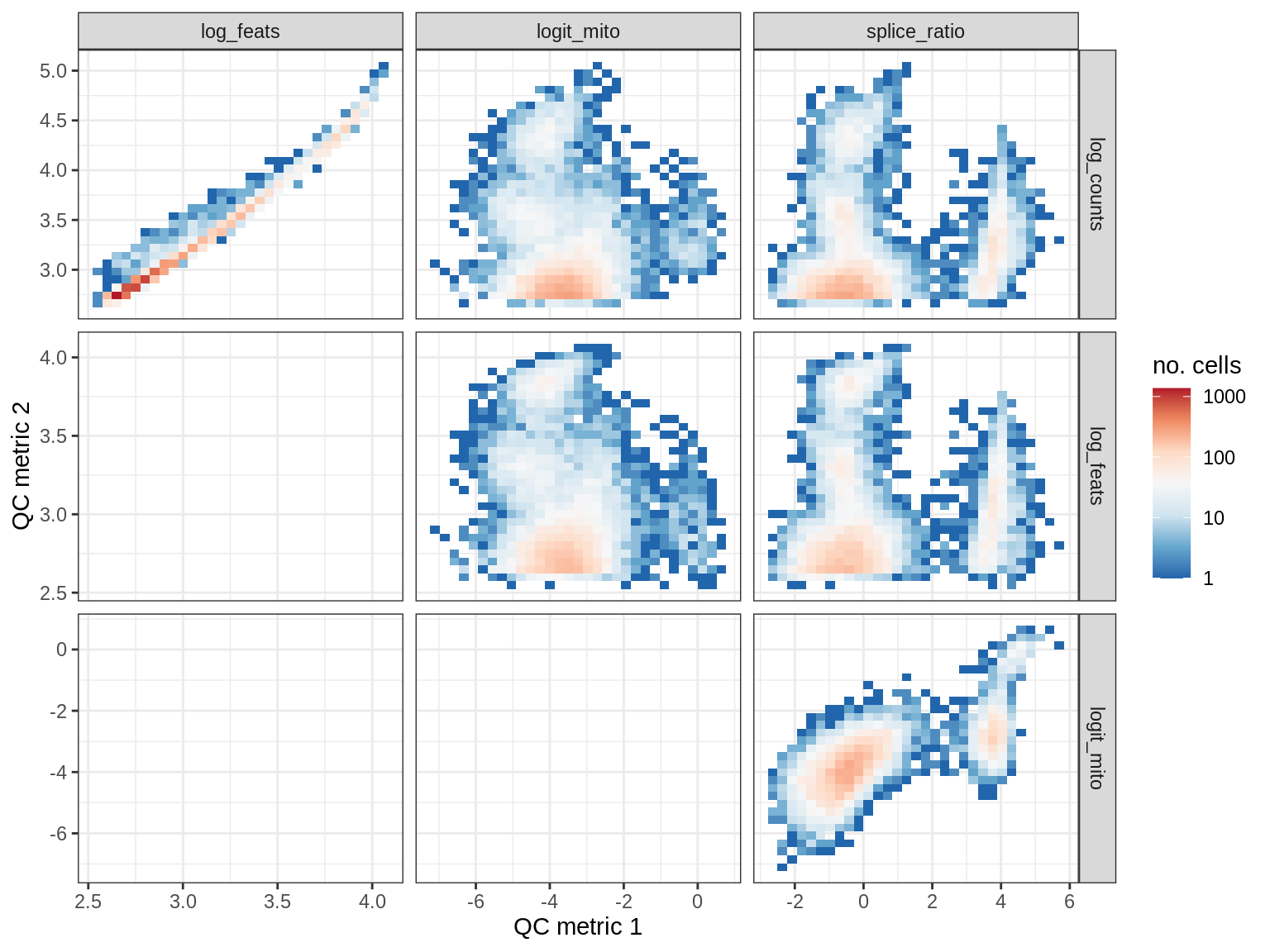
EU023
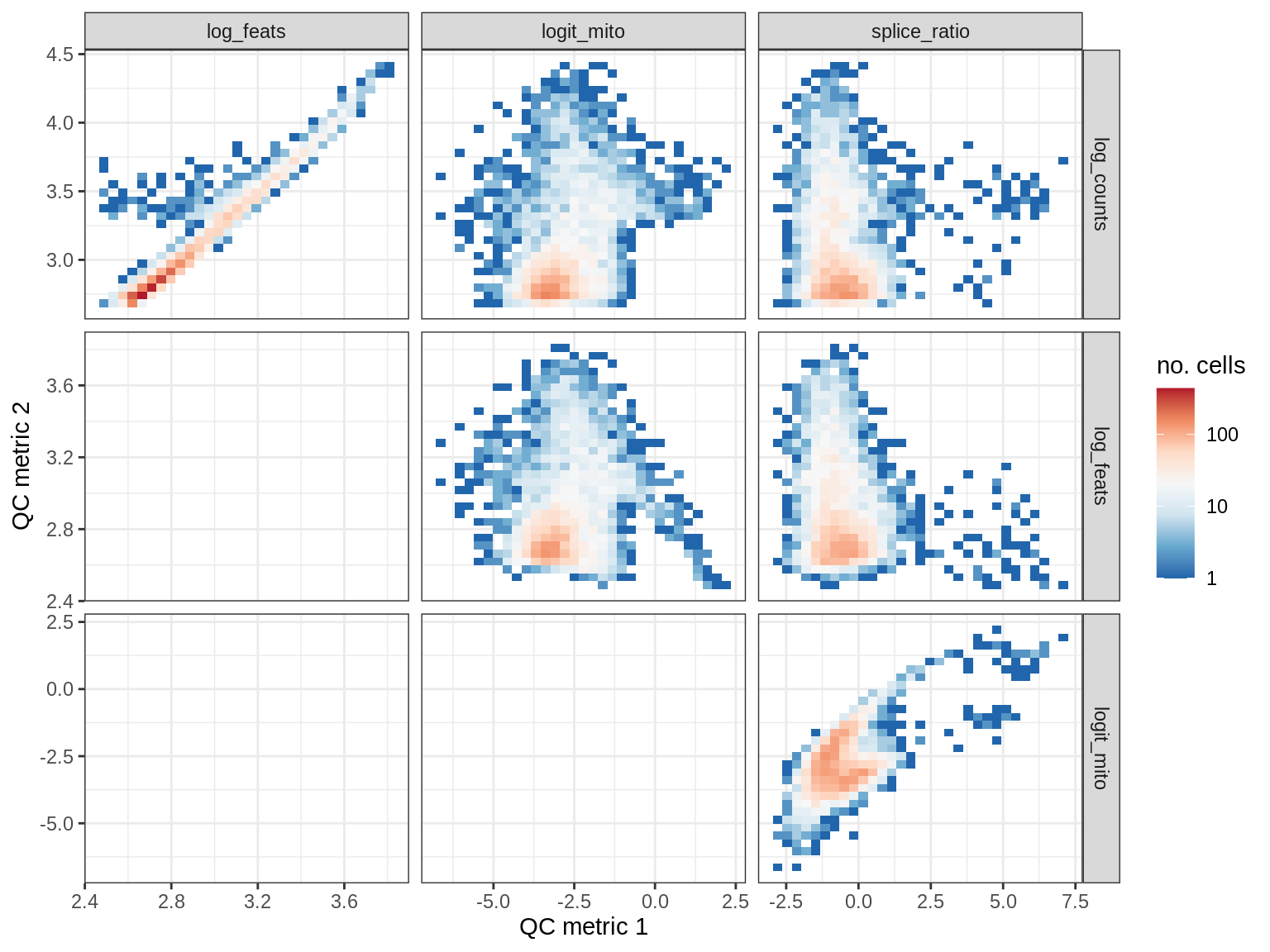
EU024
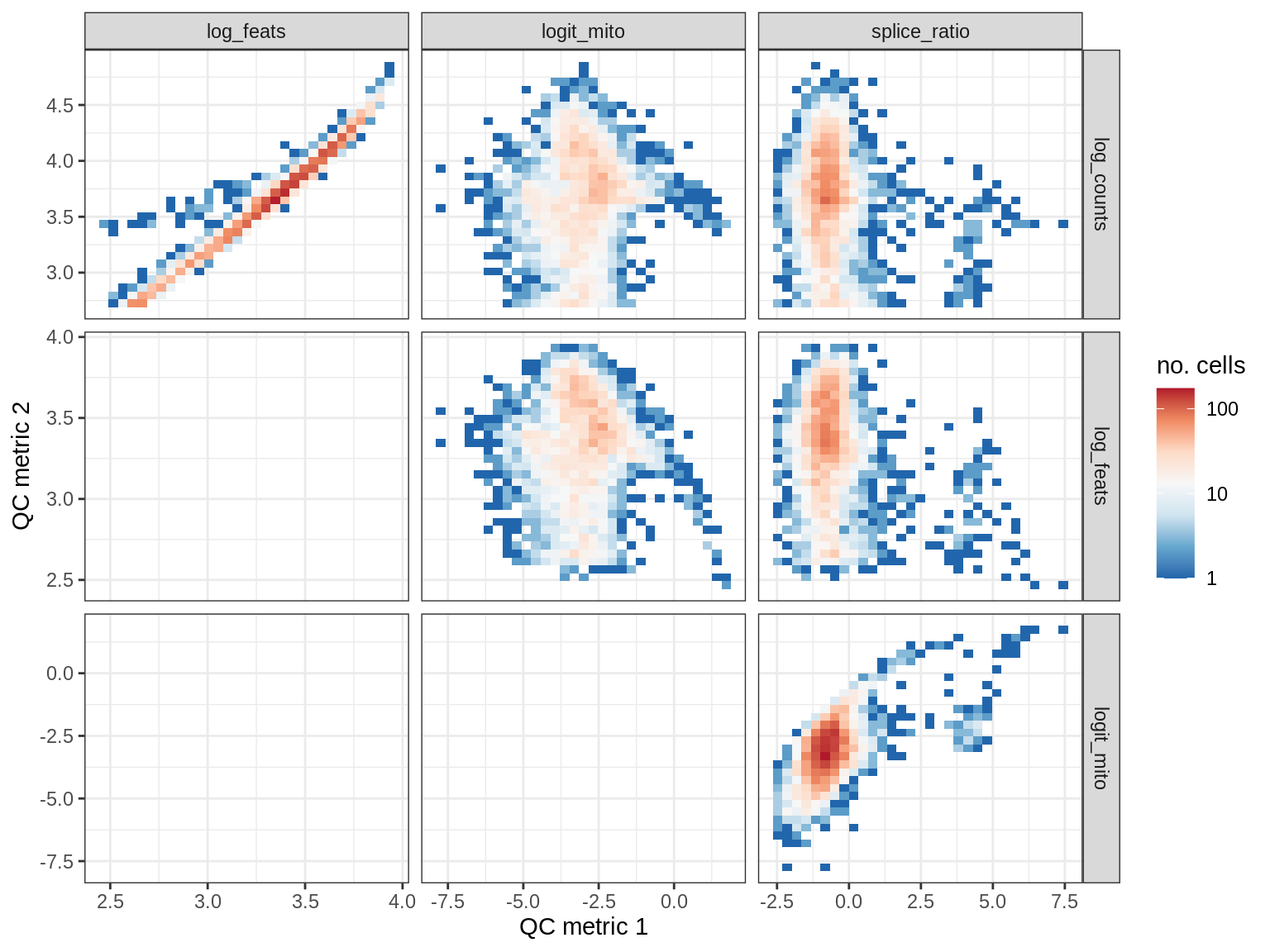
EU025
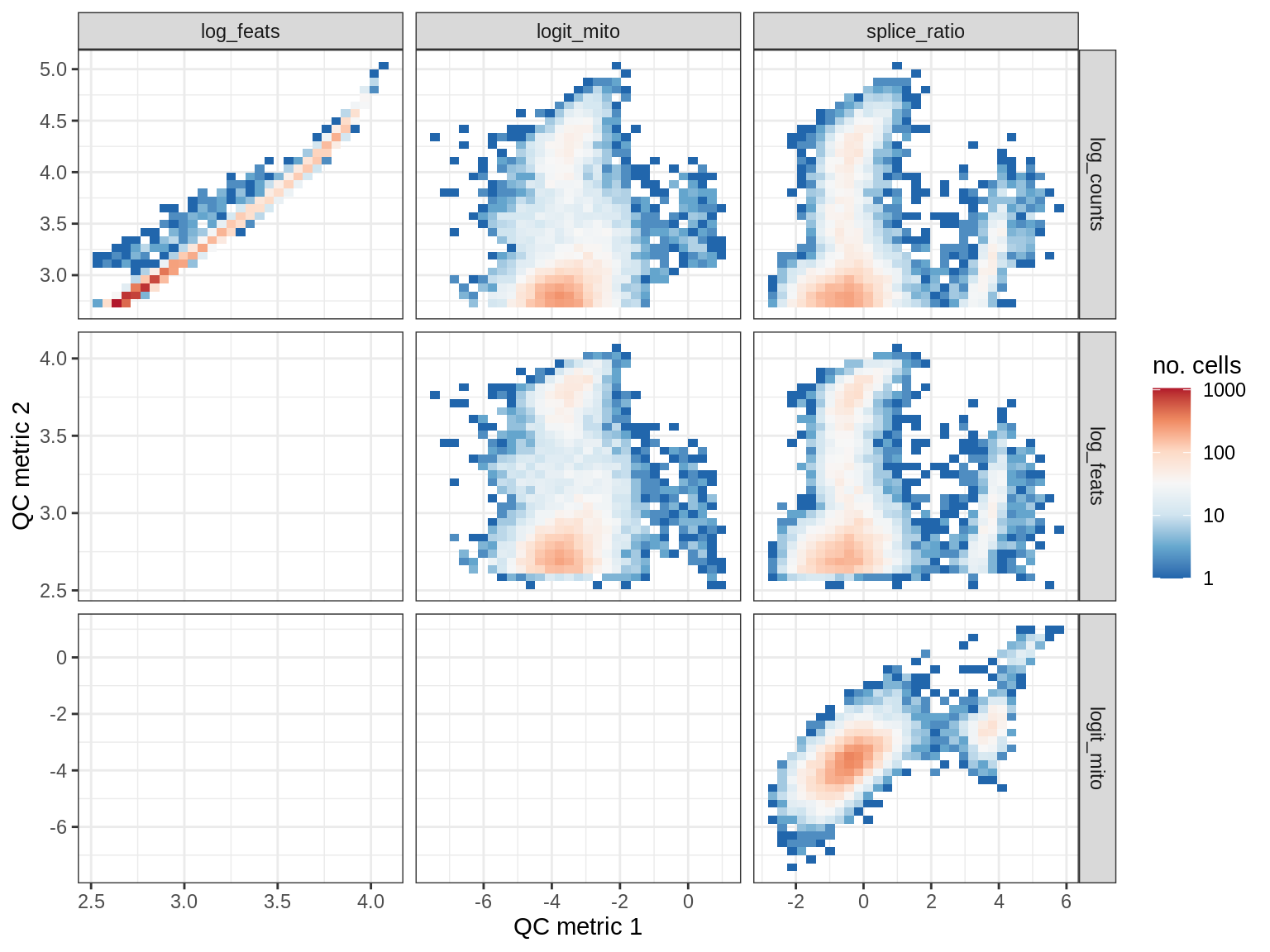
EU026
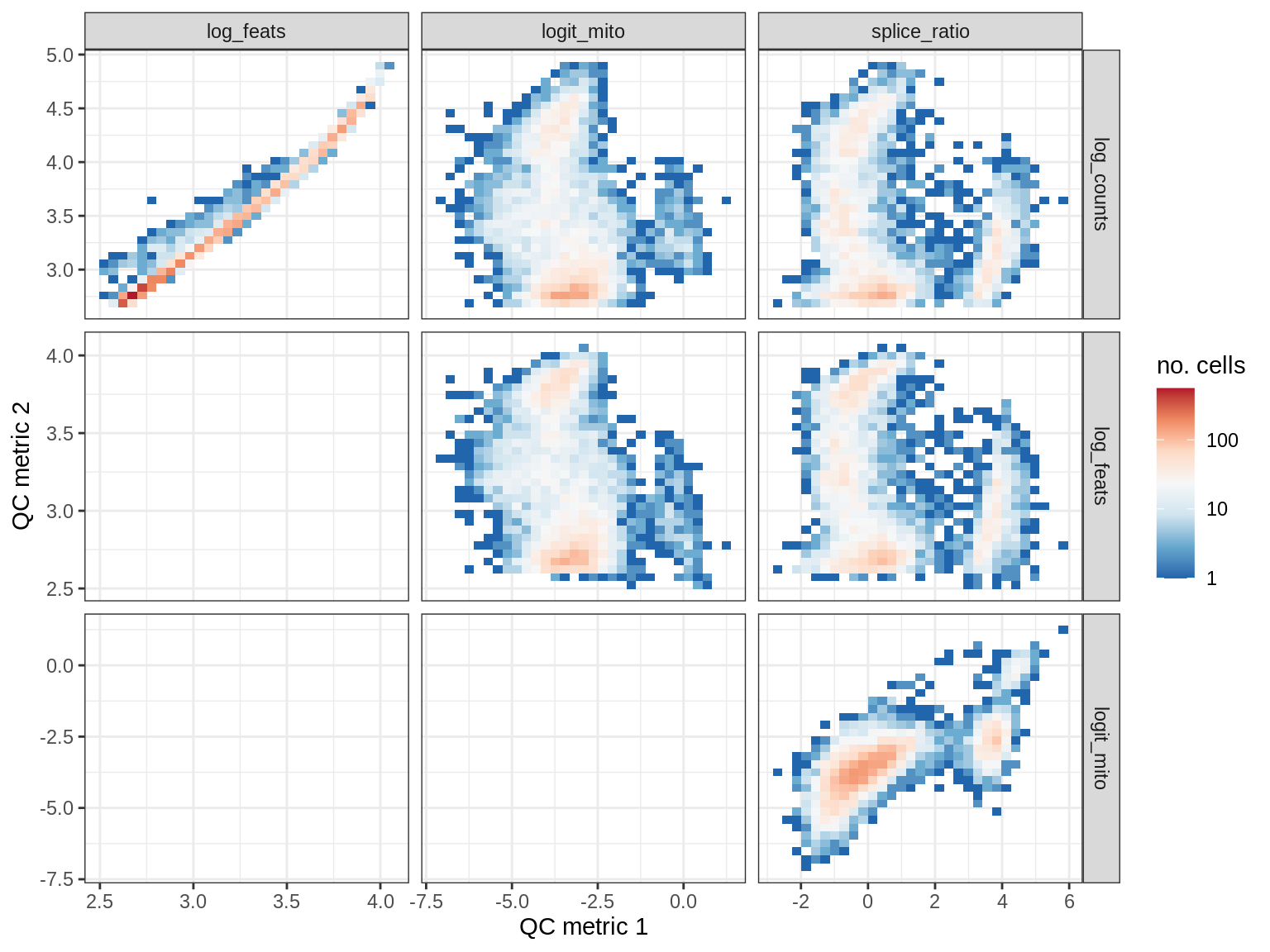
EU027
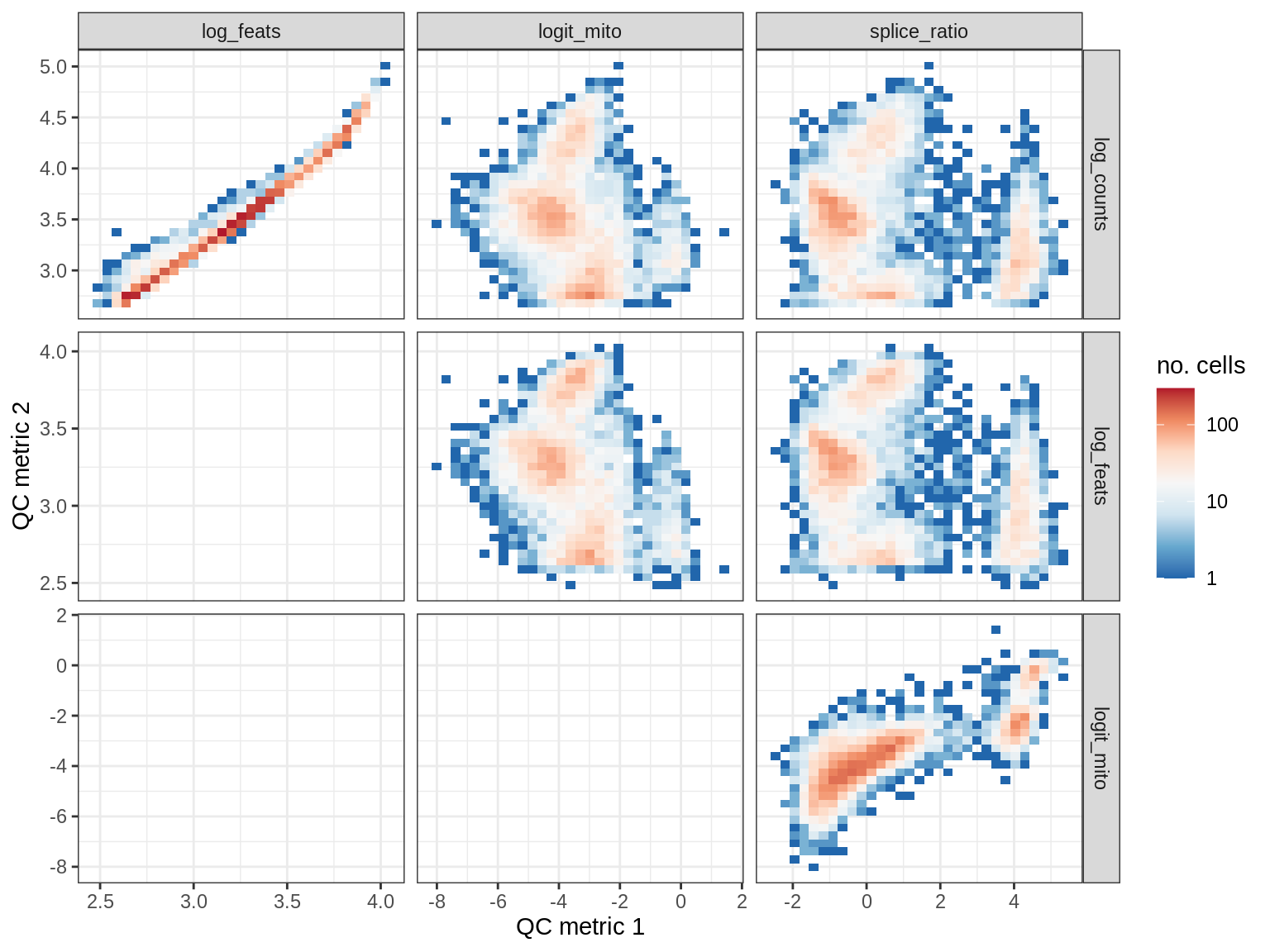
EU028
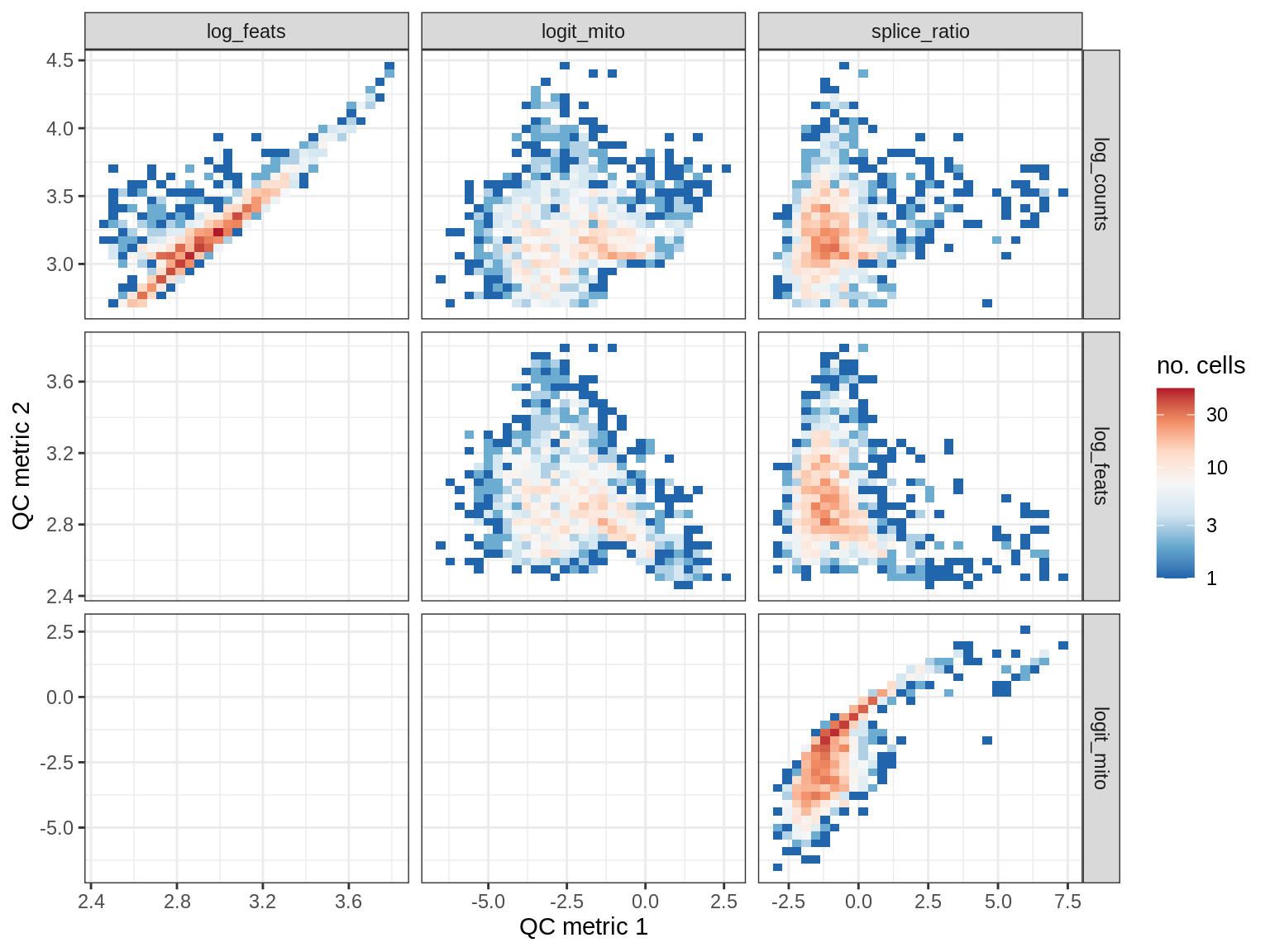
EU029
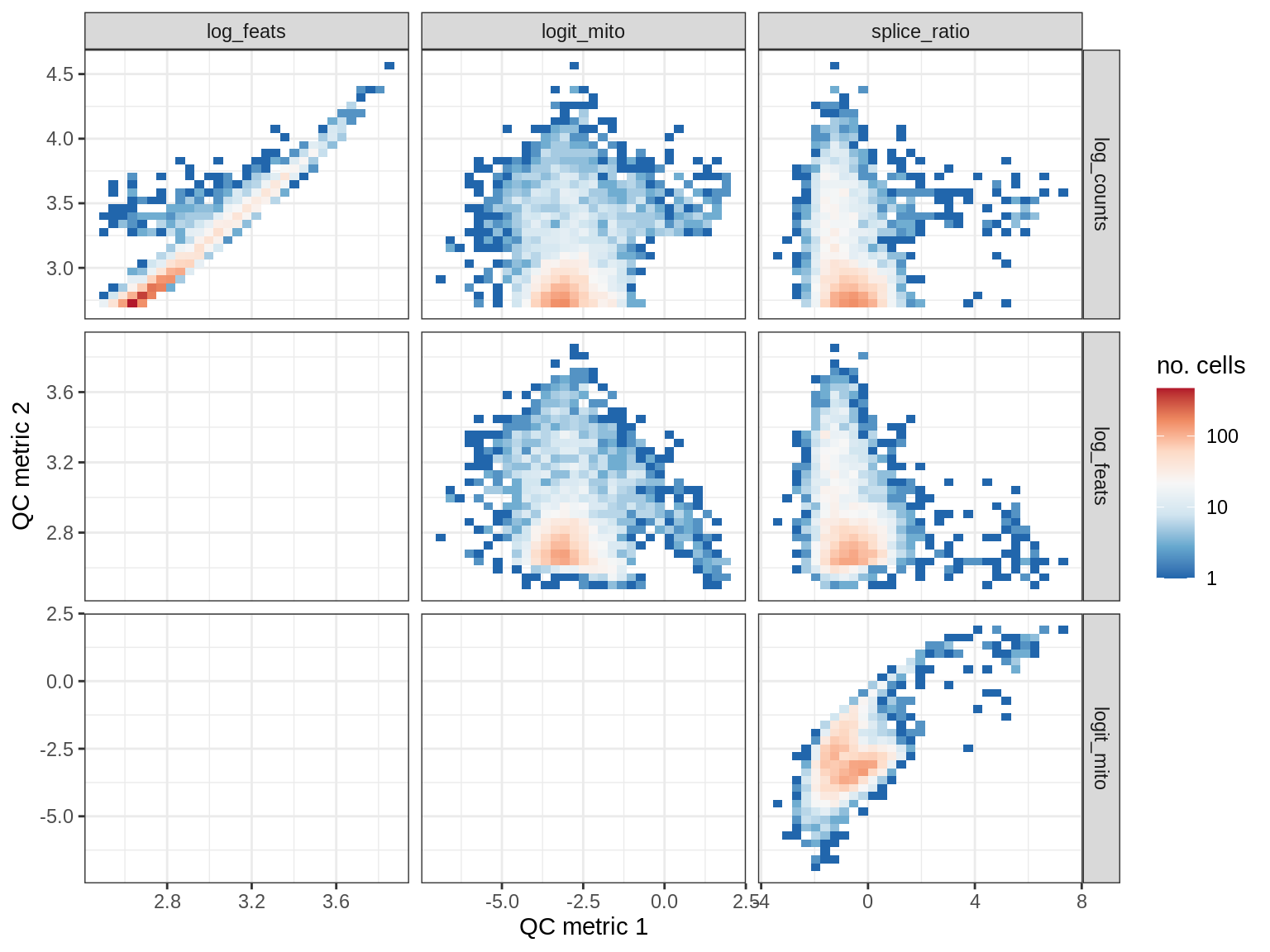
EU030
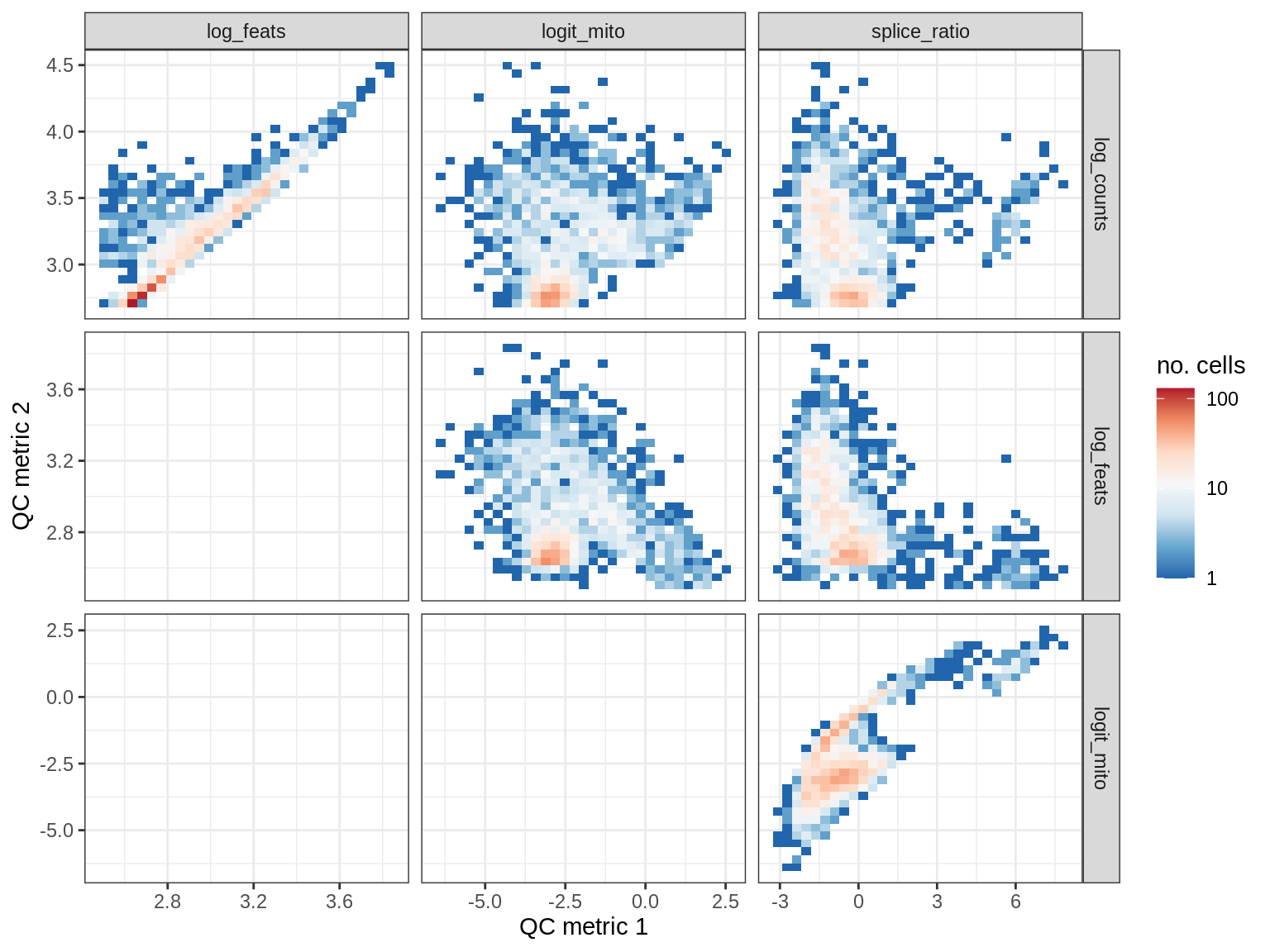
EU031
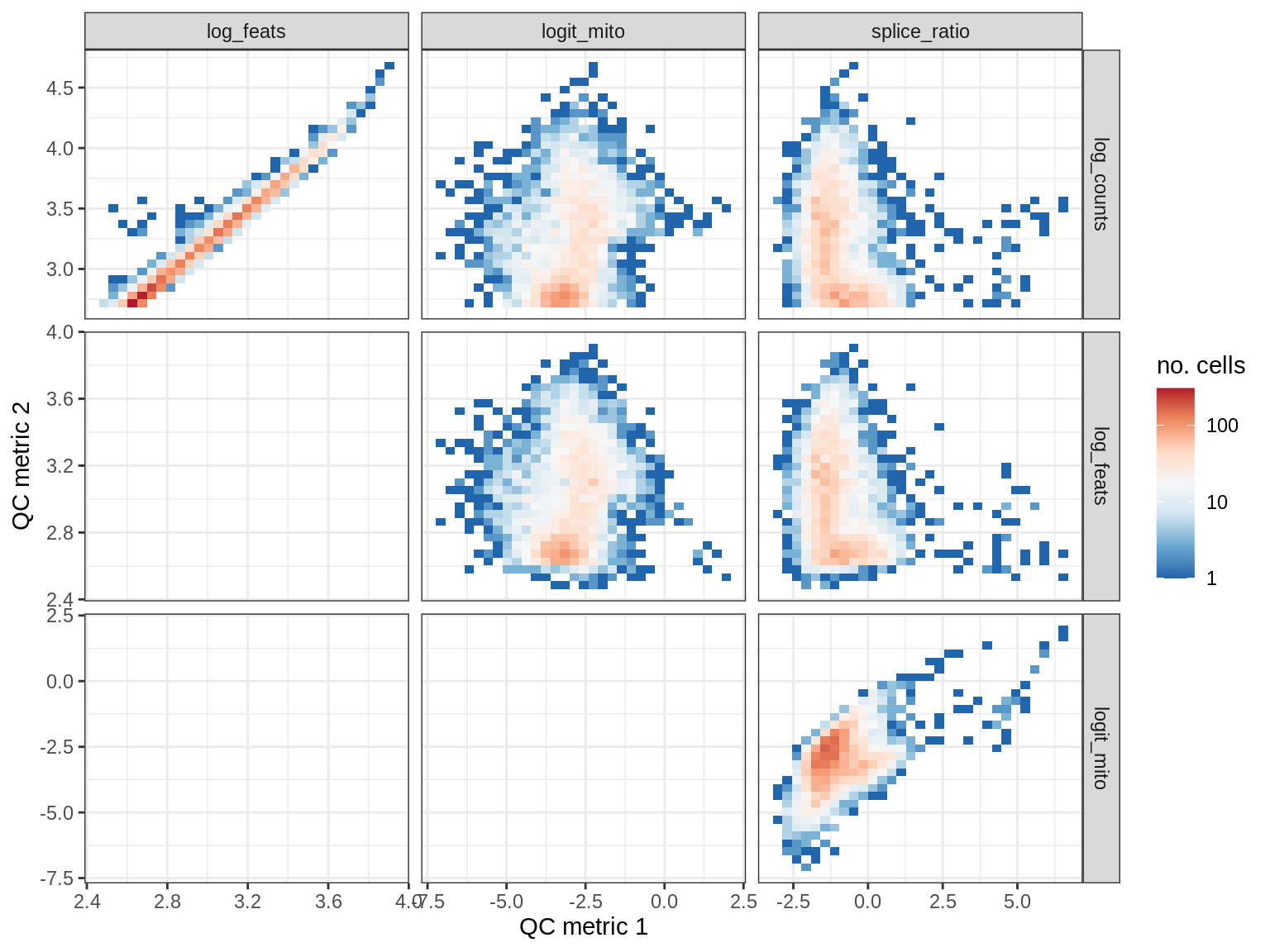
EU032
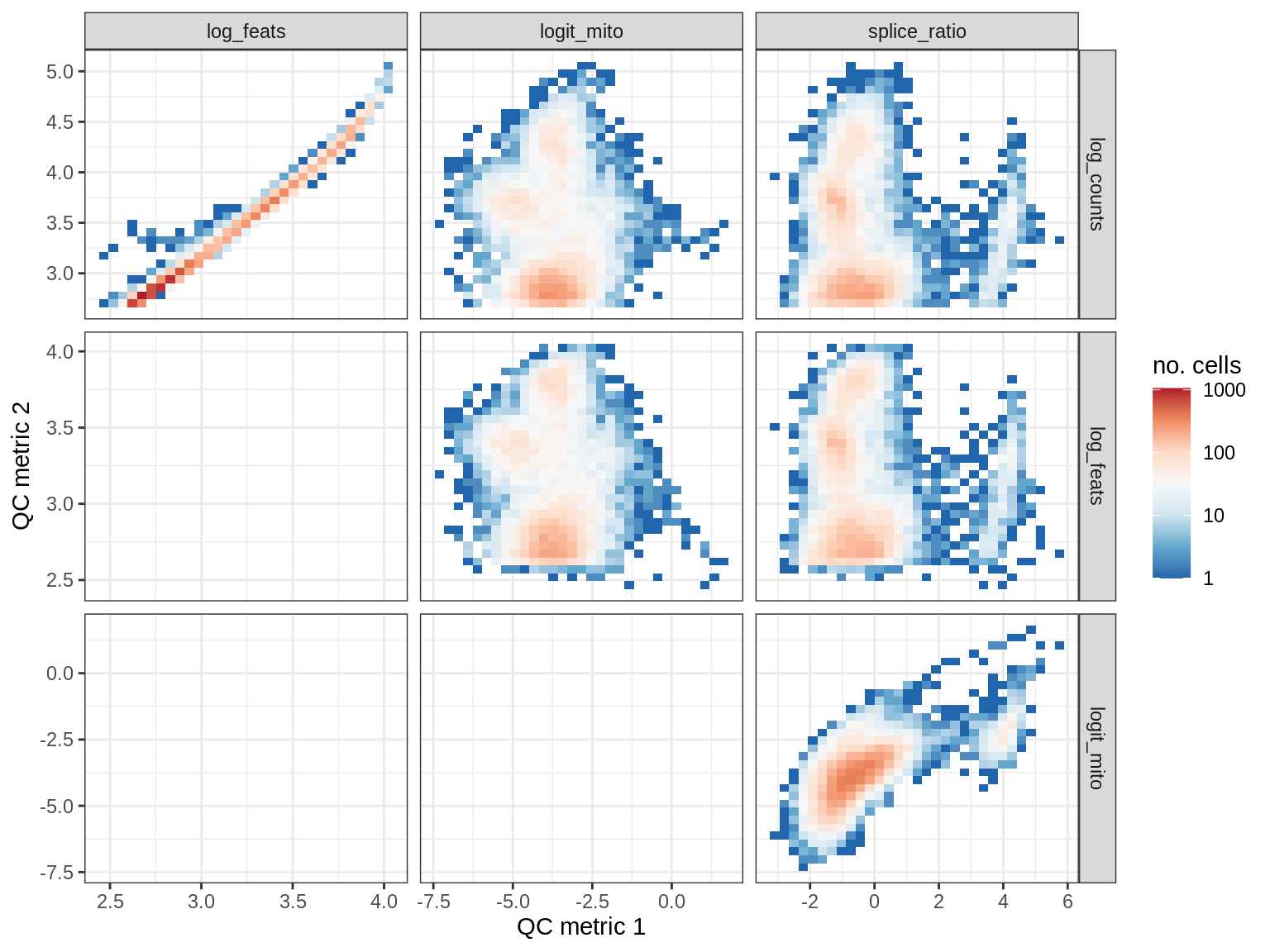
EU033
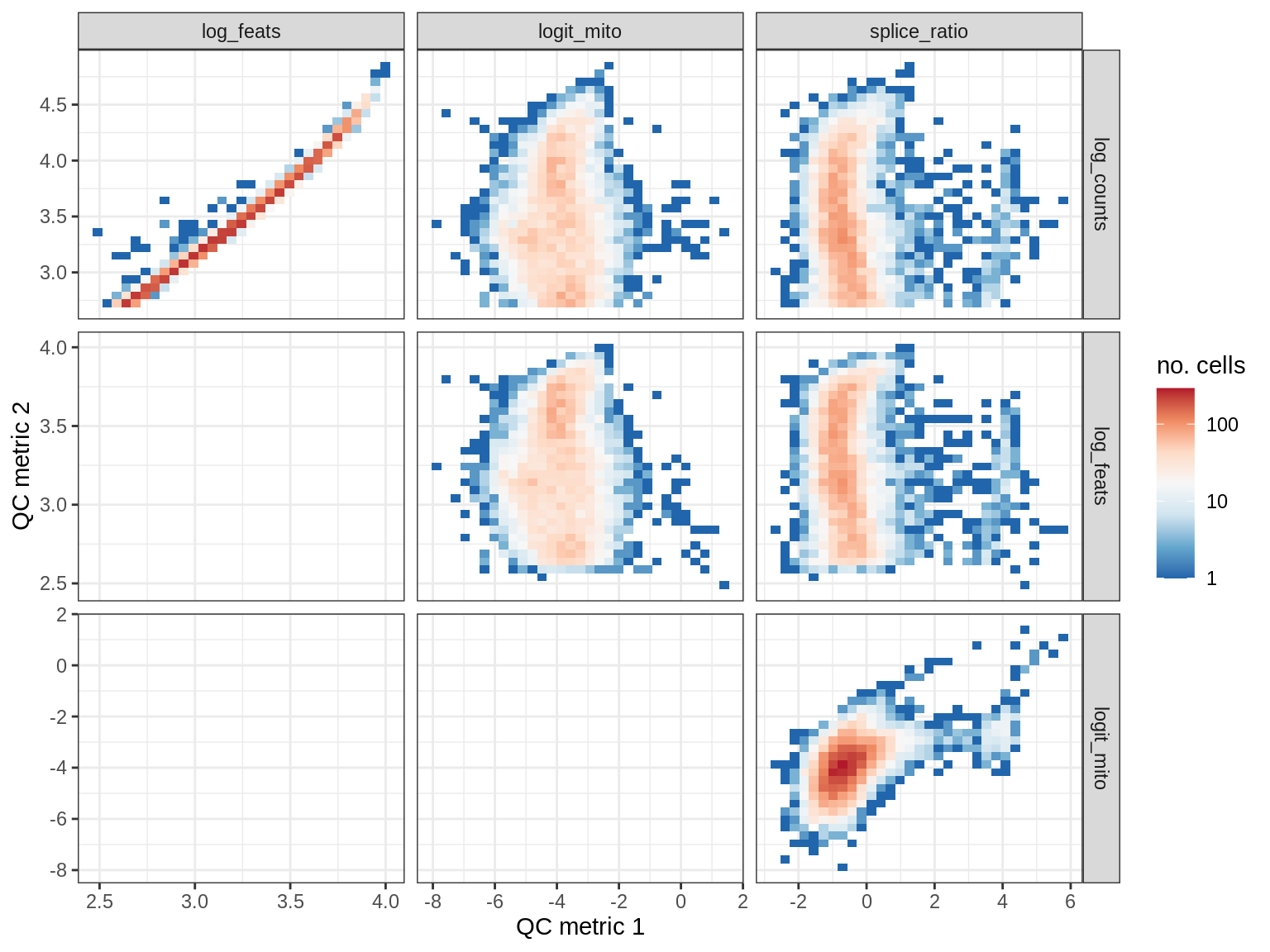
EU034
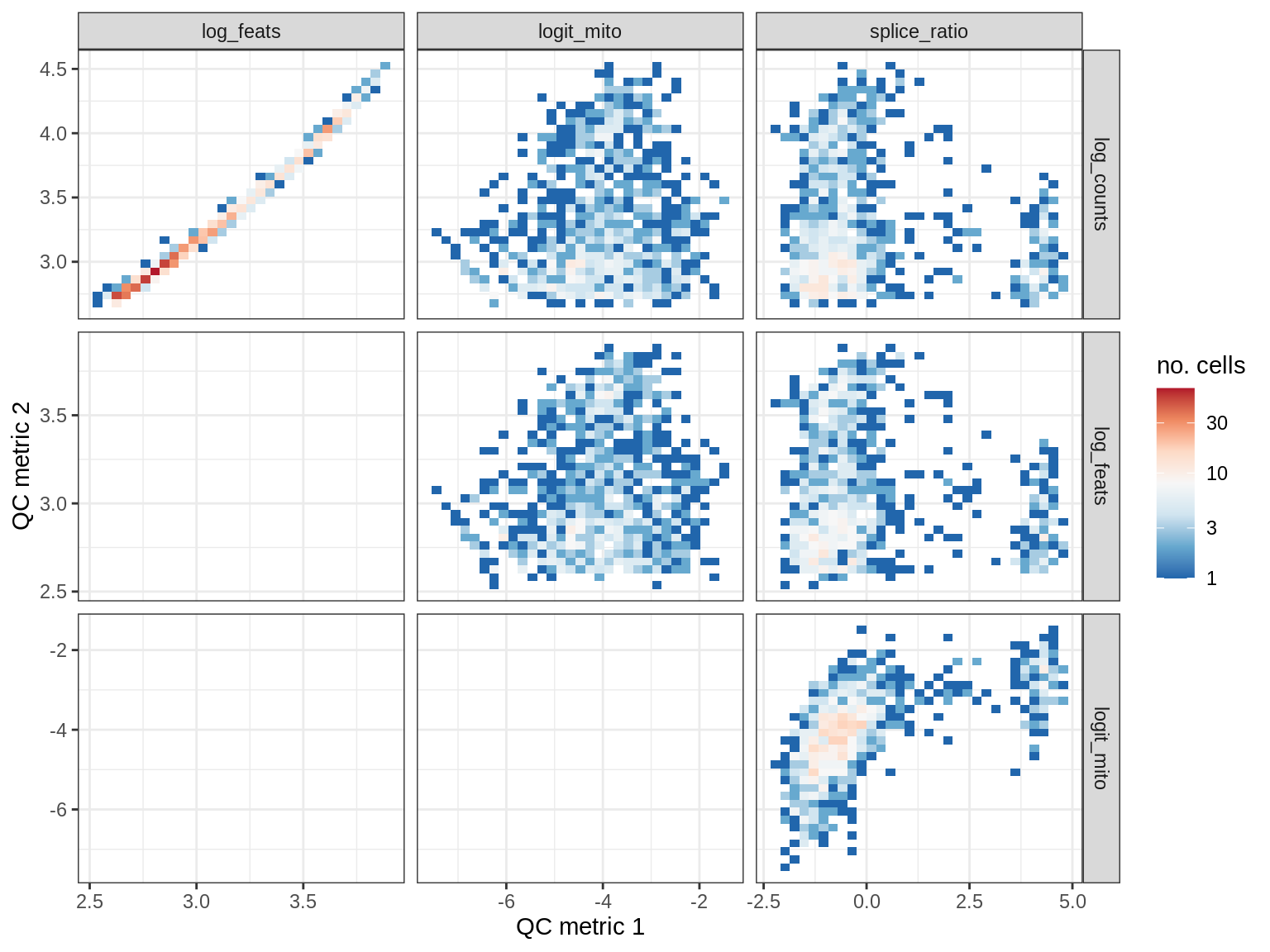
EU035
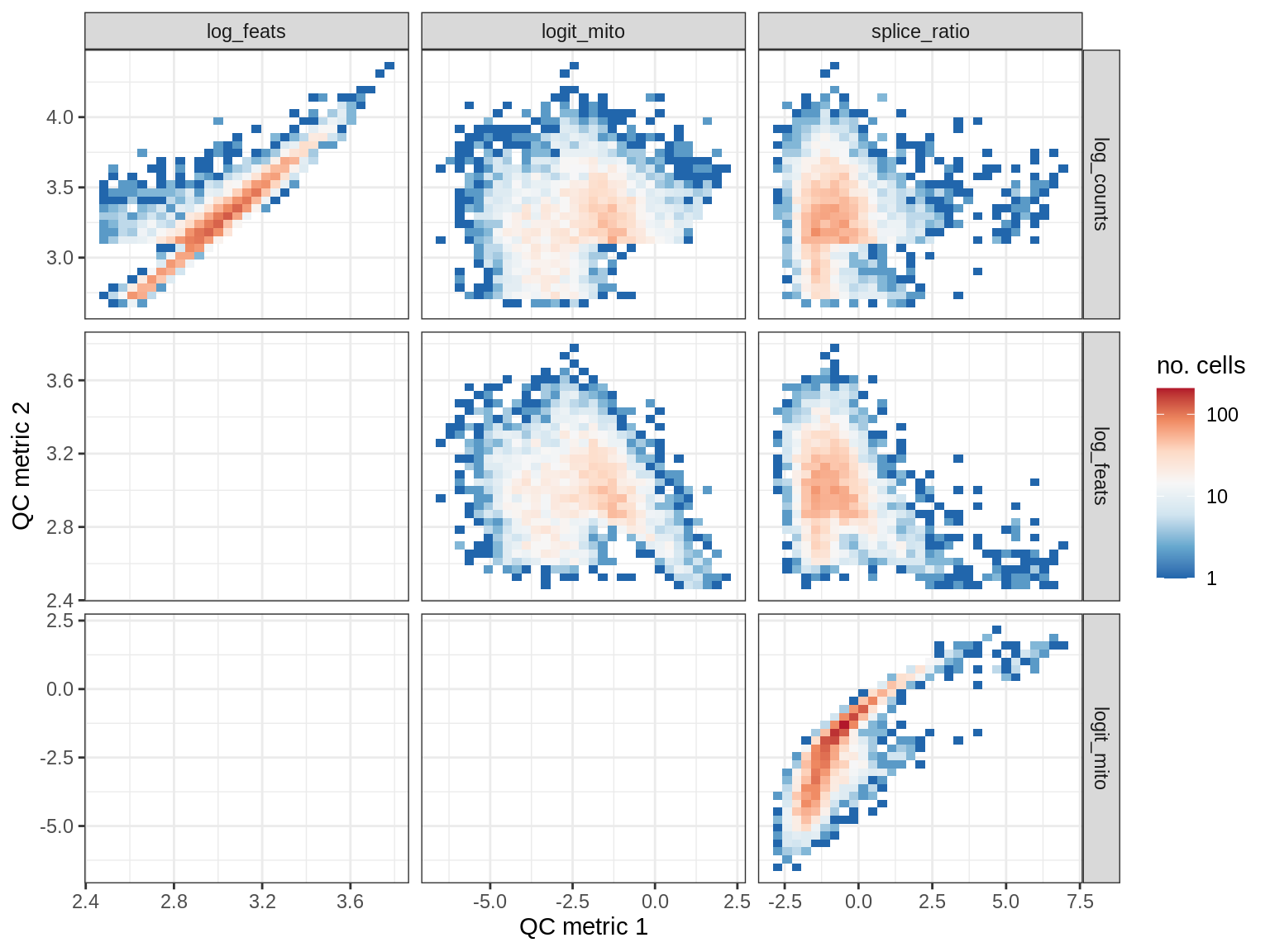
EU036
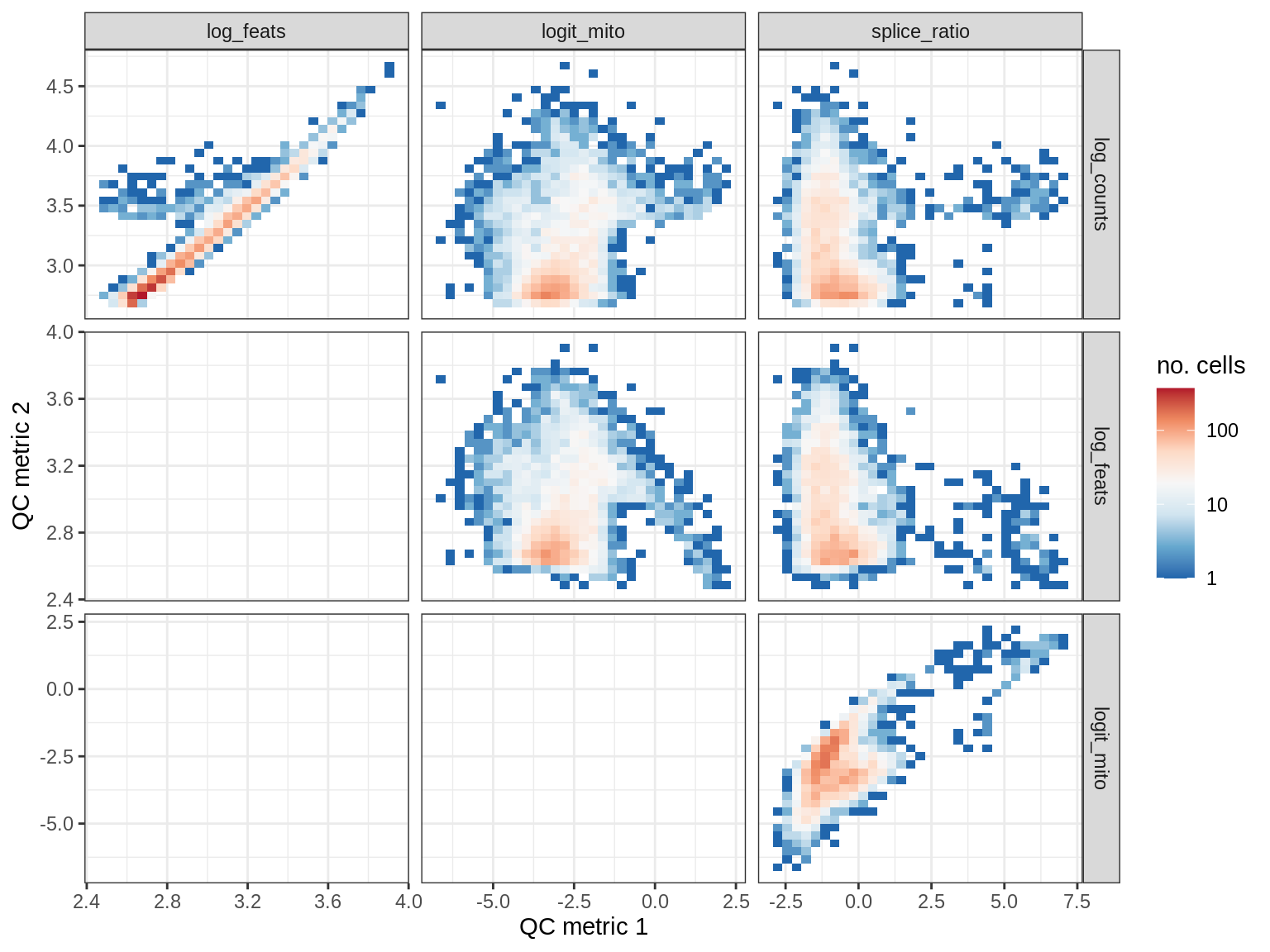
EU037
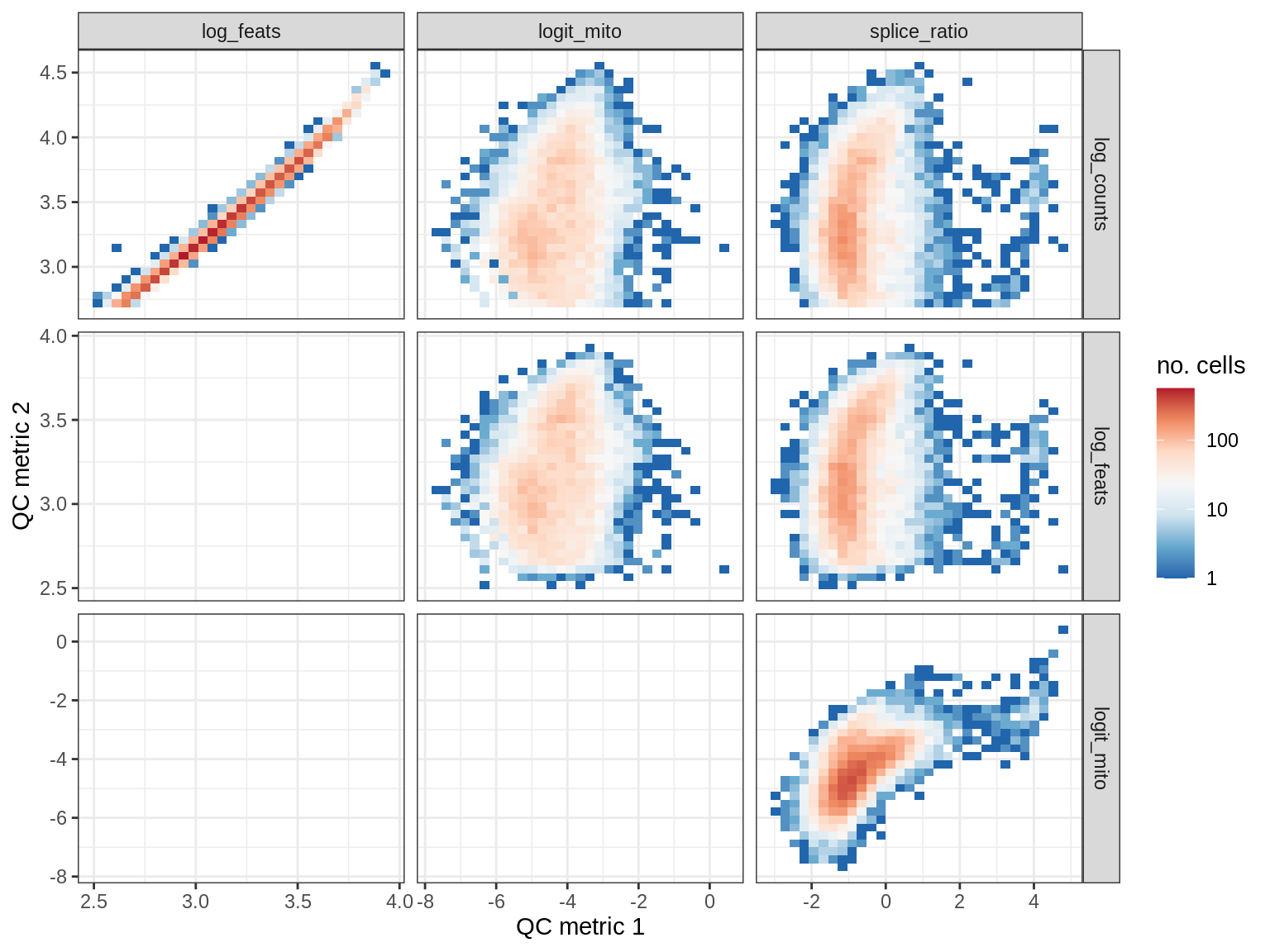
EU038
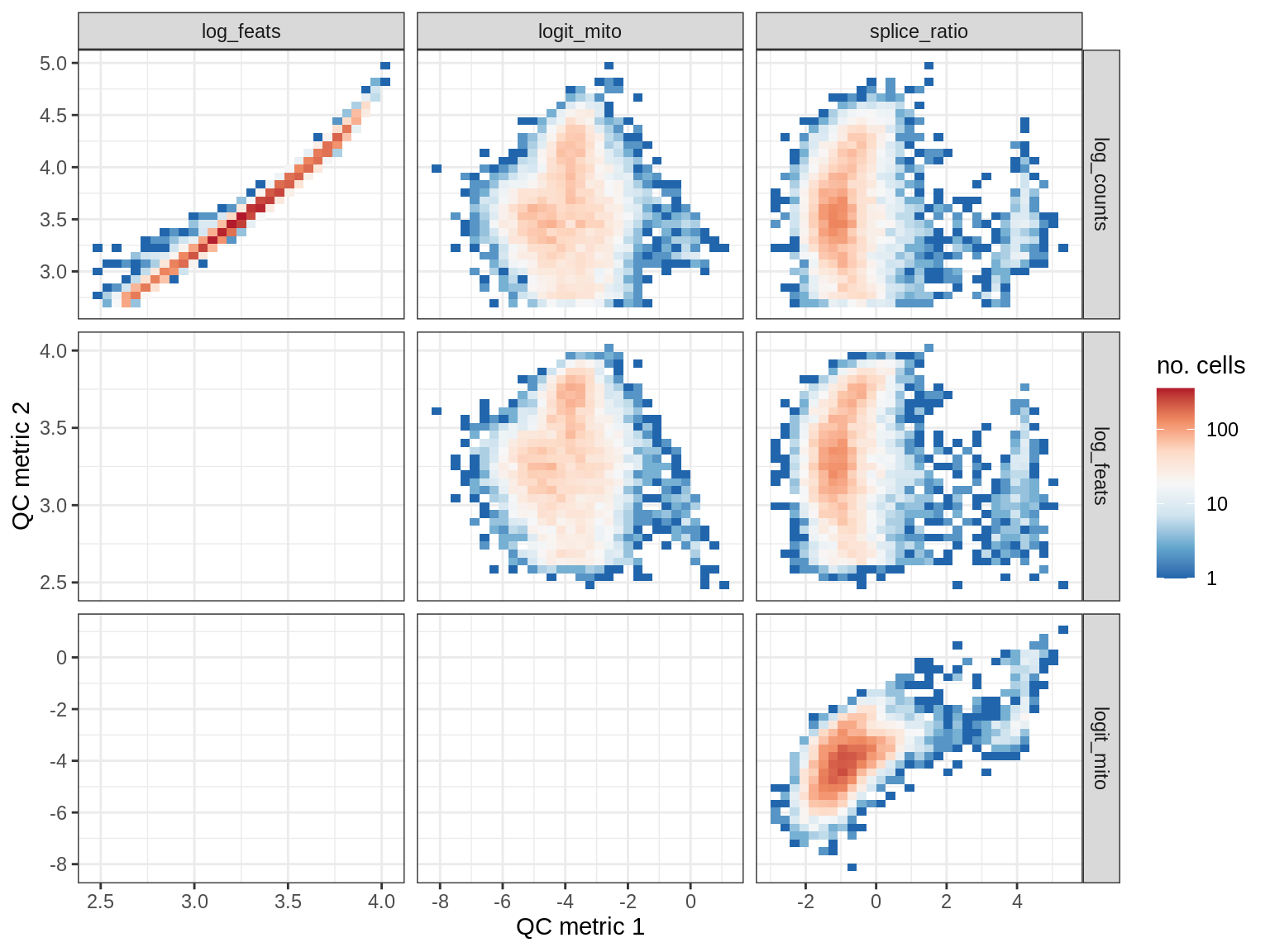
EU039
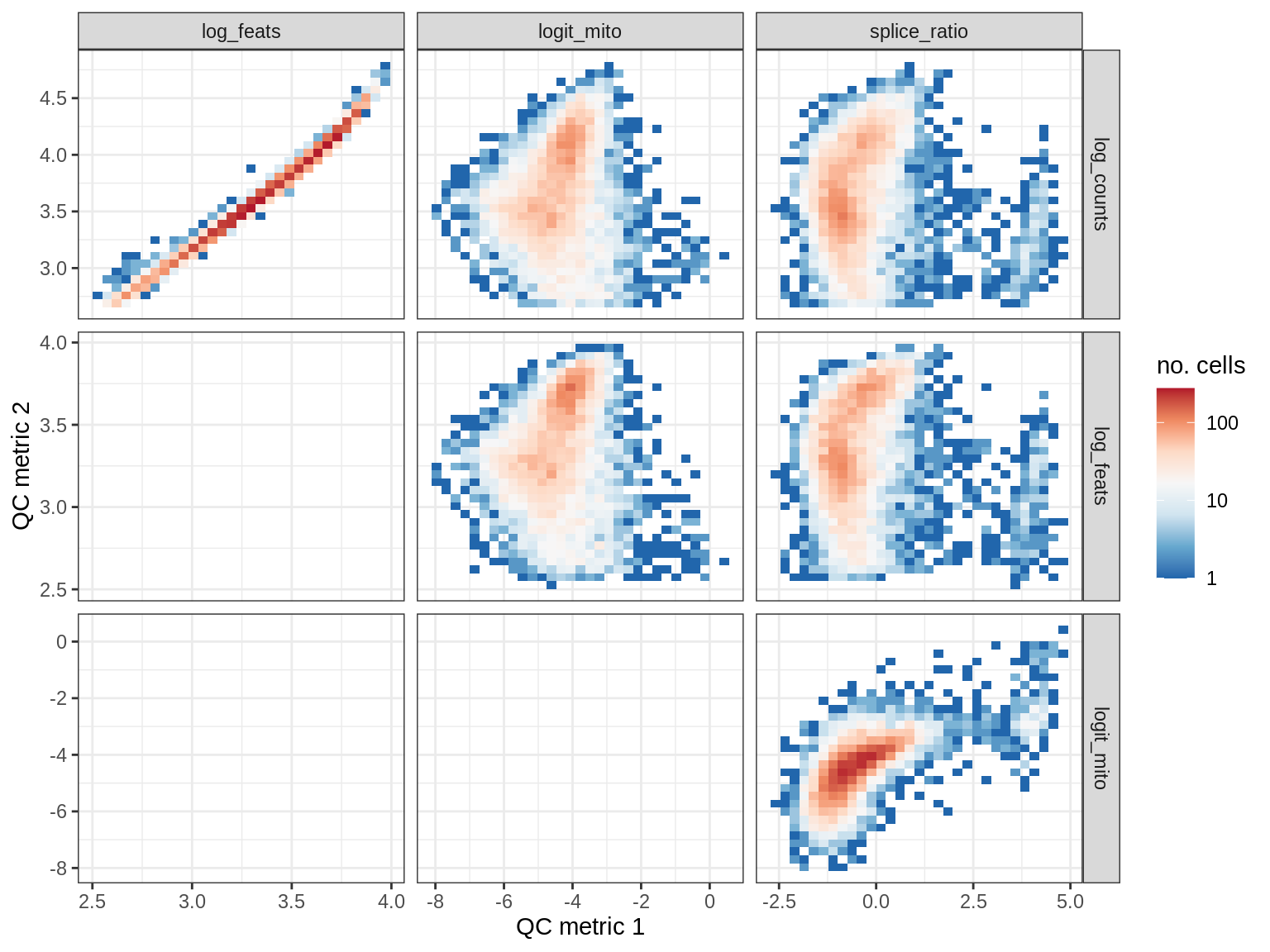
EU040
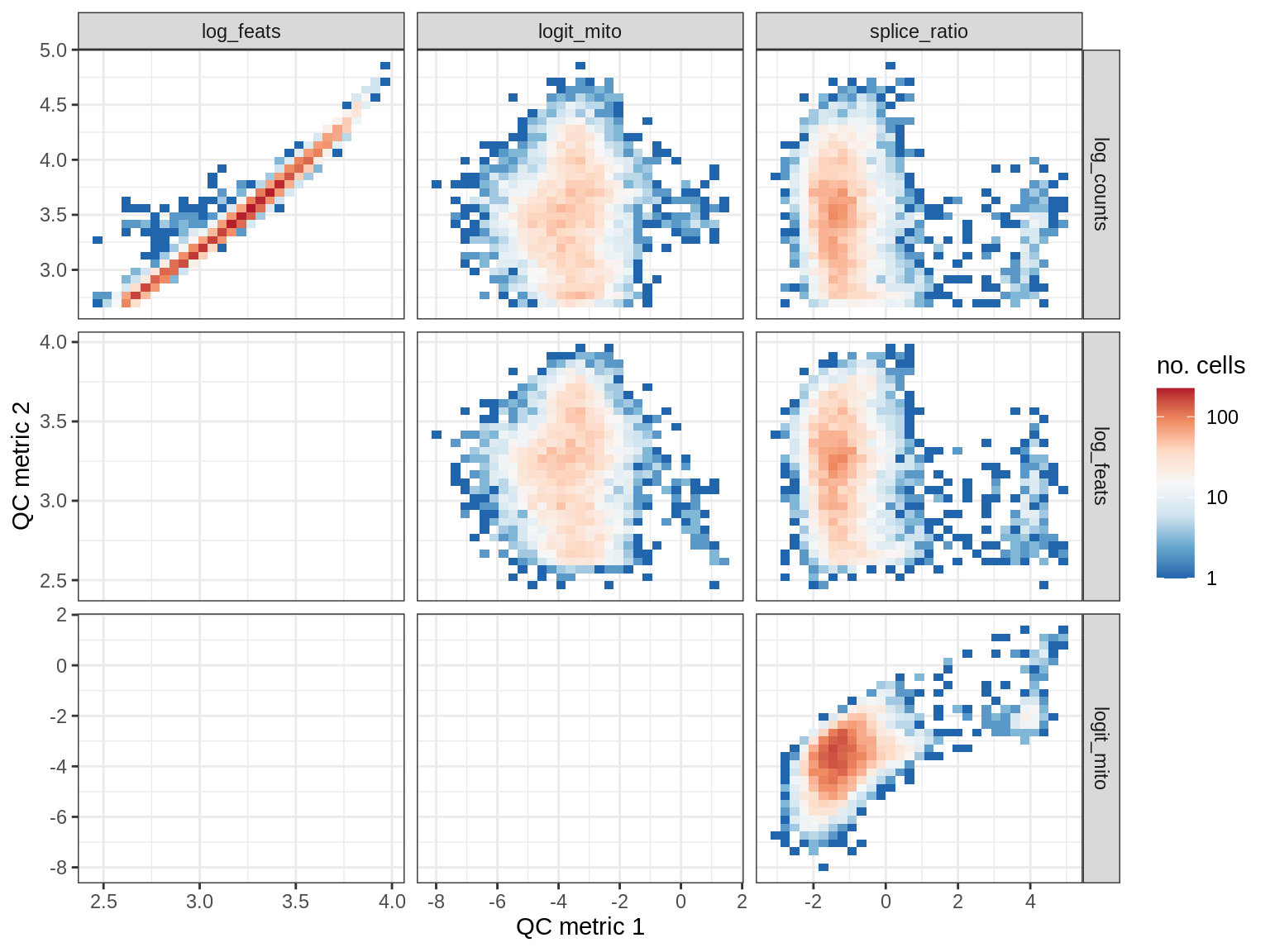
EU041
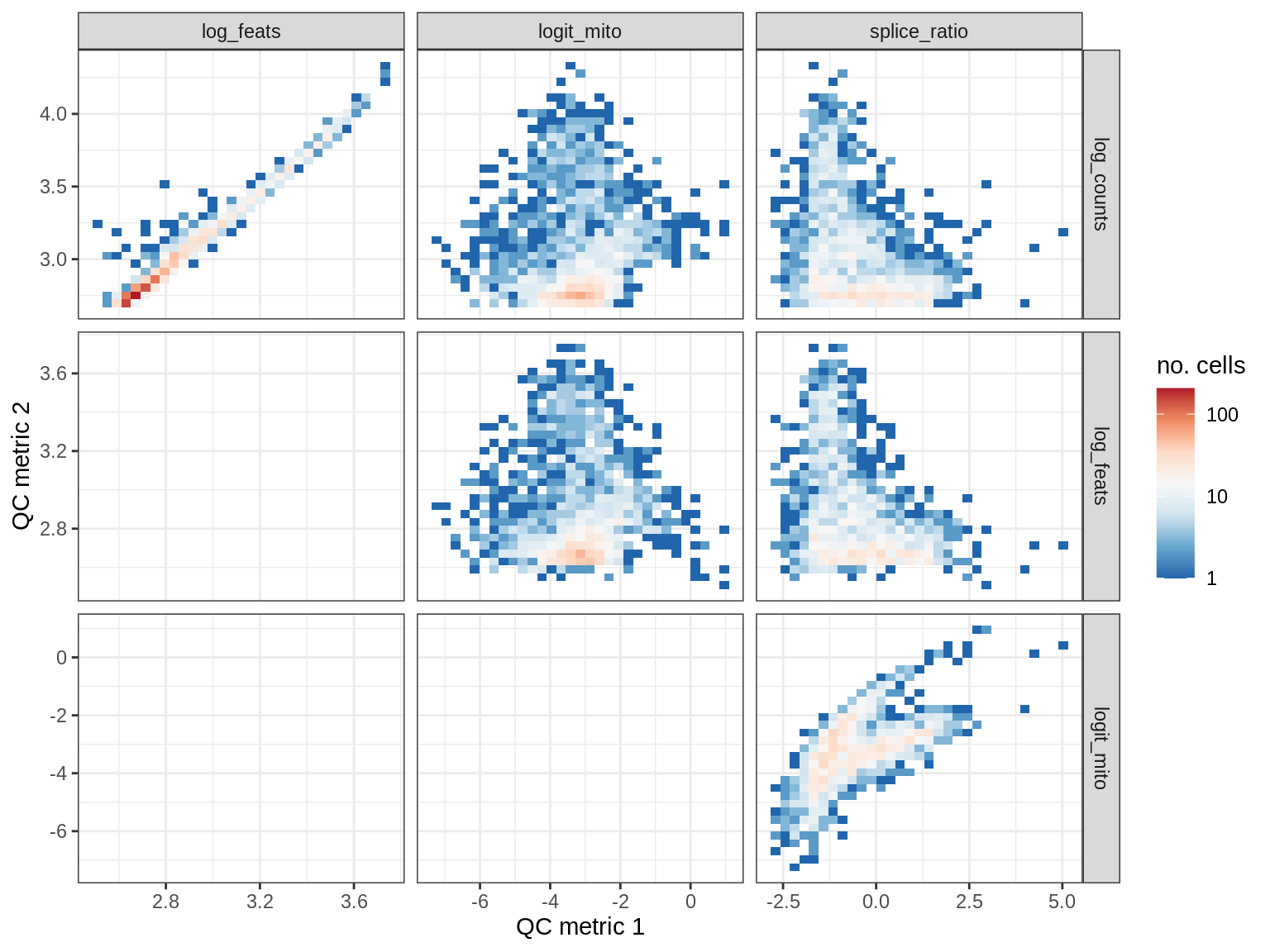
EU042
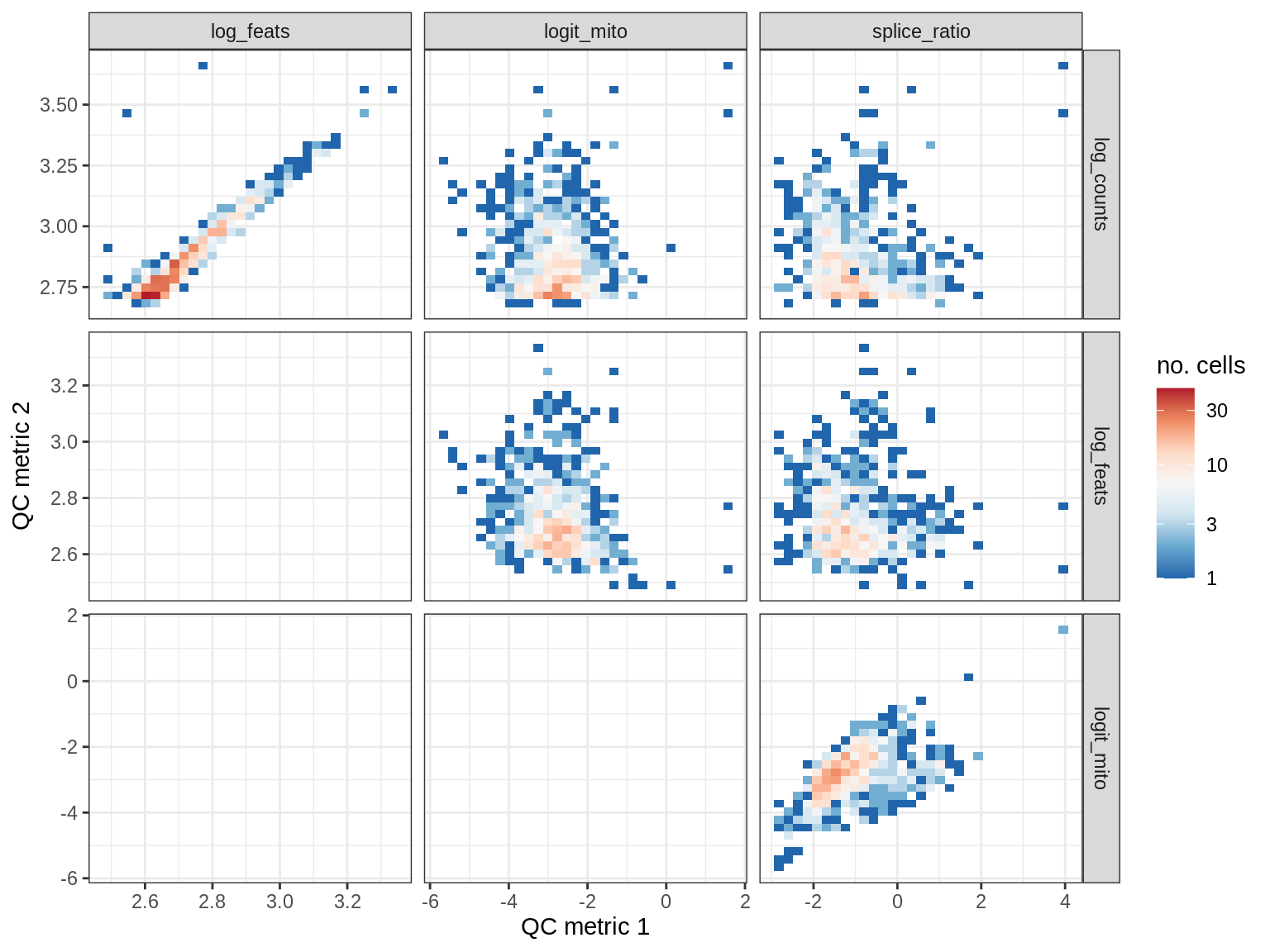
EU043
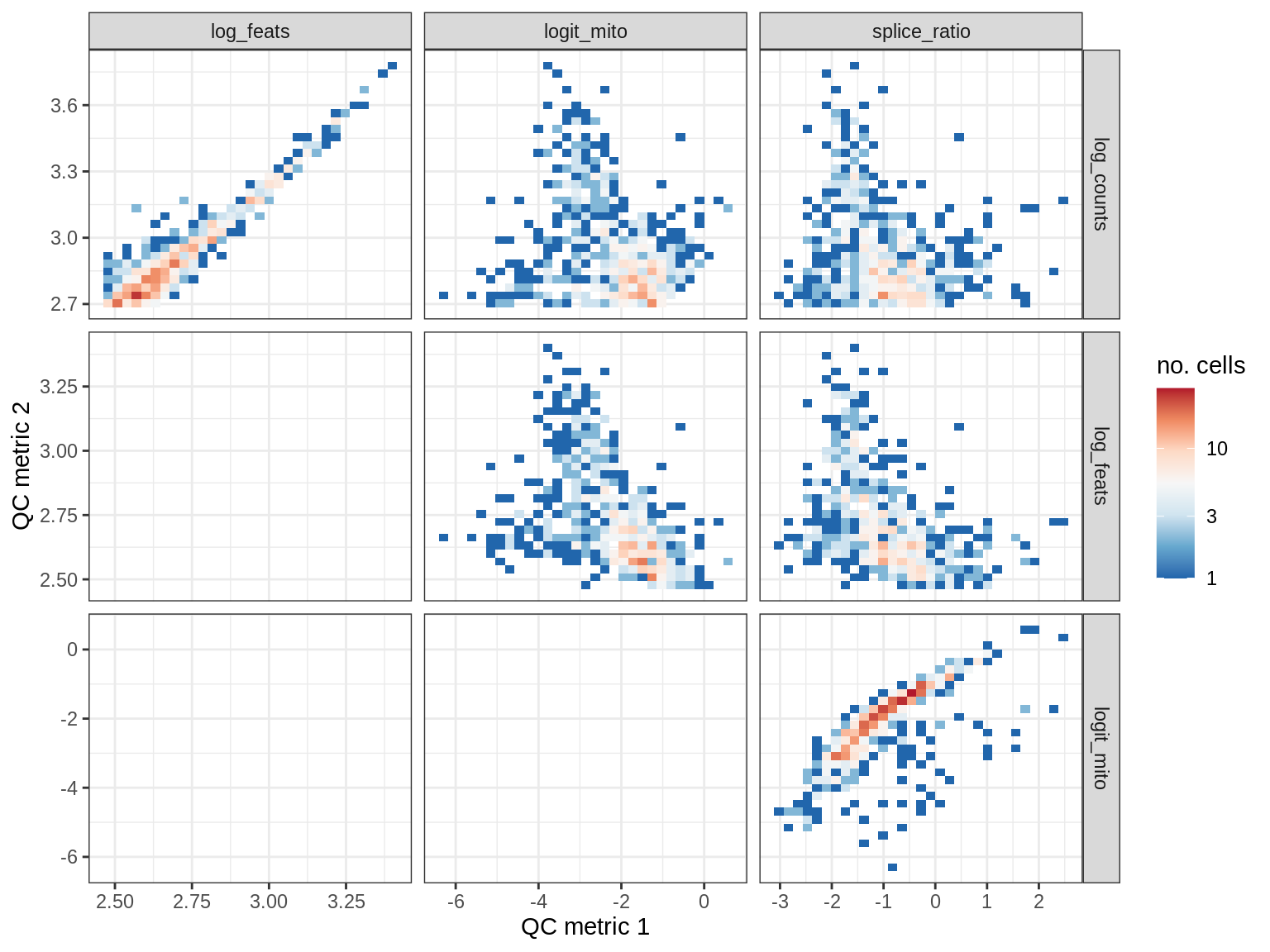
EU044
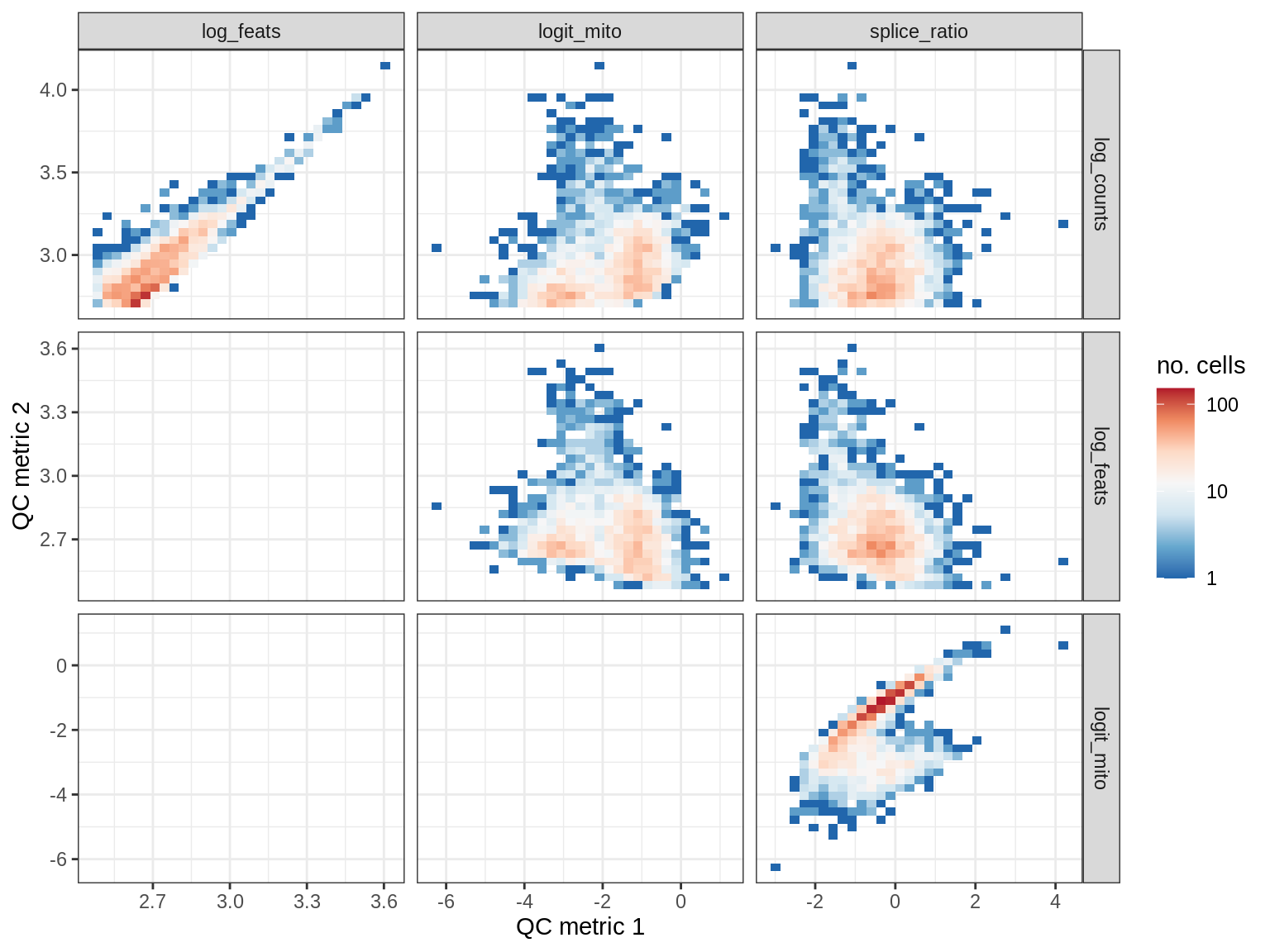
EU045
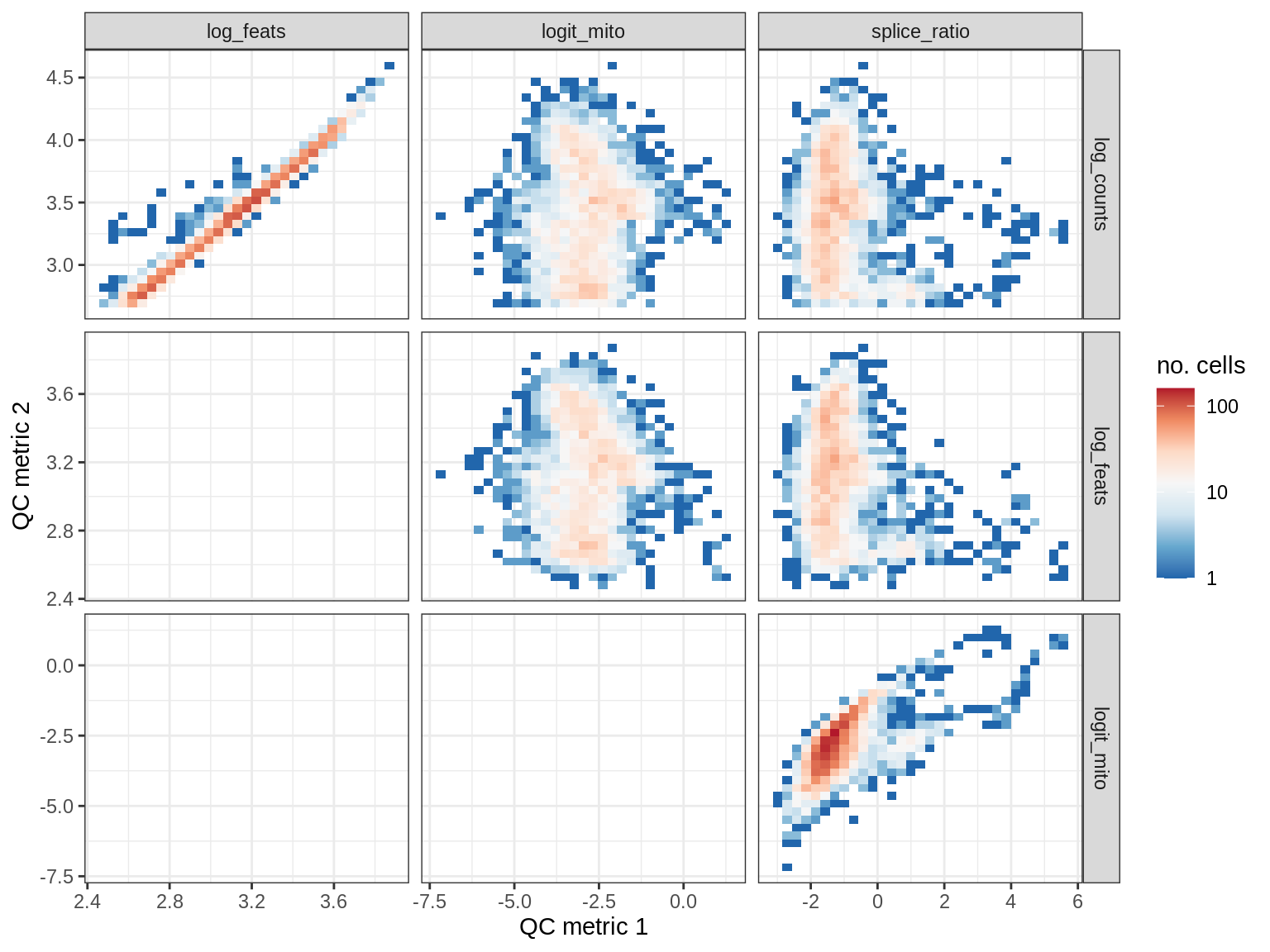
EU046
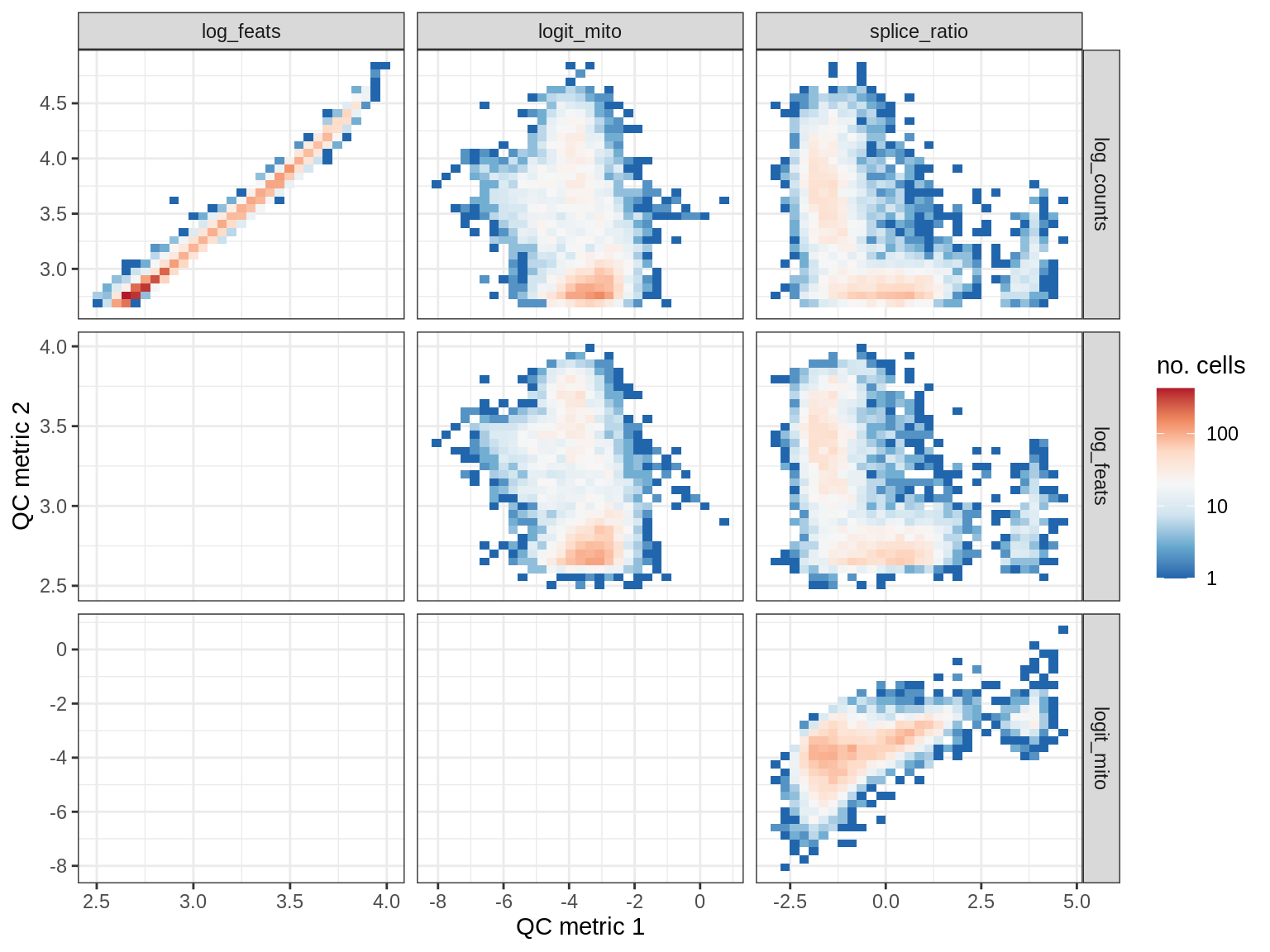
EU047
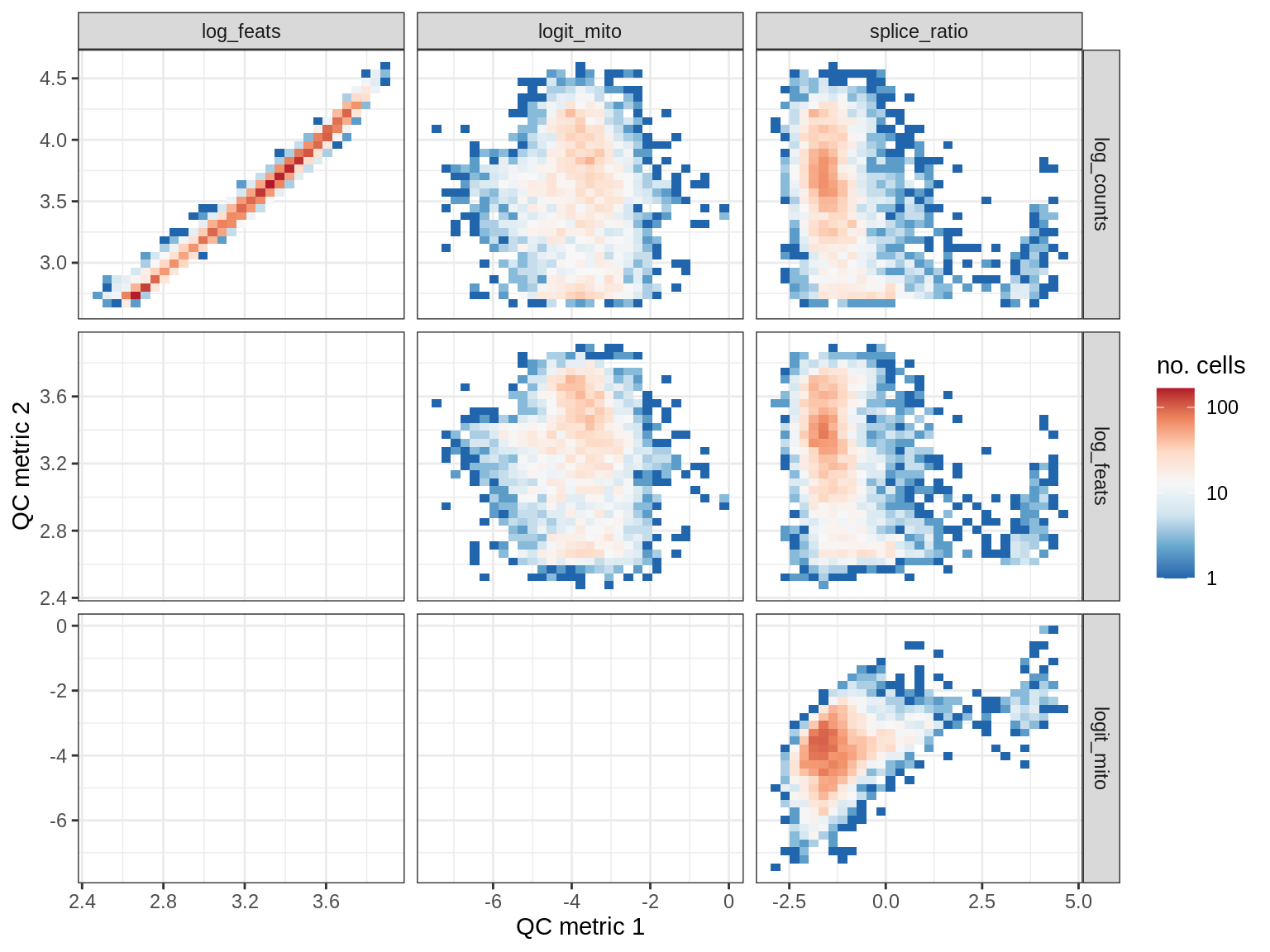
EU048
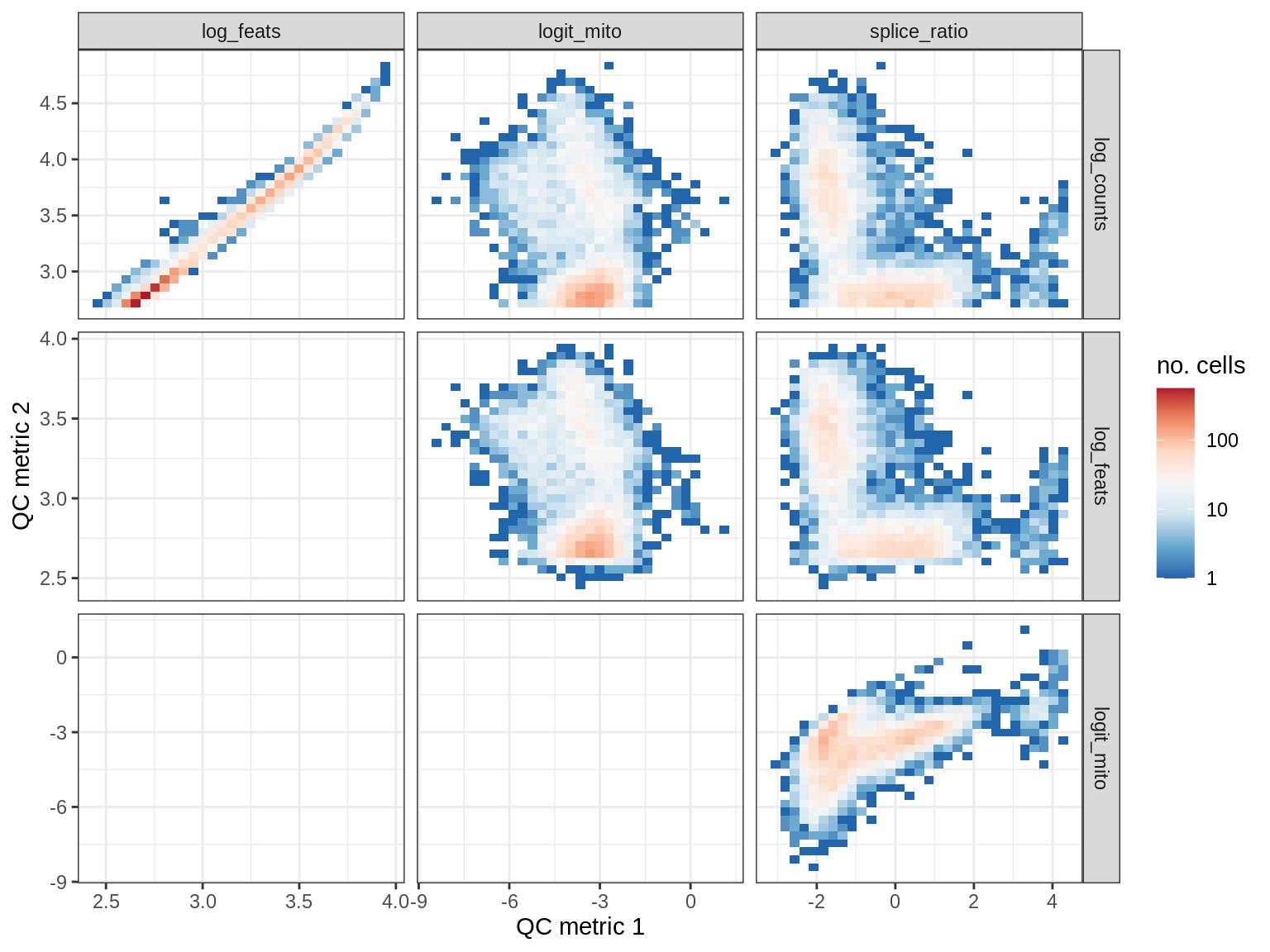
EU050
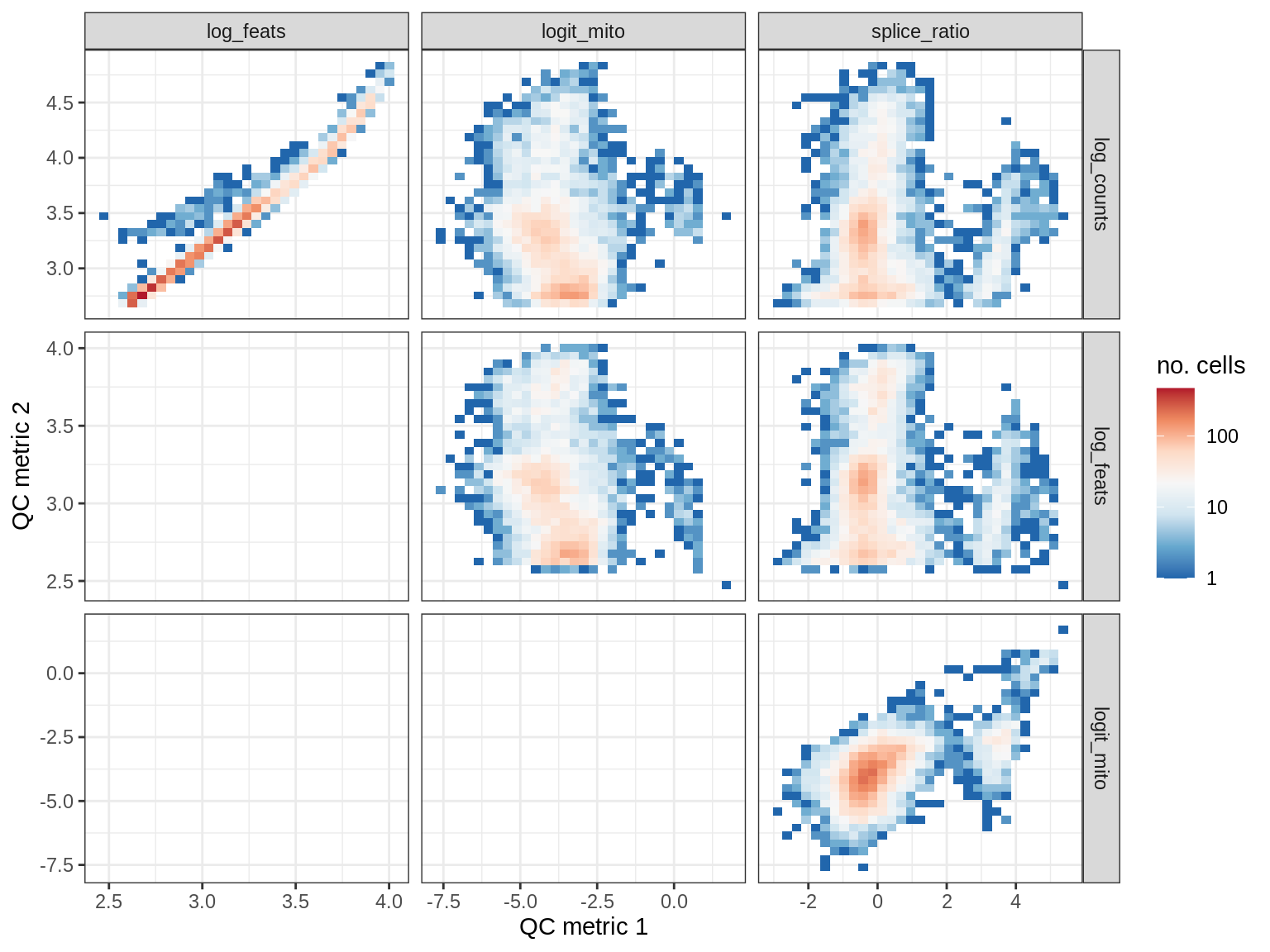
EU051
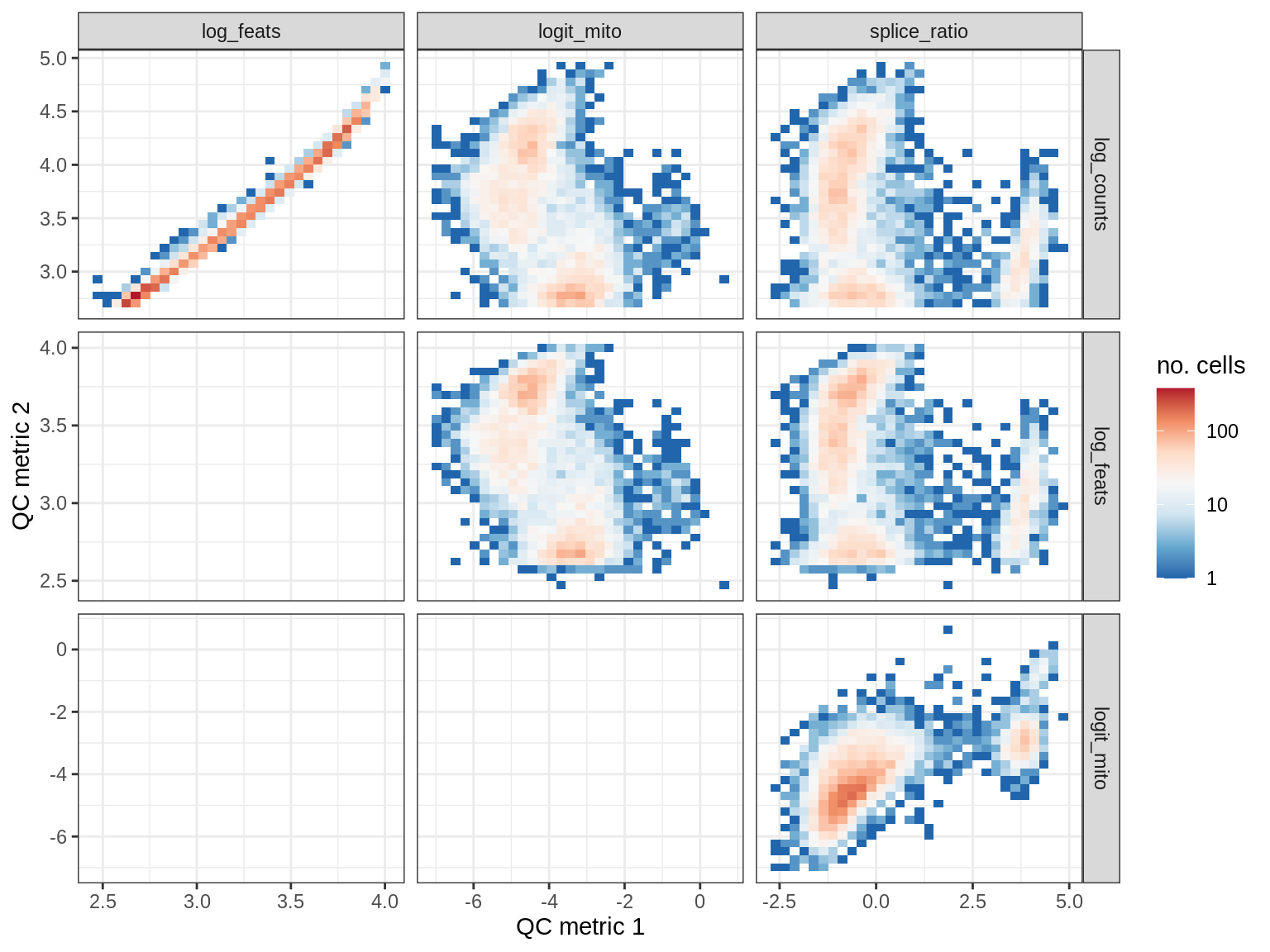
EU052
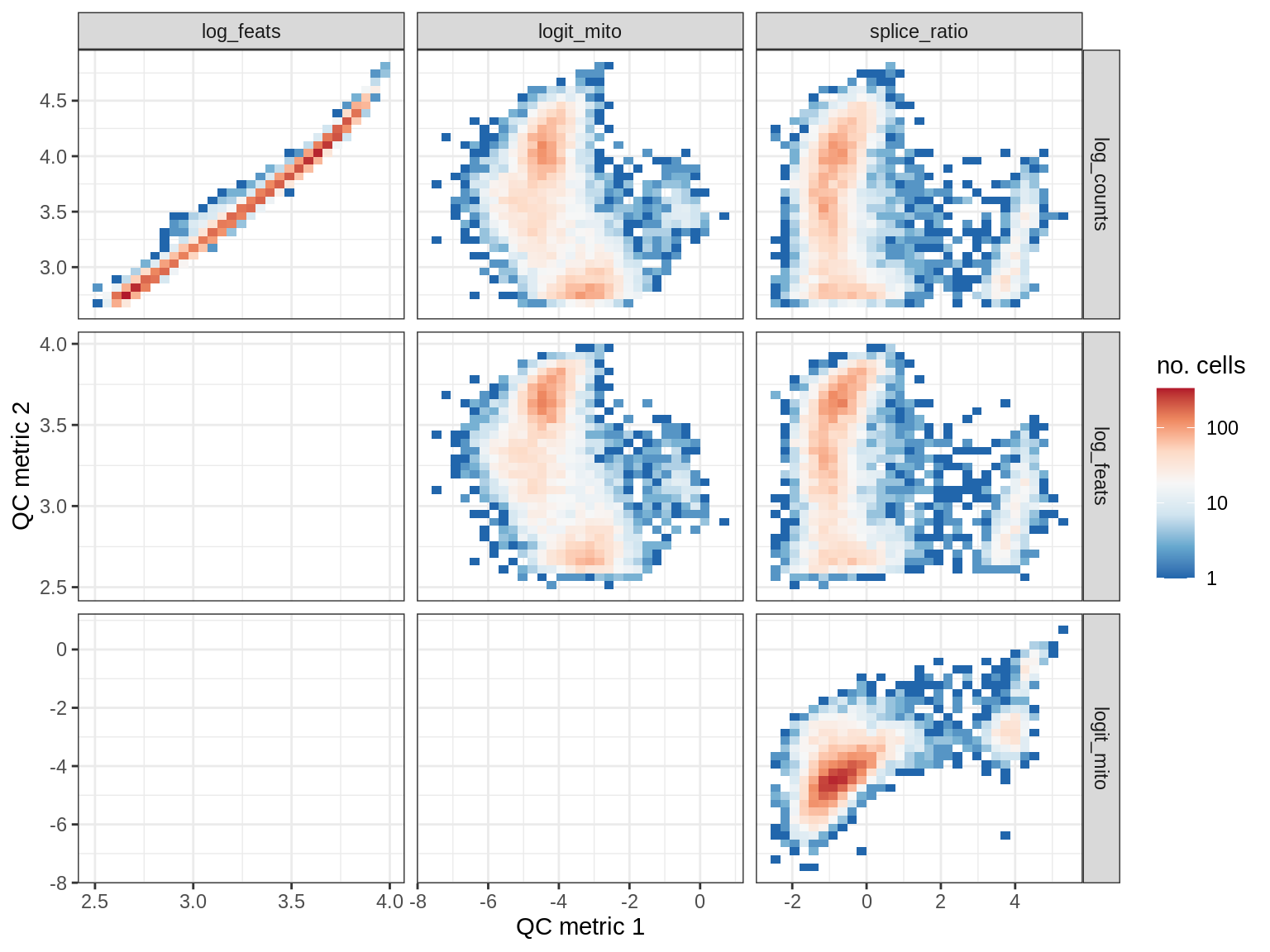
EU053
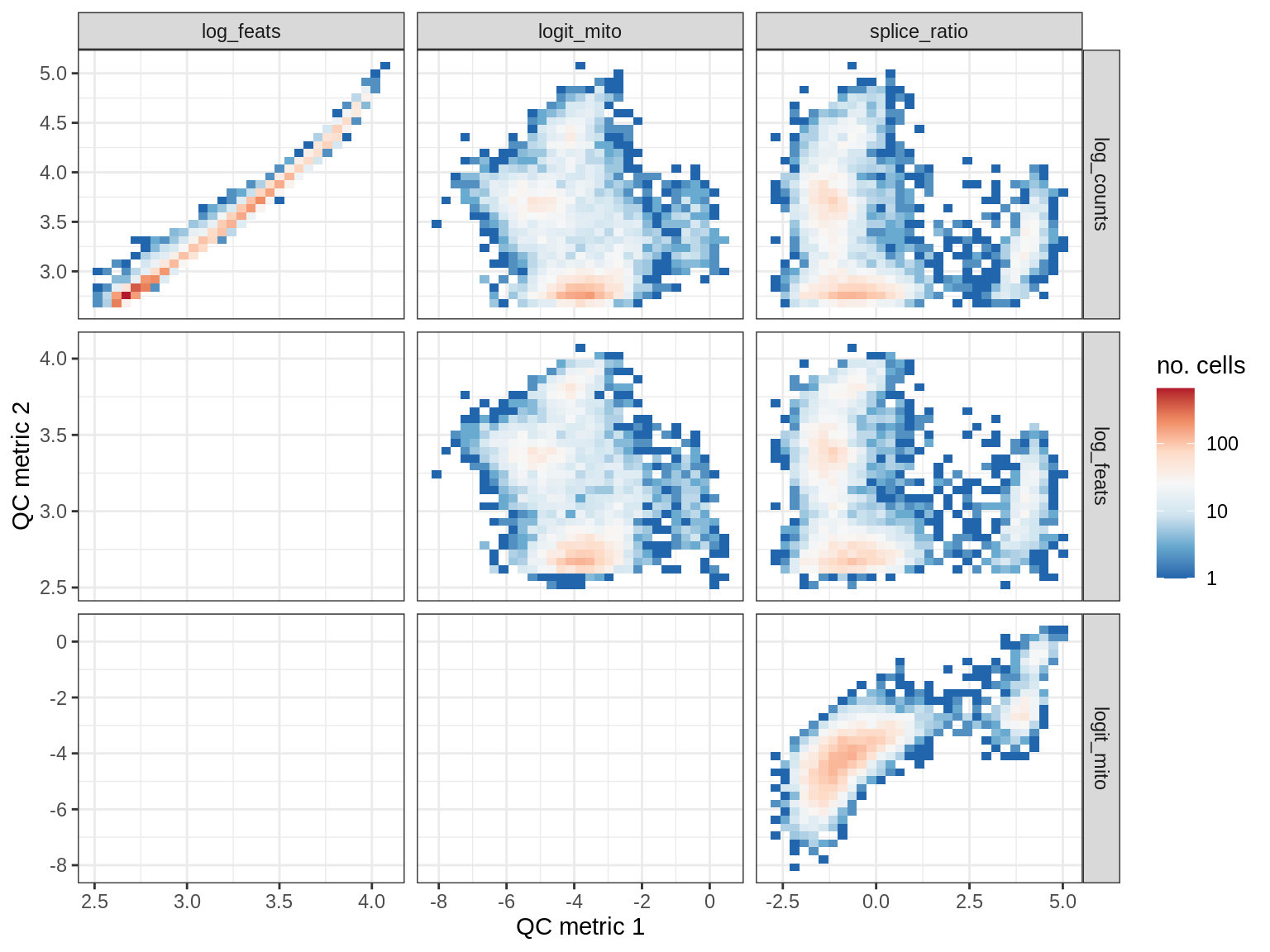
EU054
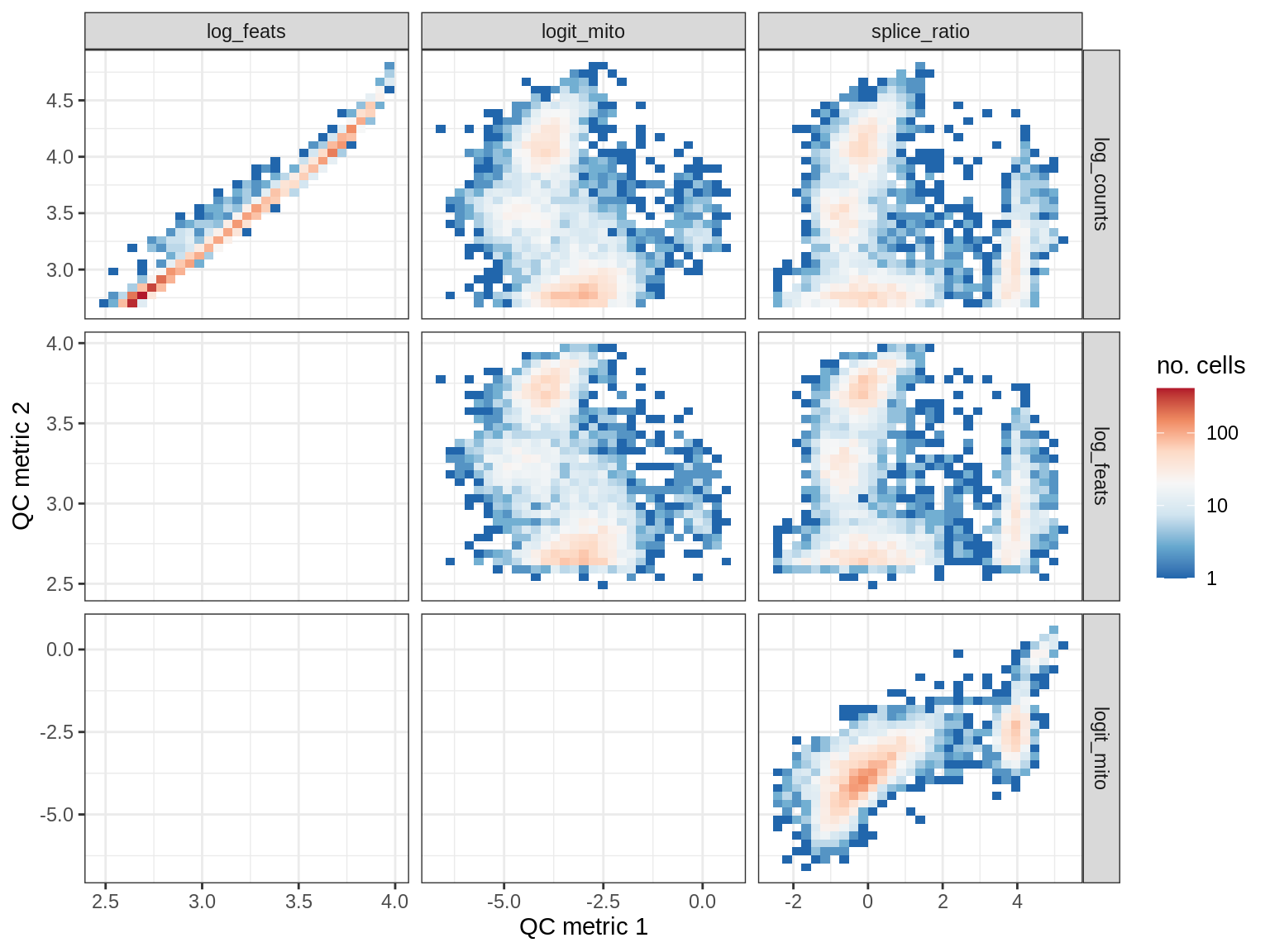
EU055
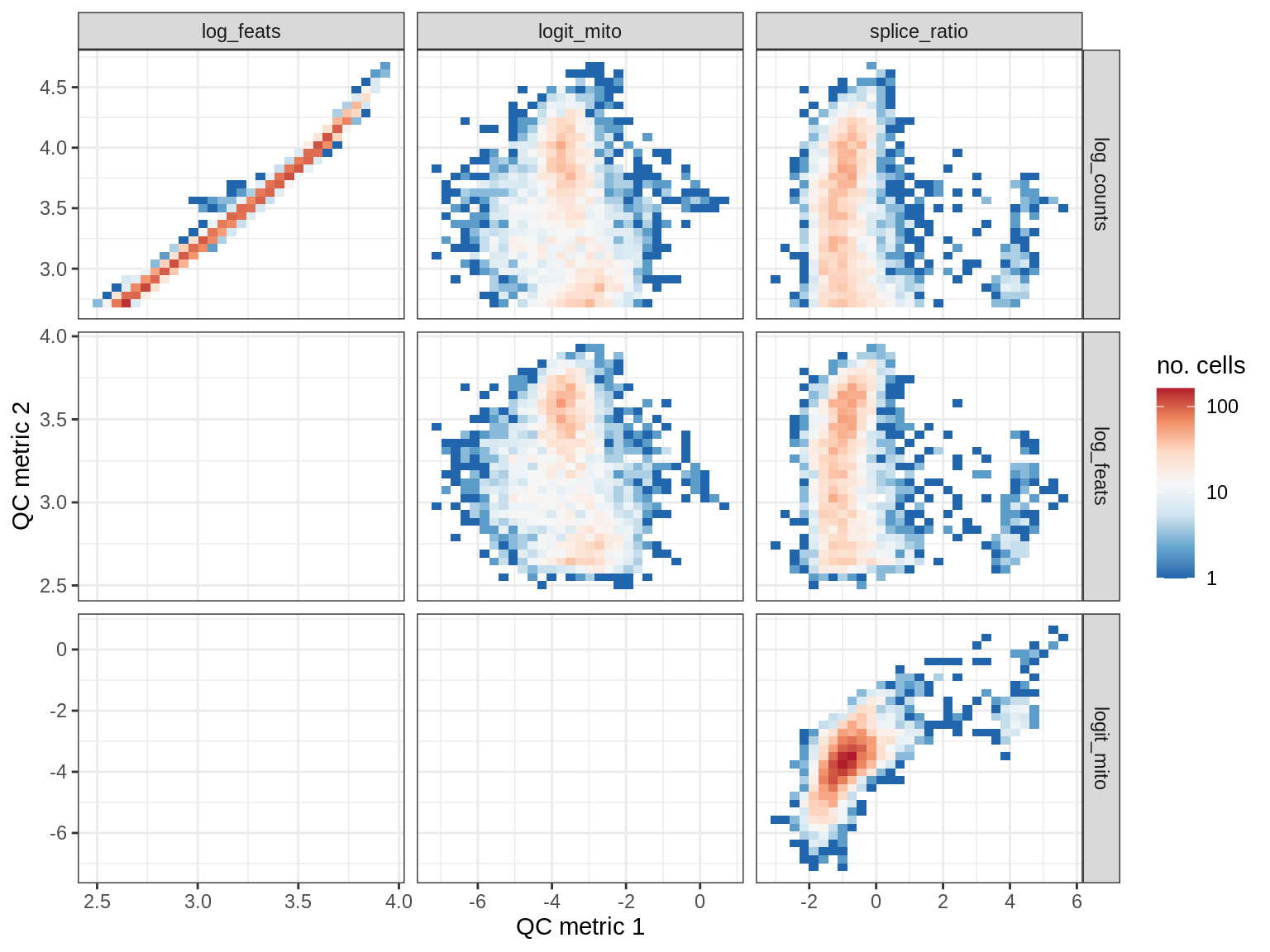
EU056
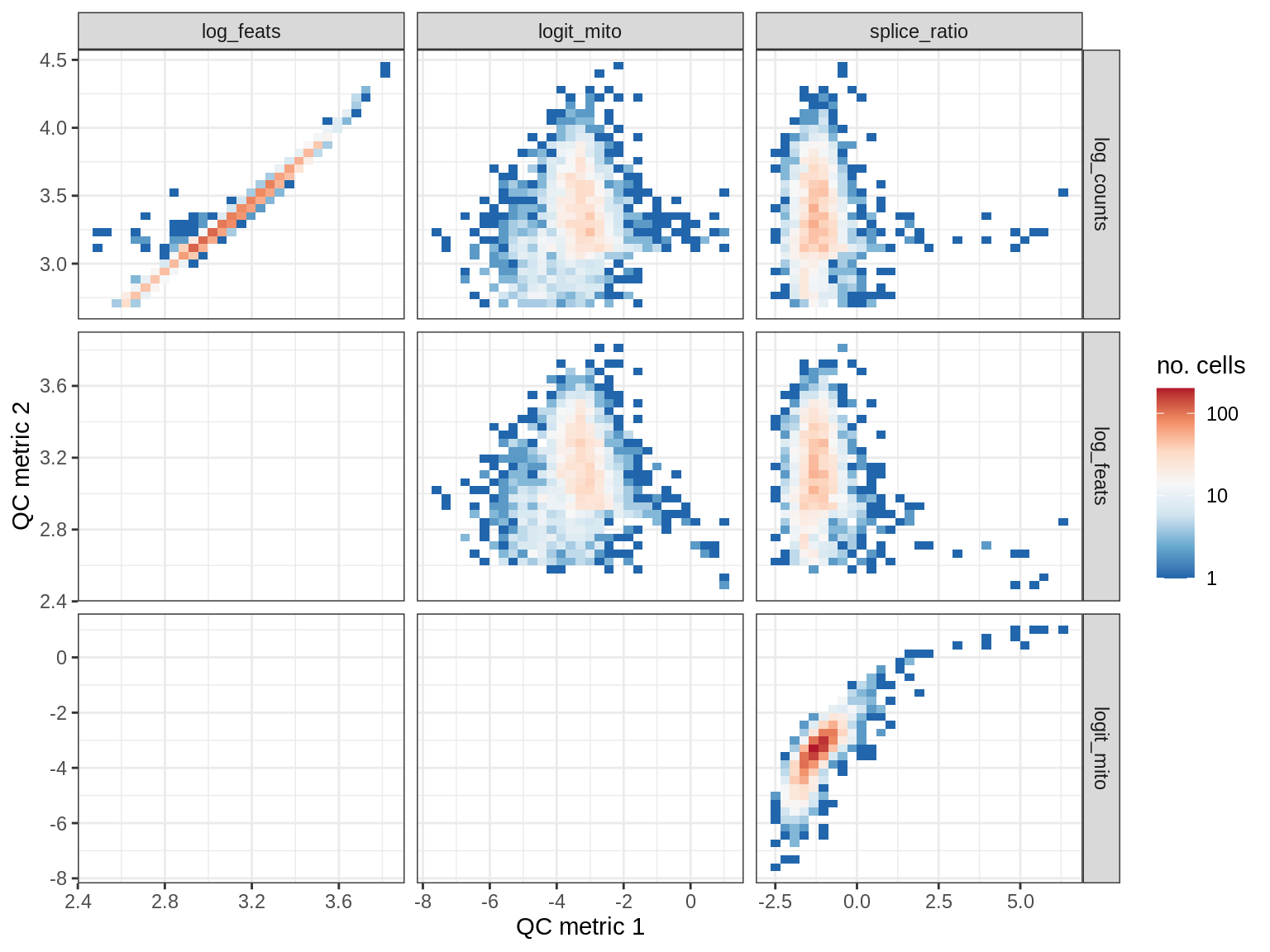
EU057
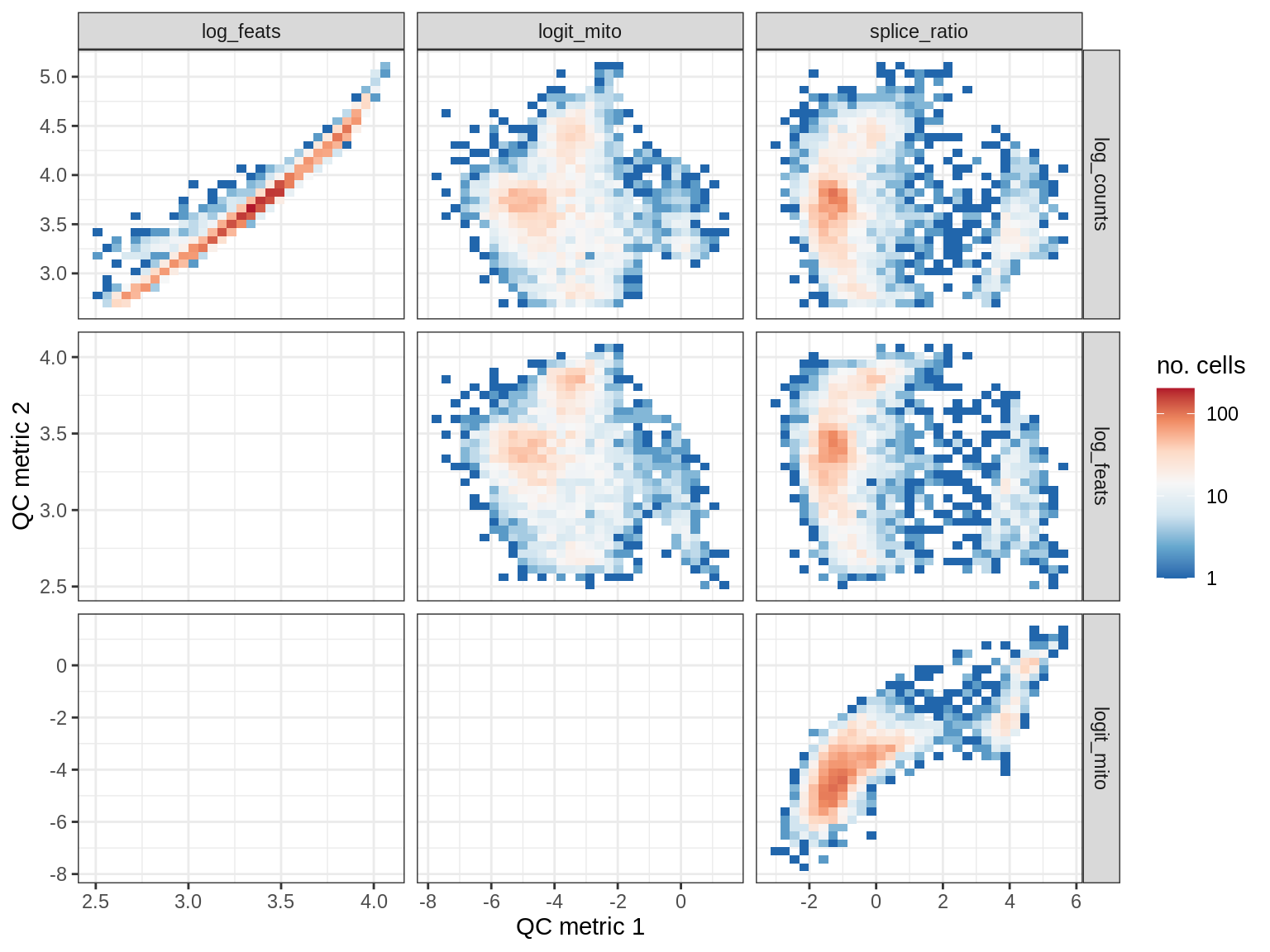
EU058
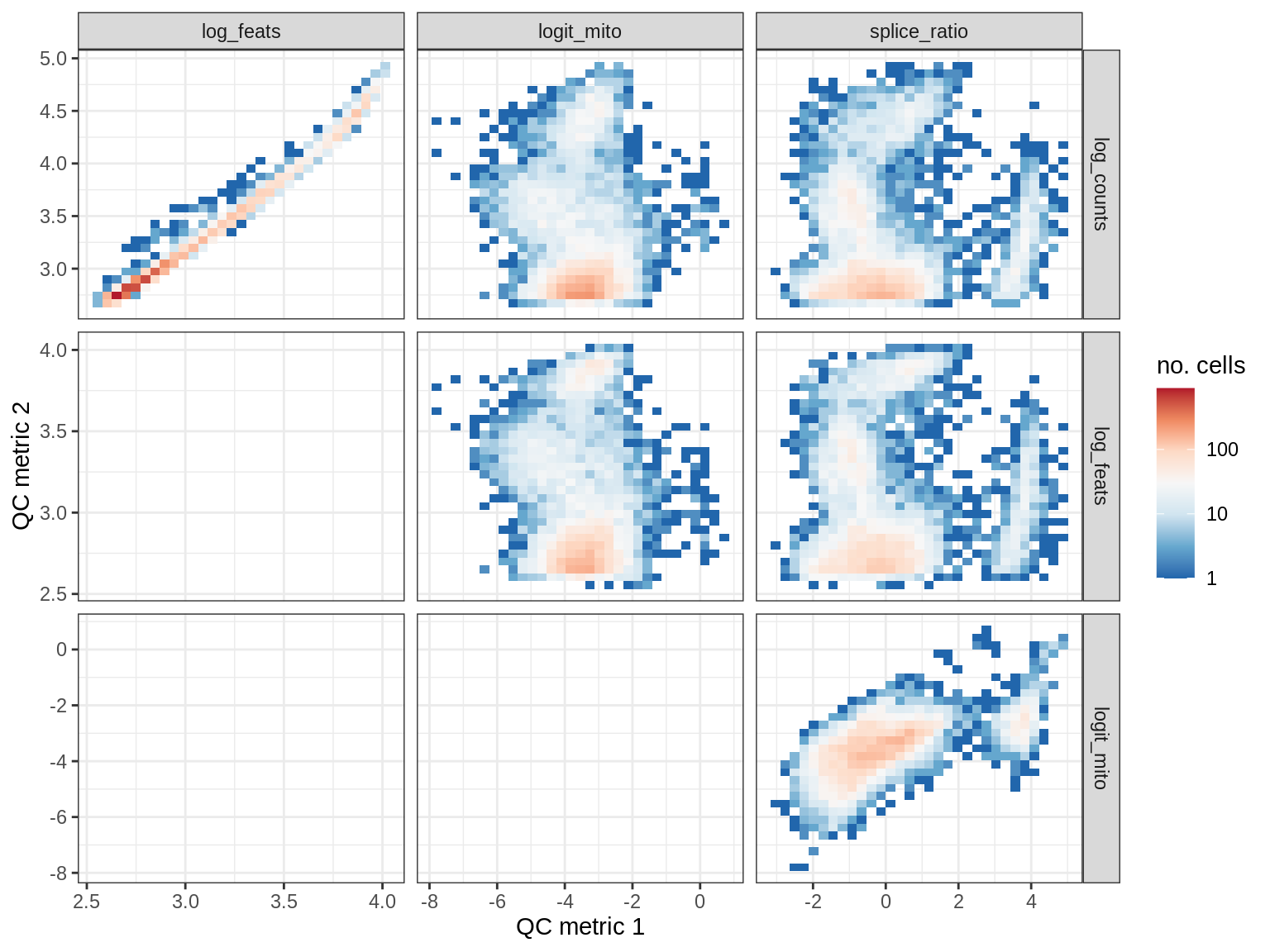
EU059
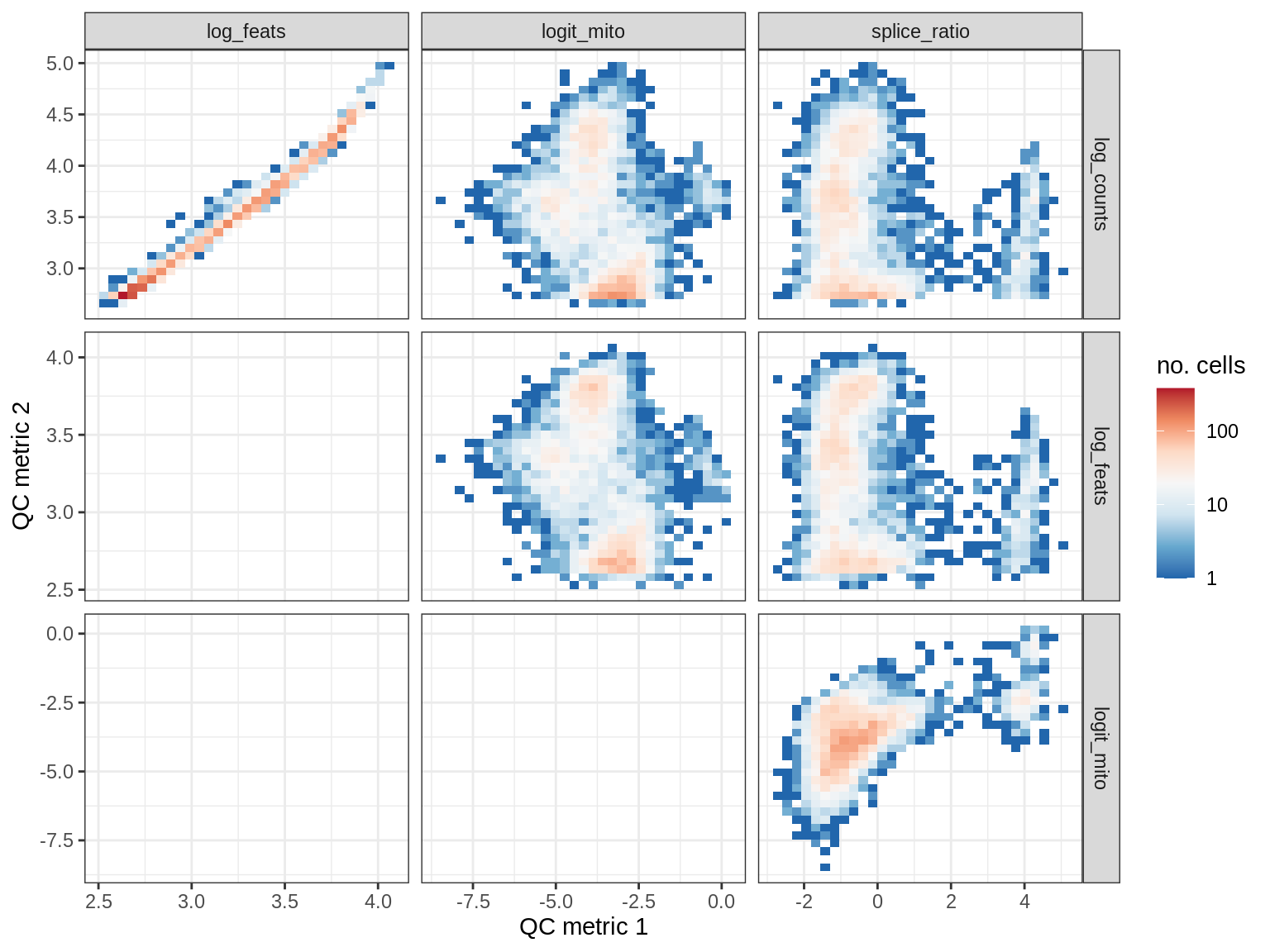
EU060
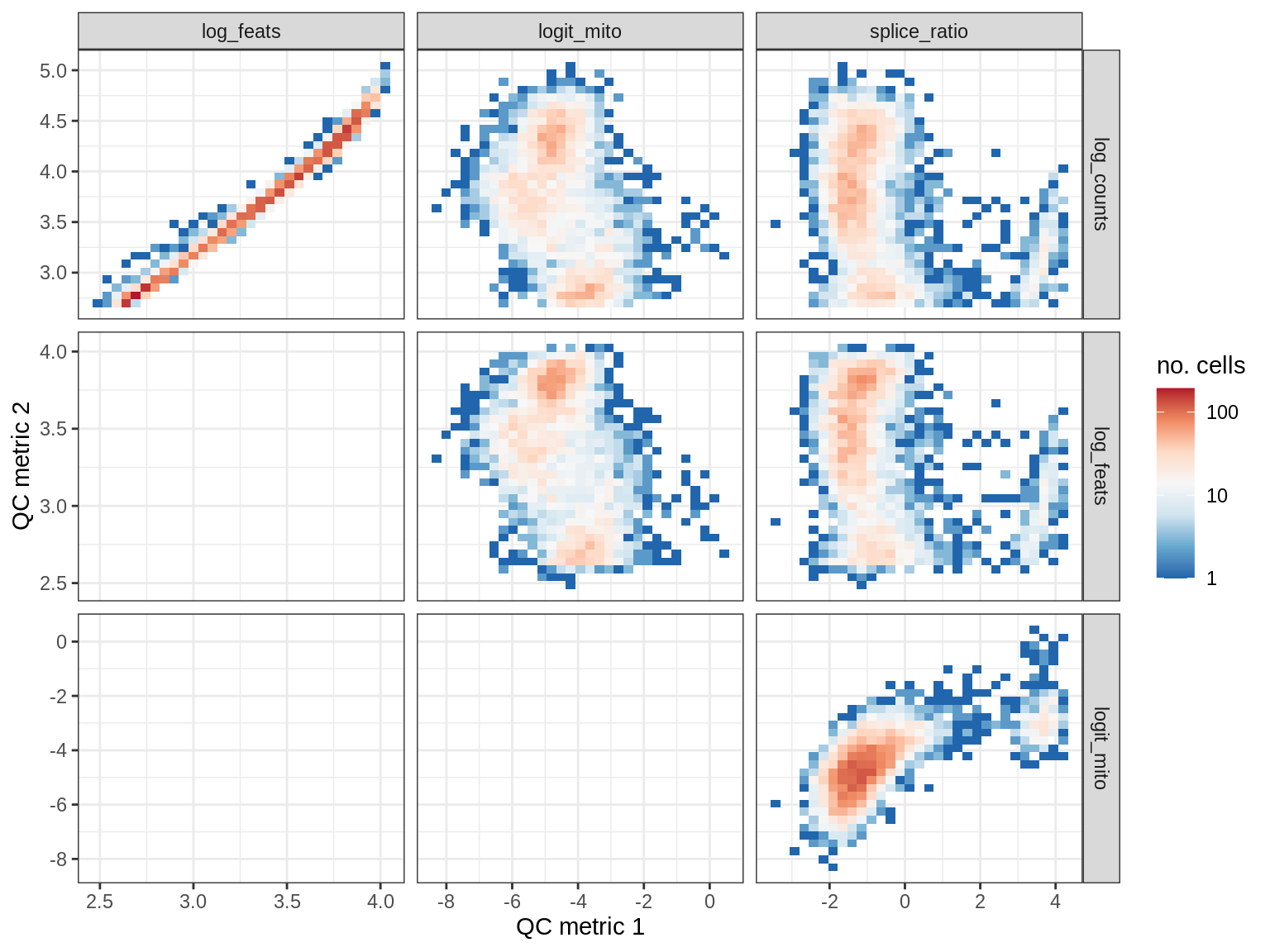
EU061
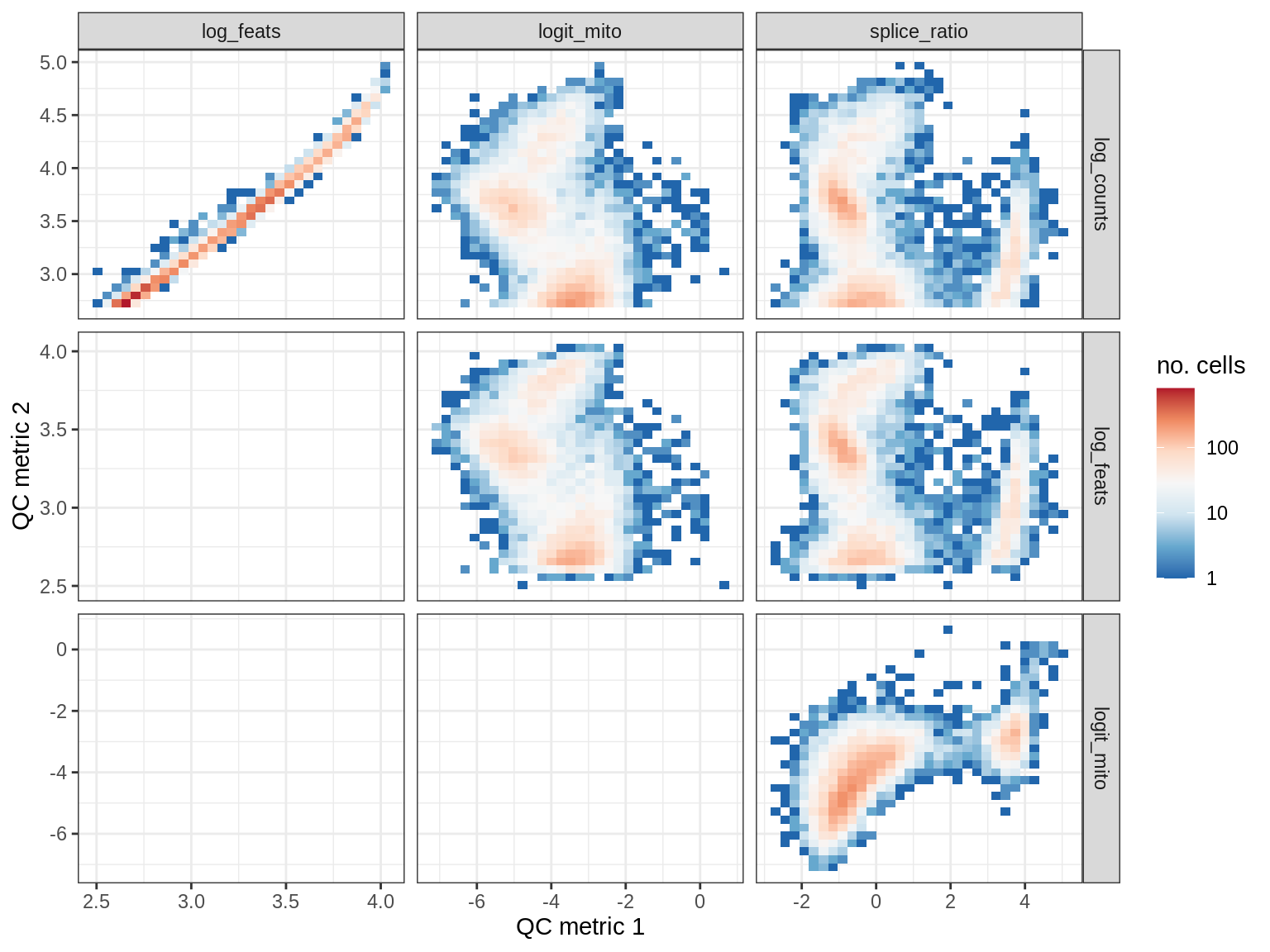
EU062
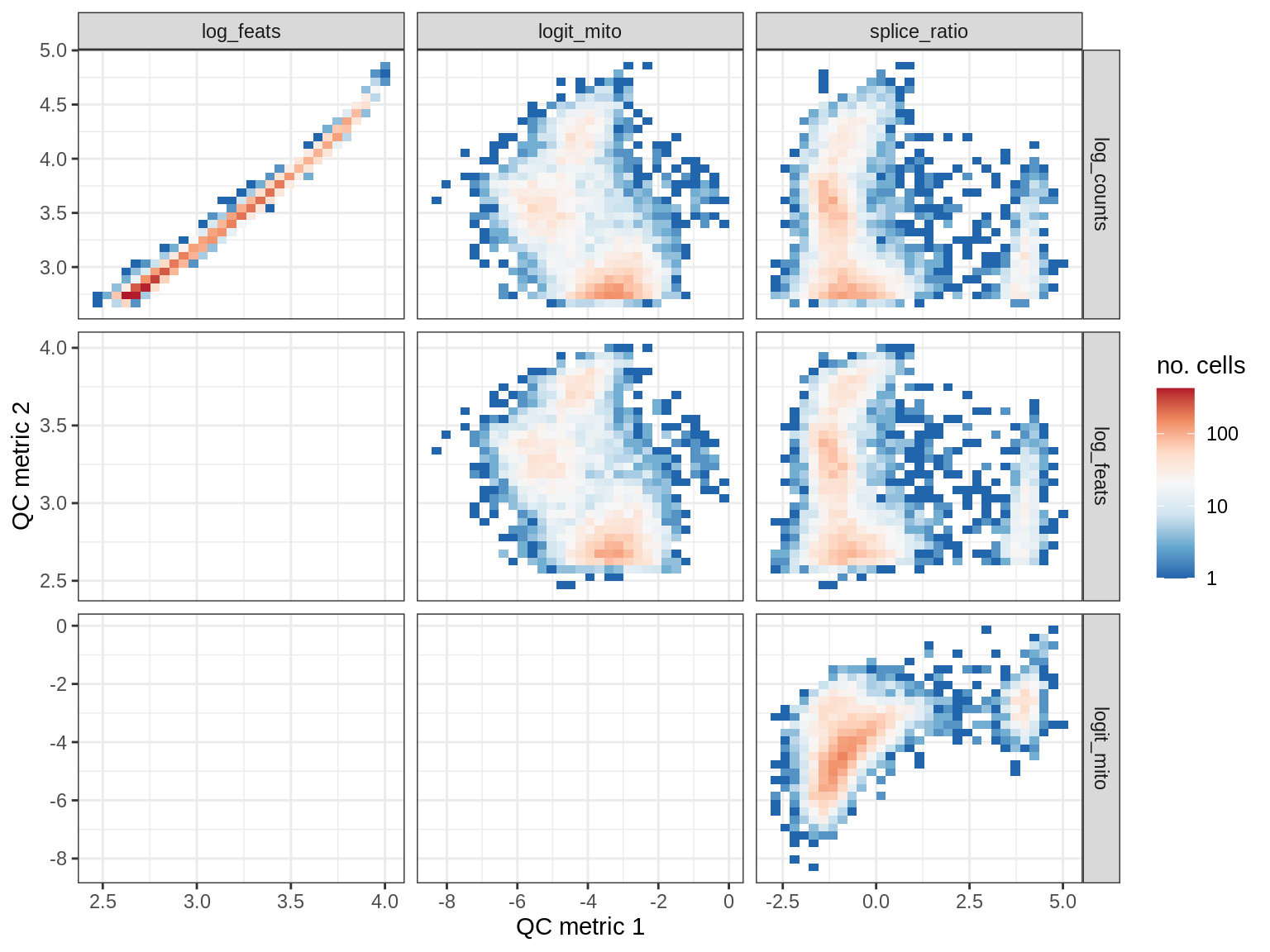
EU063
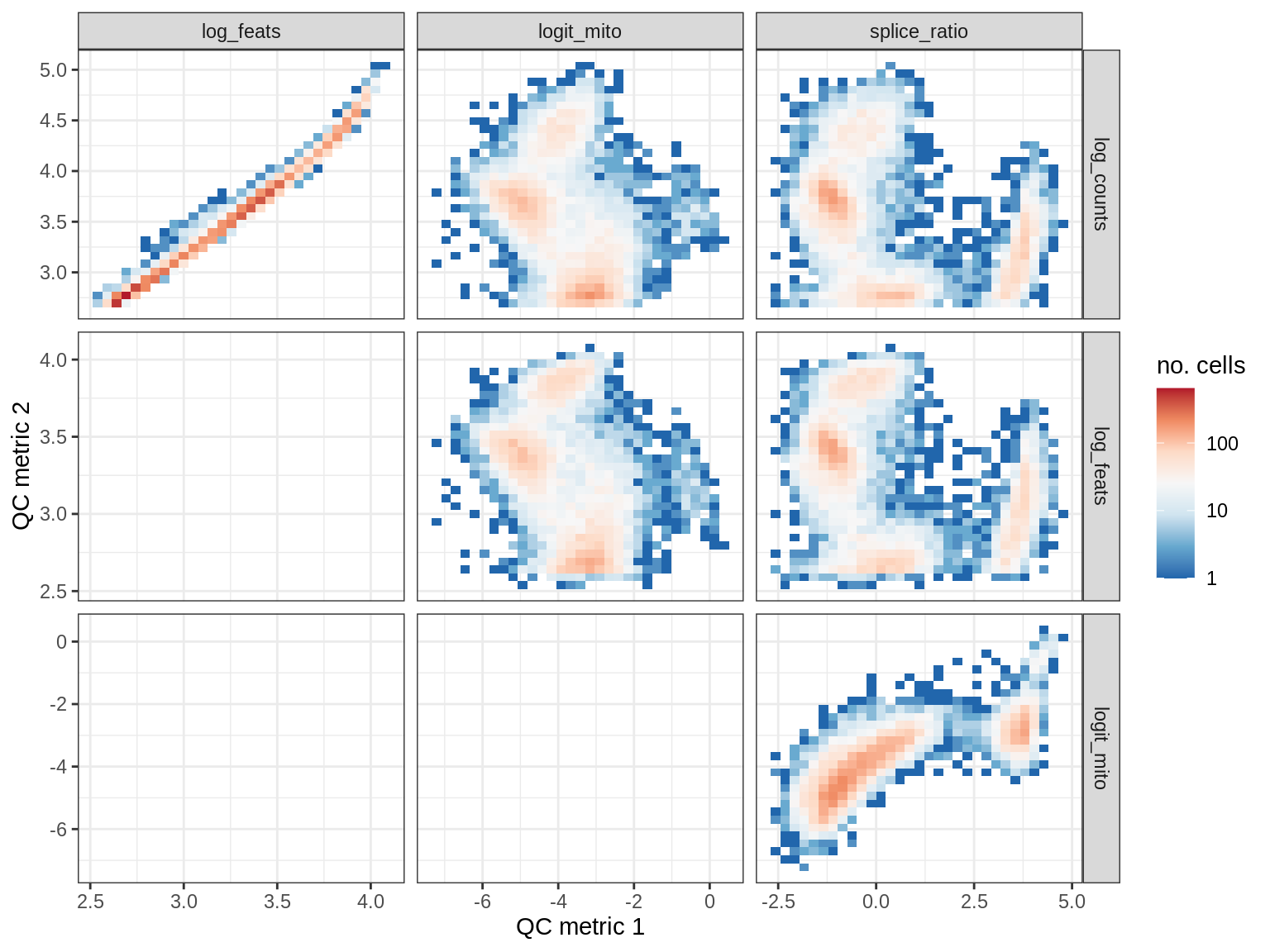
EU064
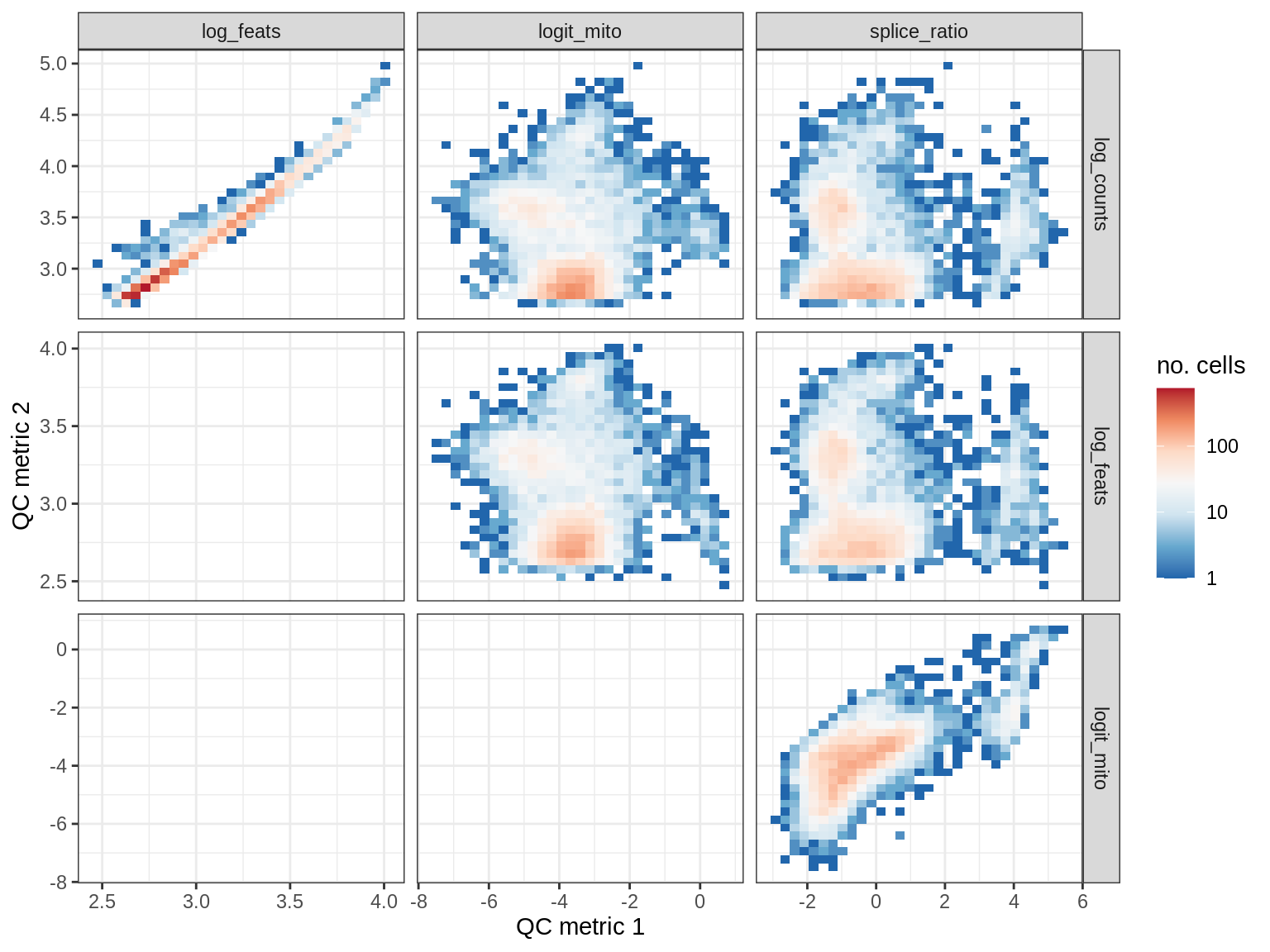
EU065
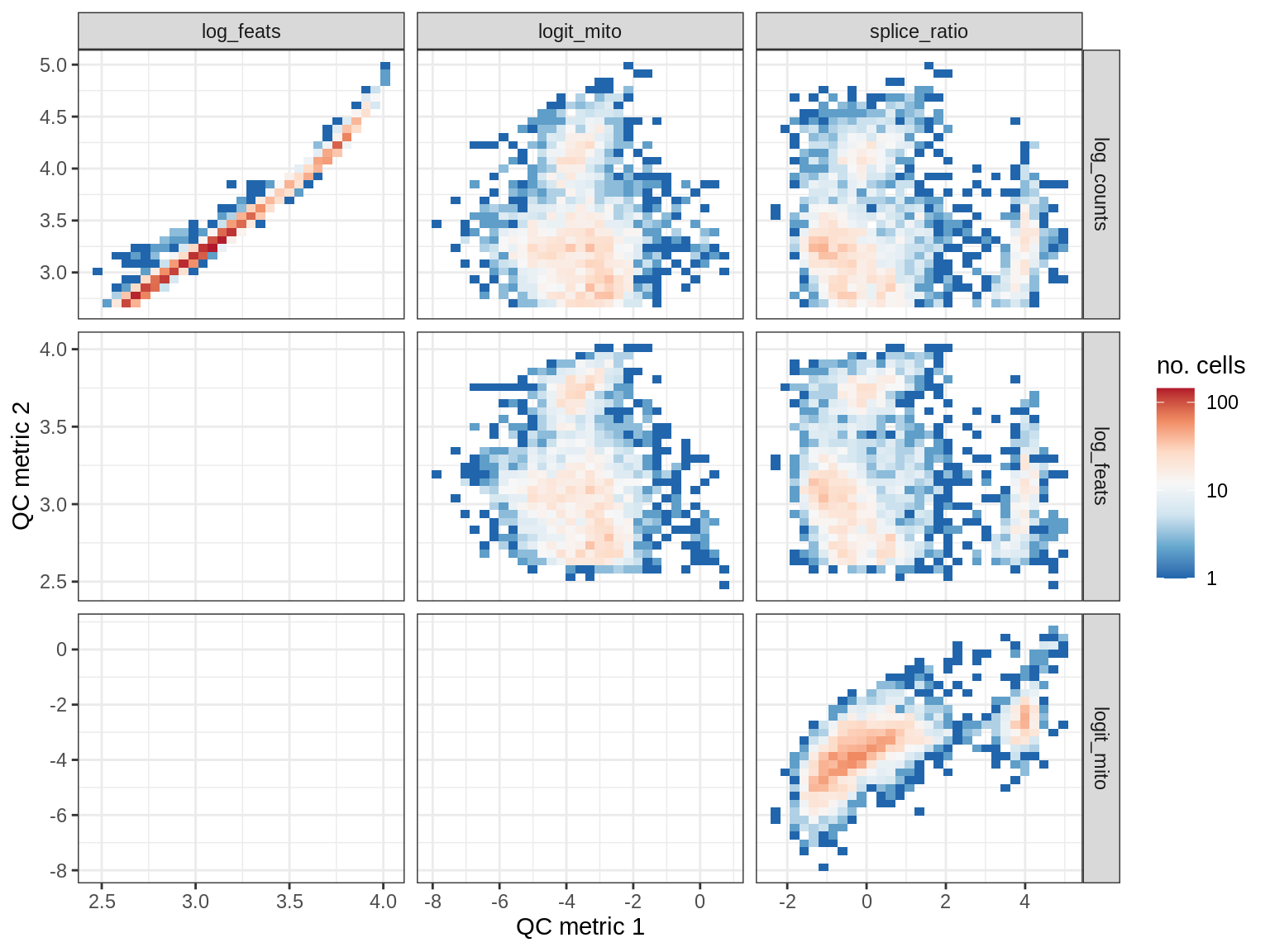
EU066
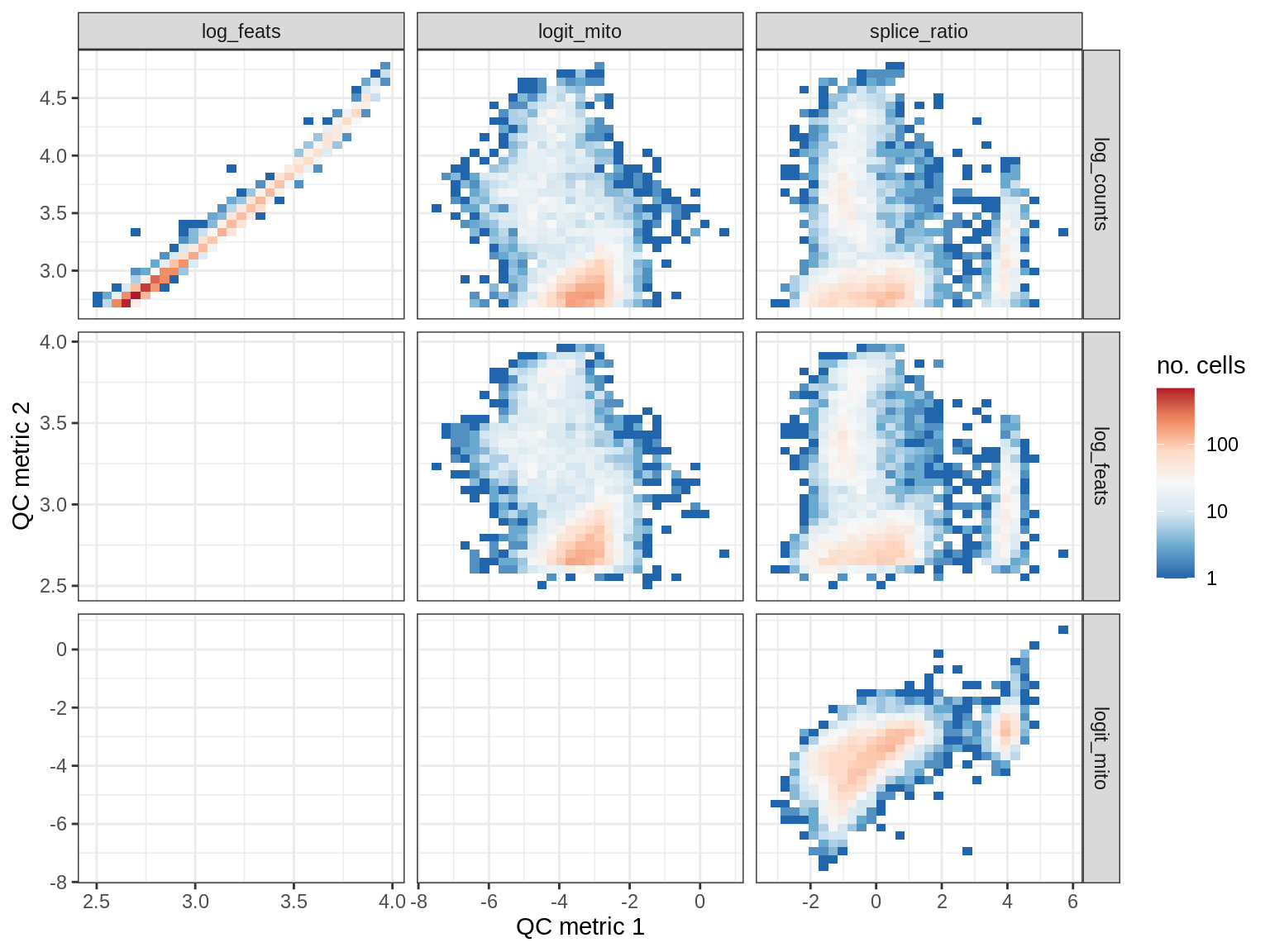
EU067
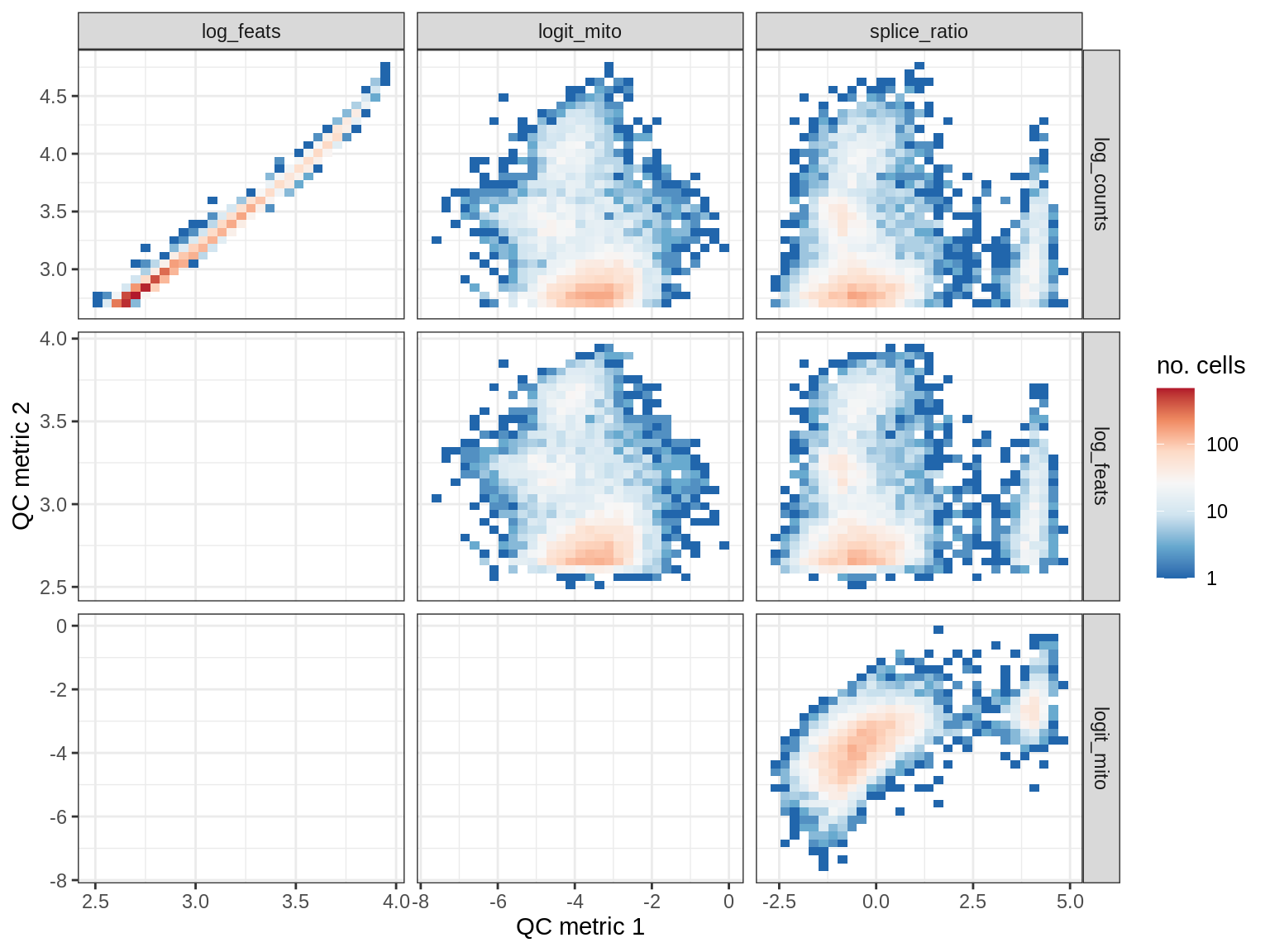
EU068
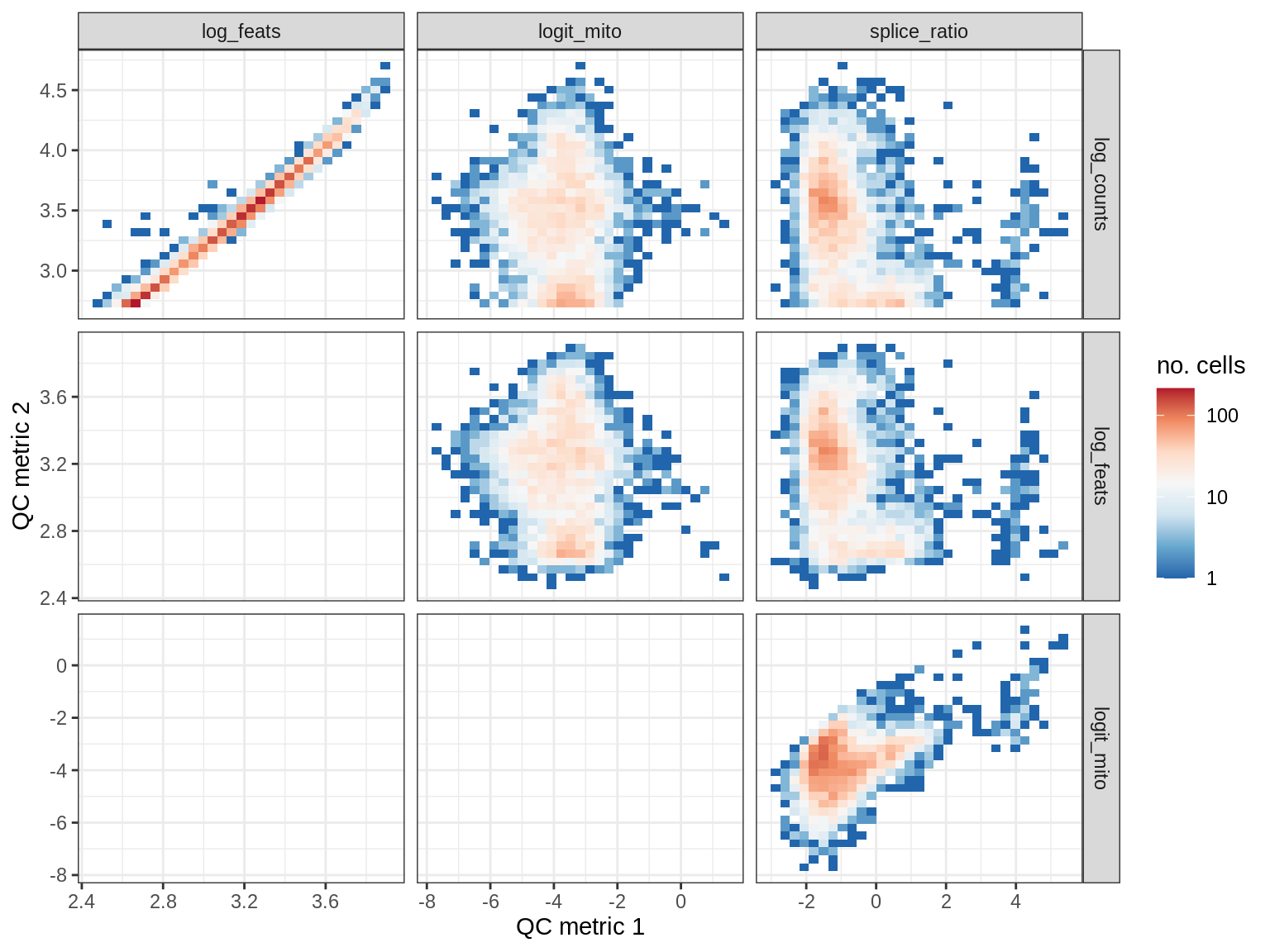
EU069
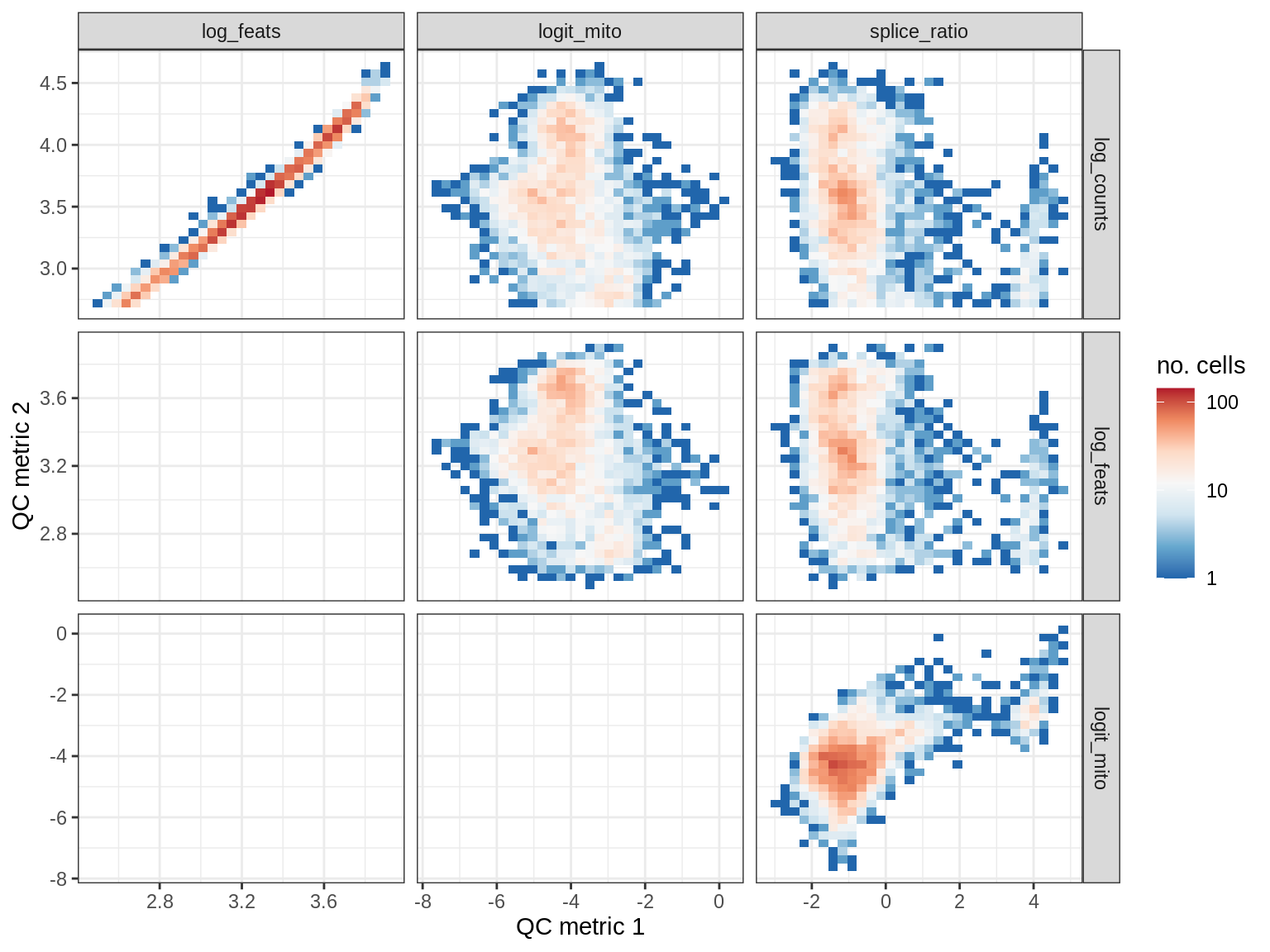
EU070
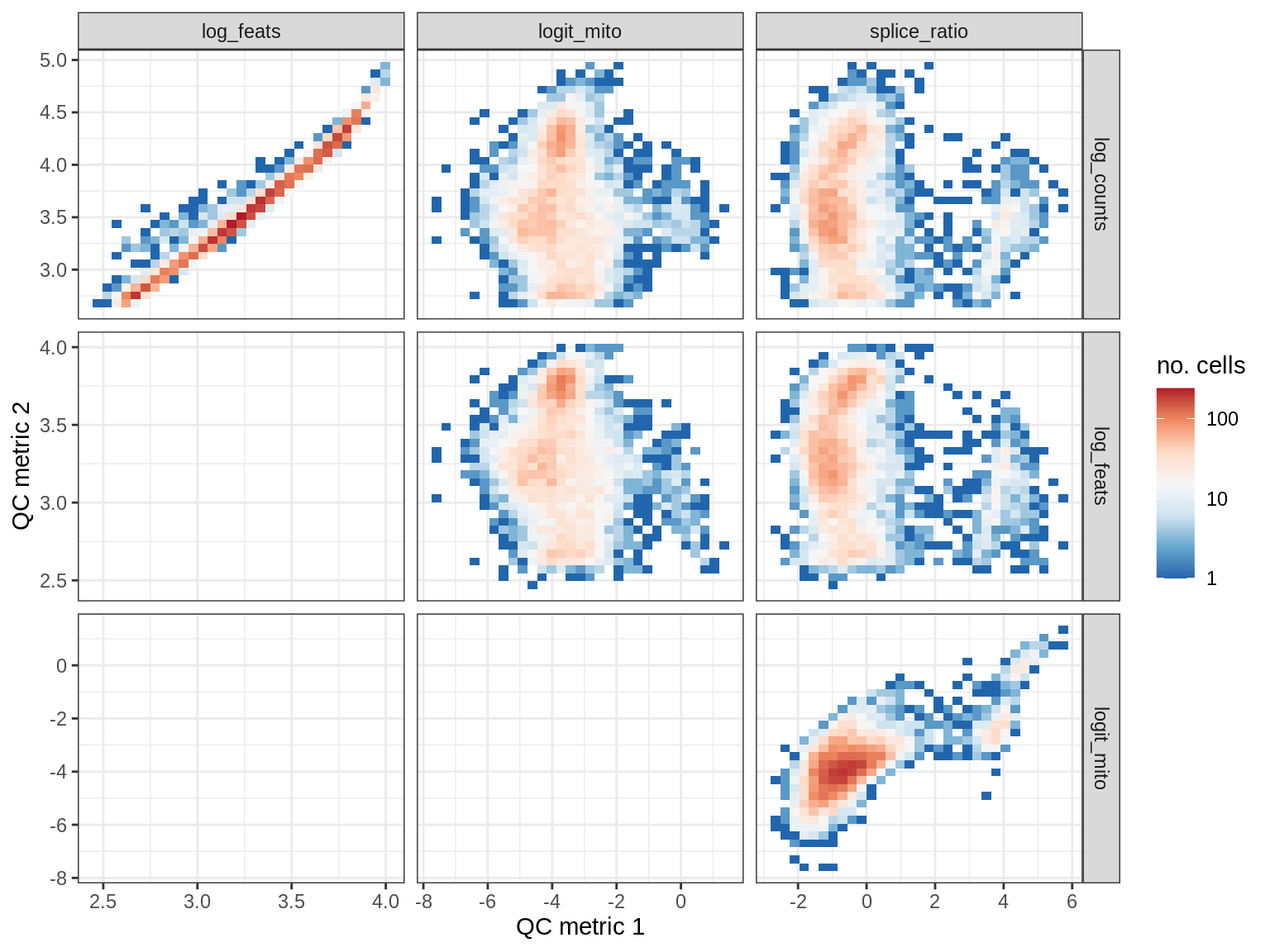
EU071
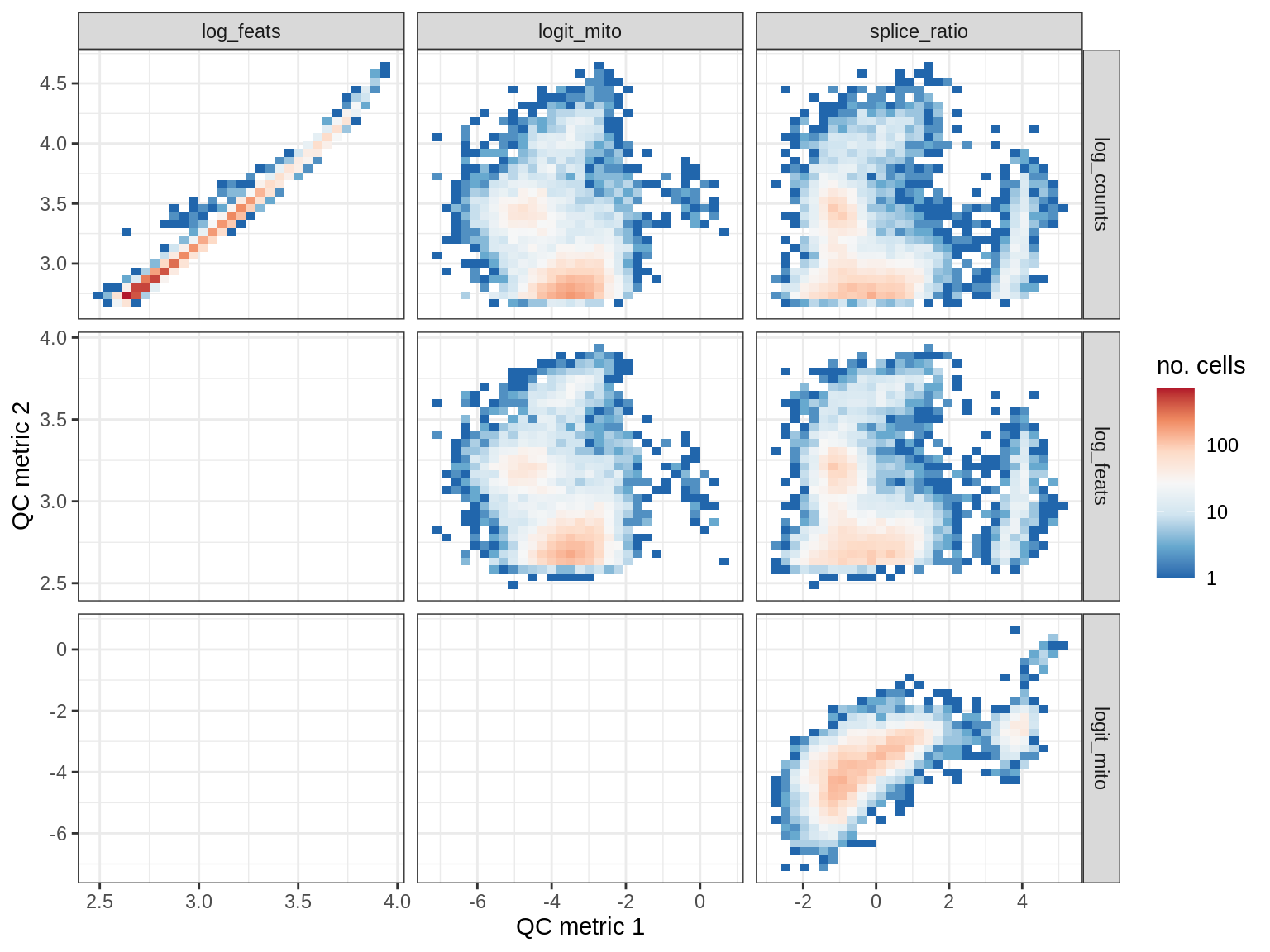
EU072
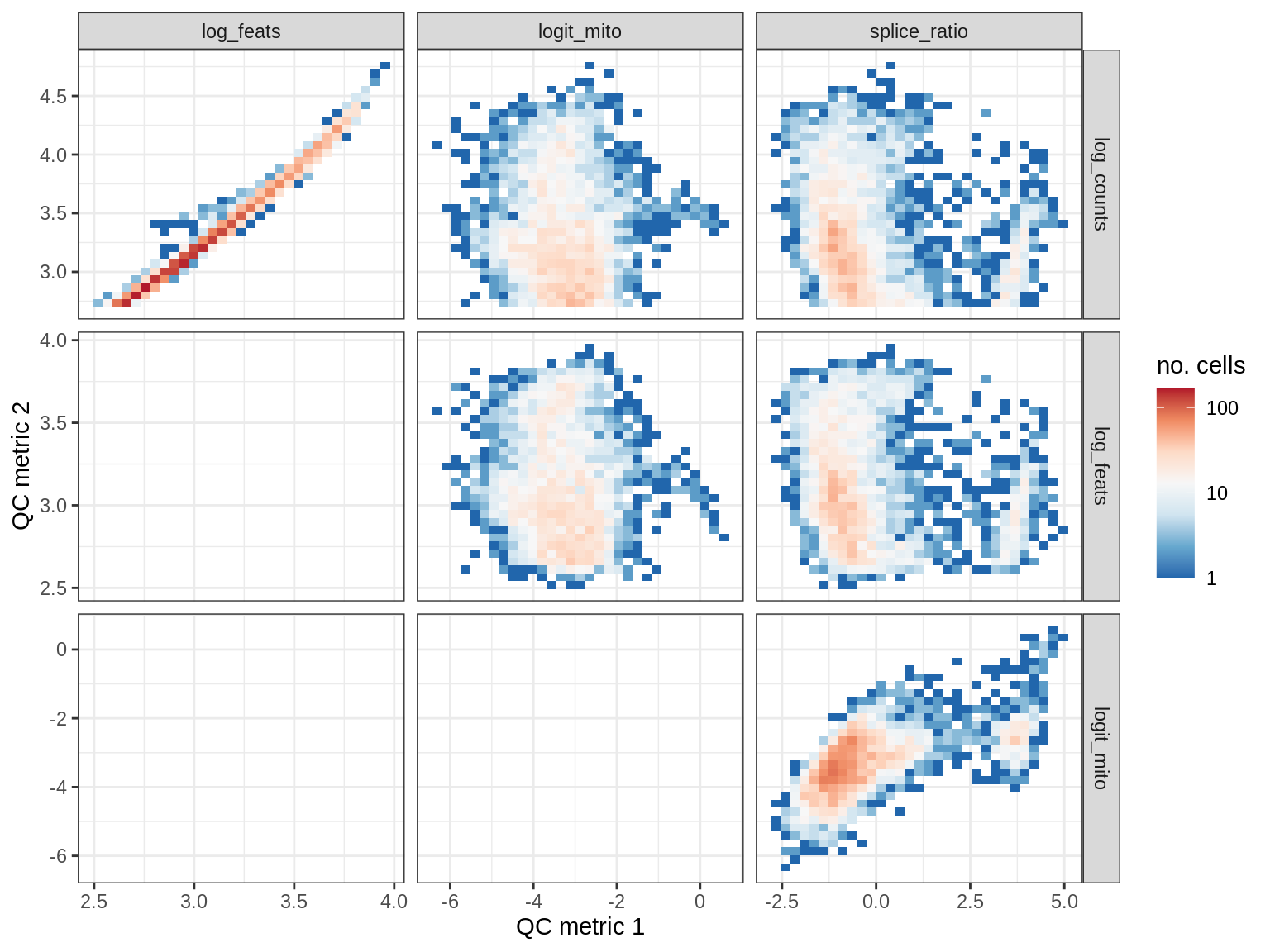
EU073
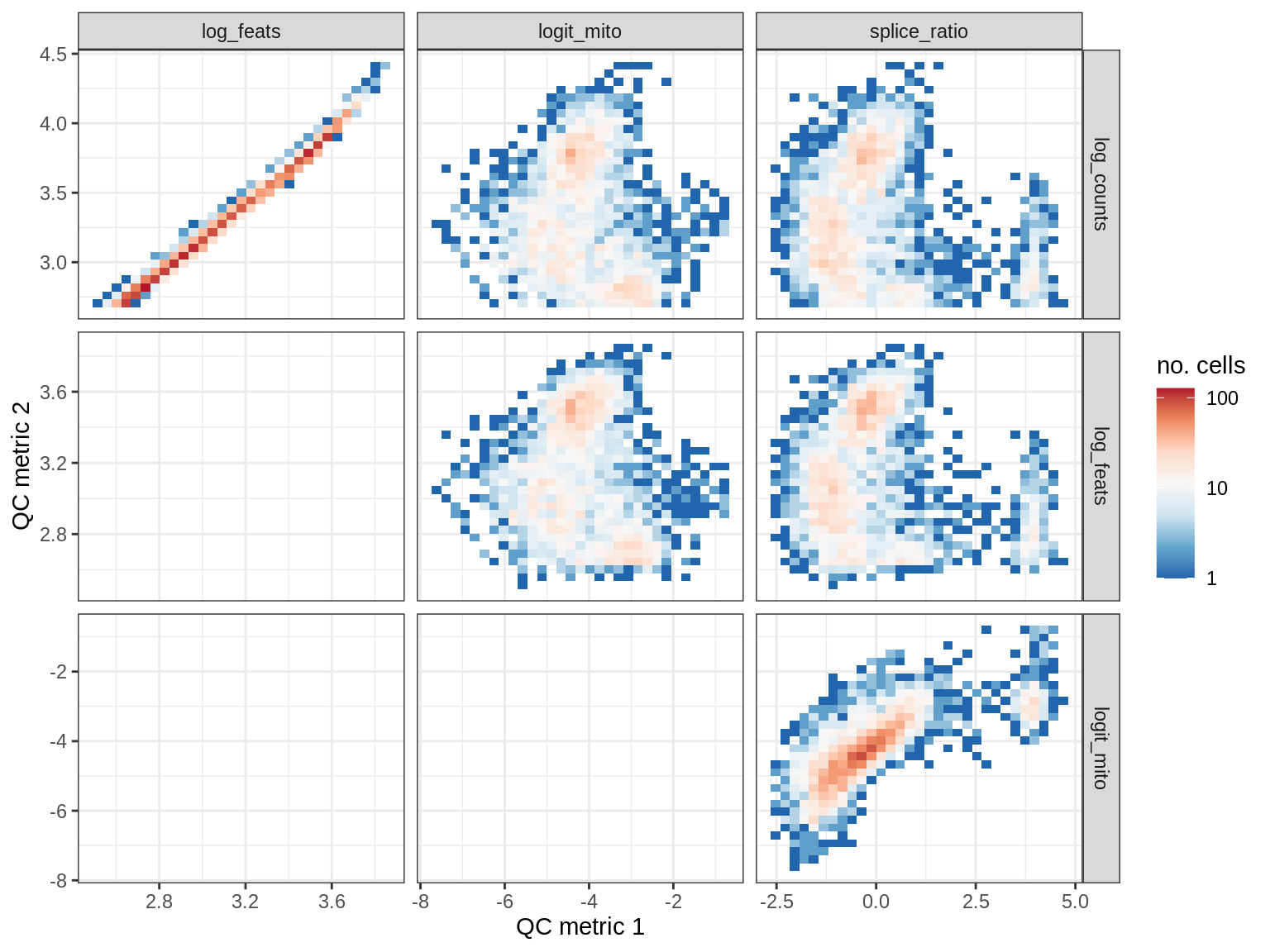
EU074
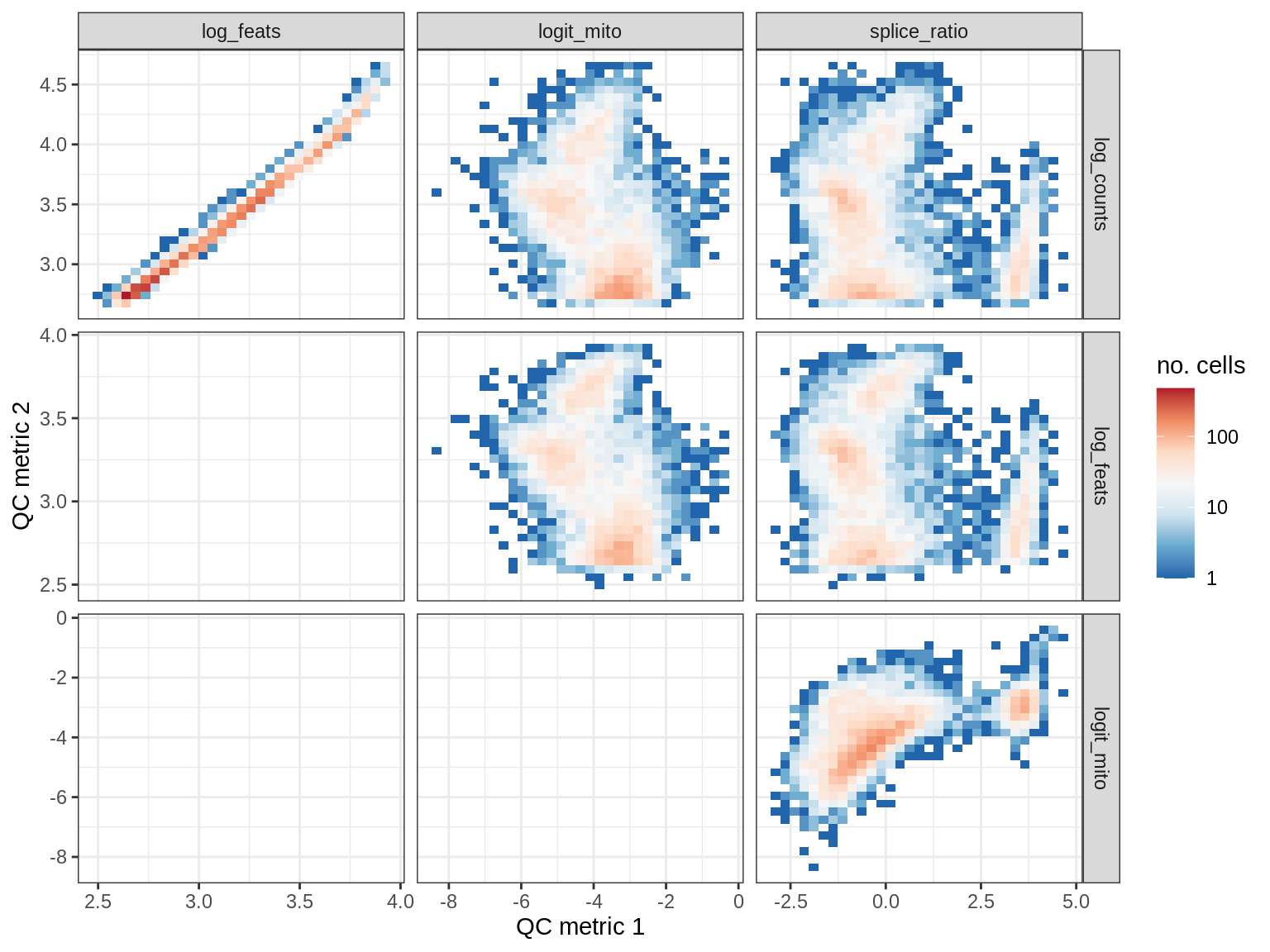
EU075
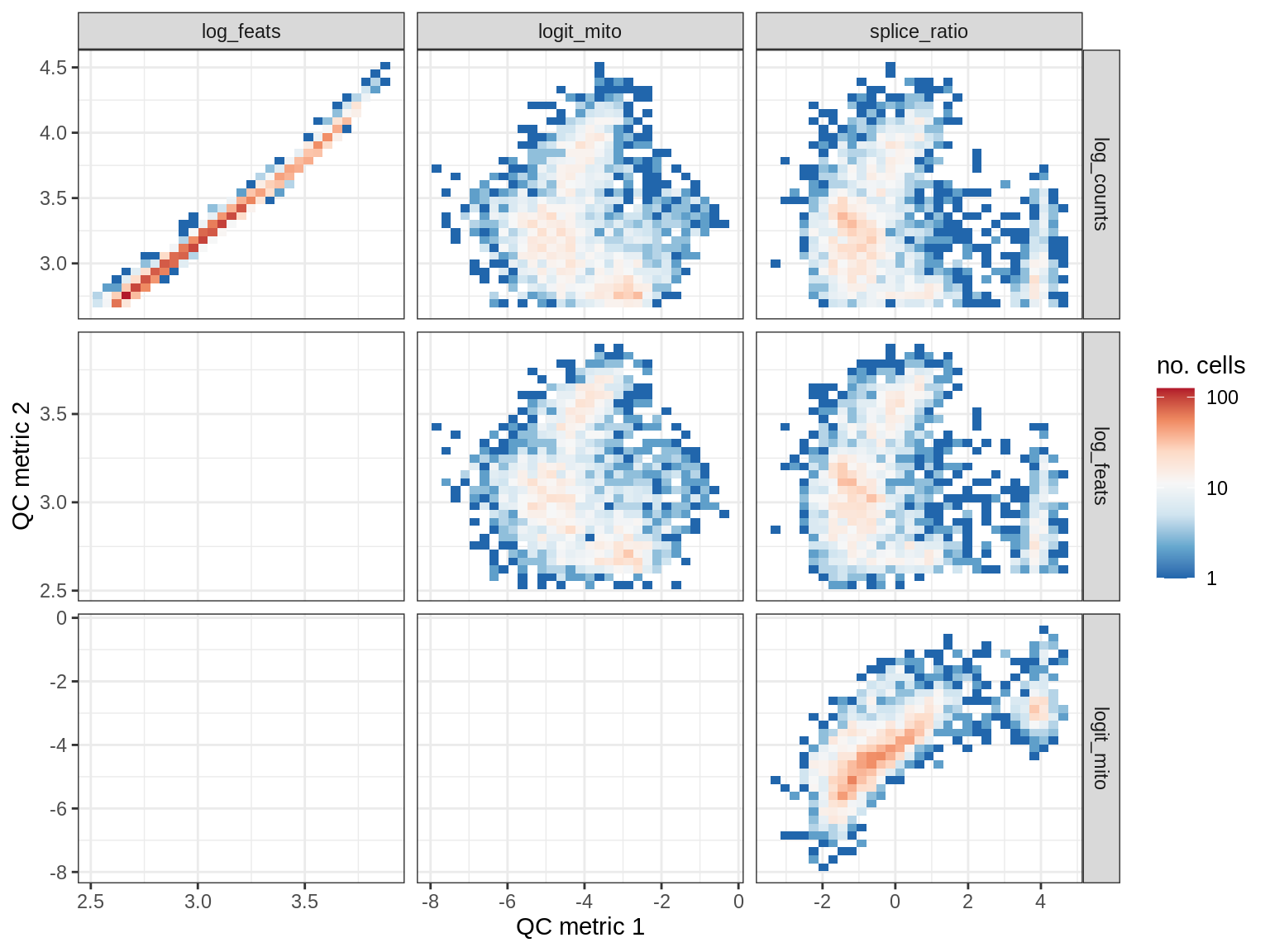
EU076
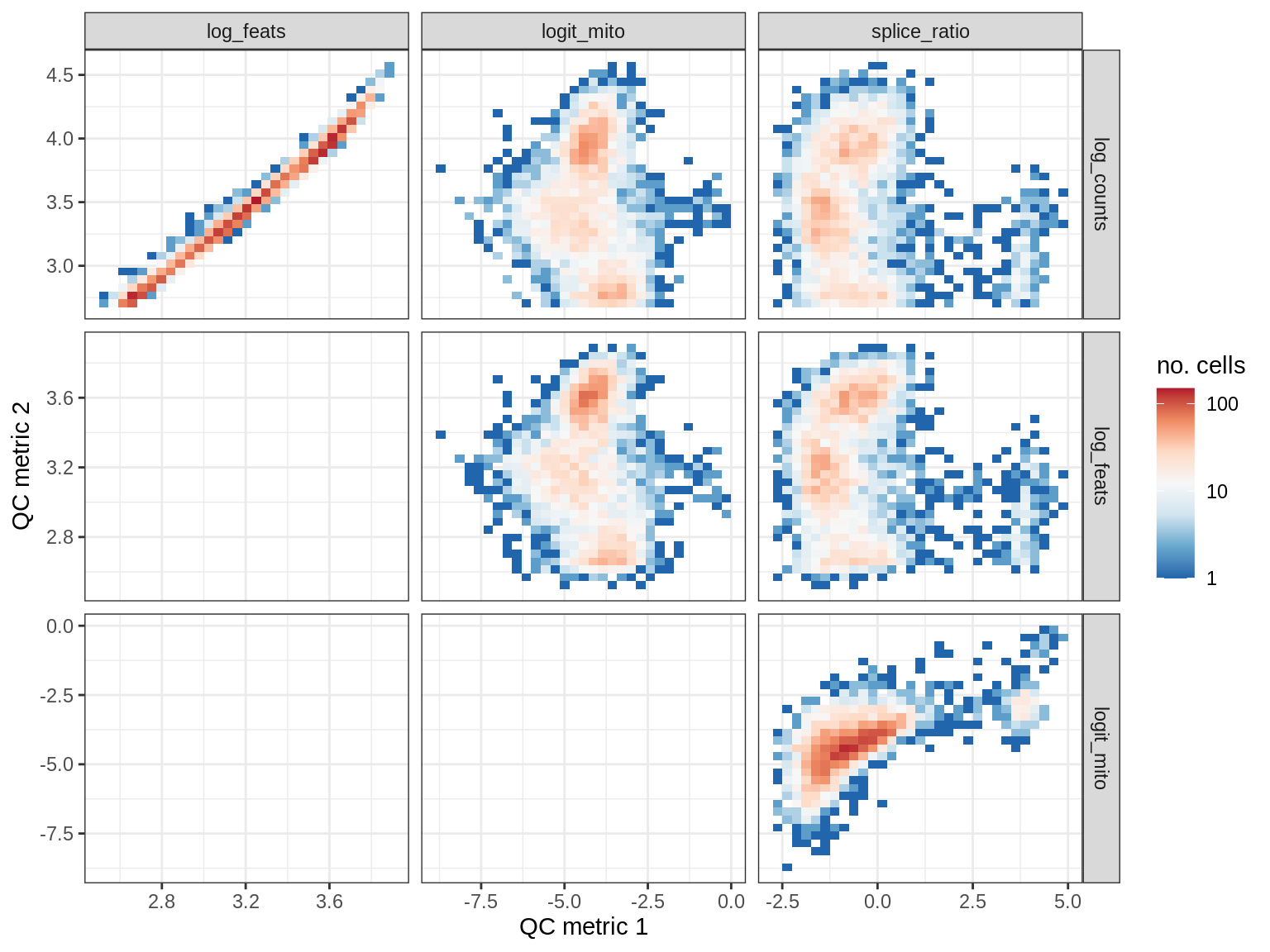
EU077
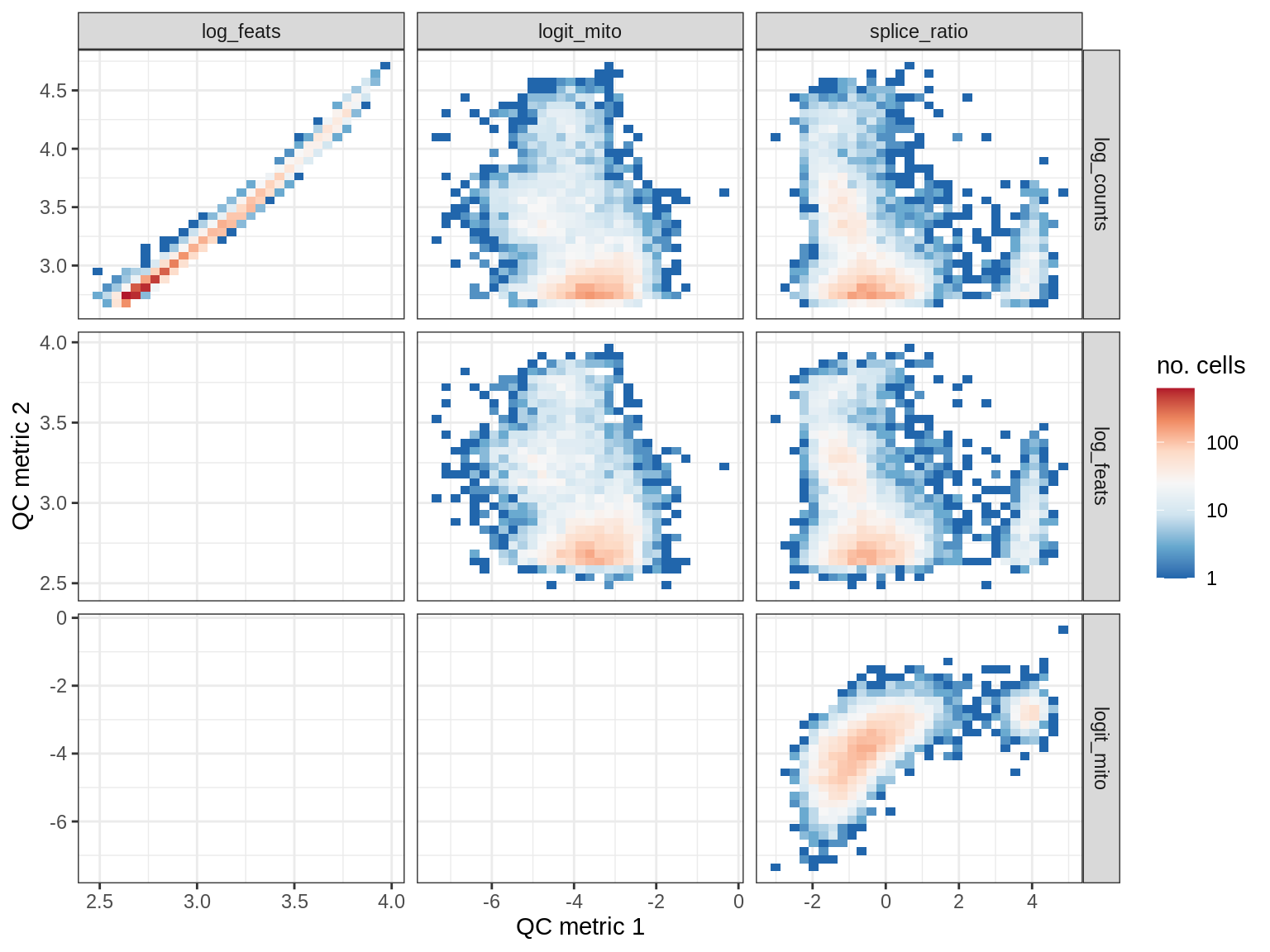
EU078
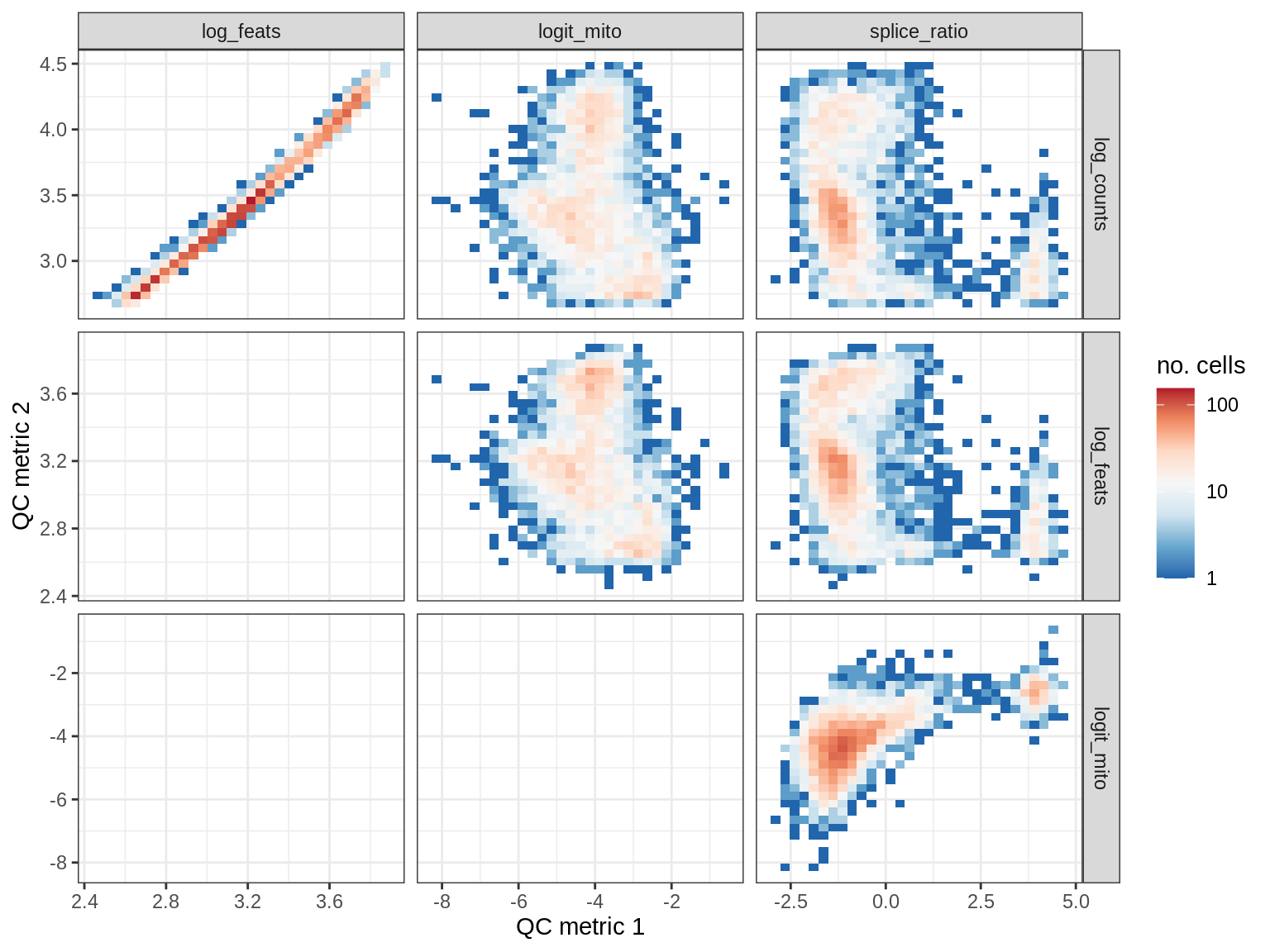
WM101
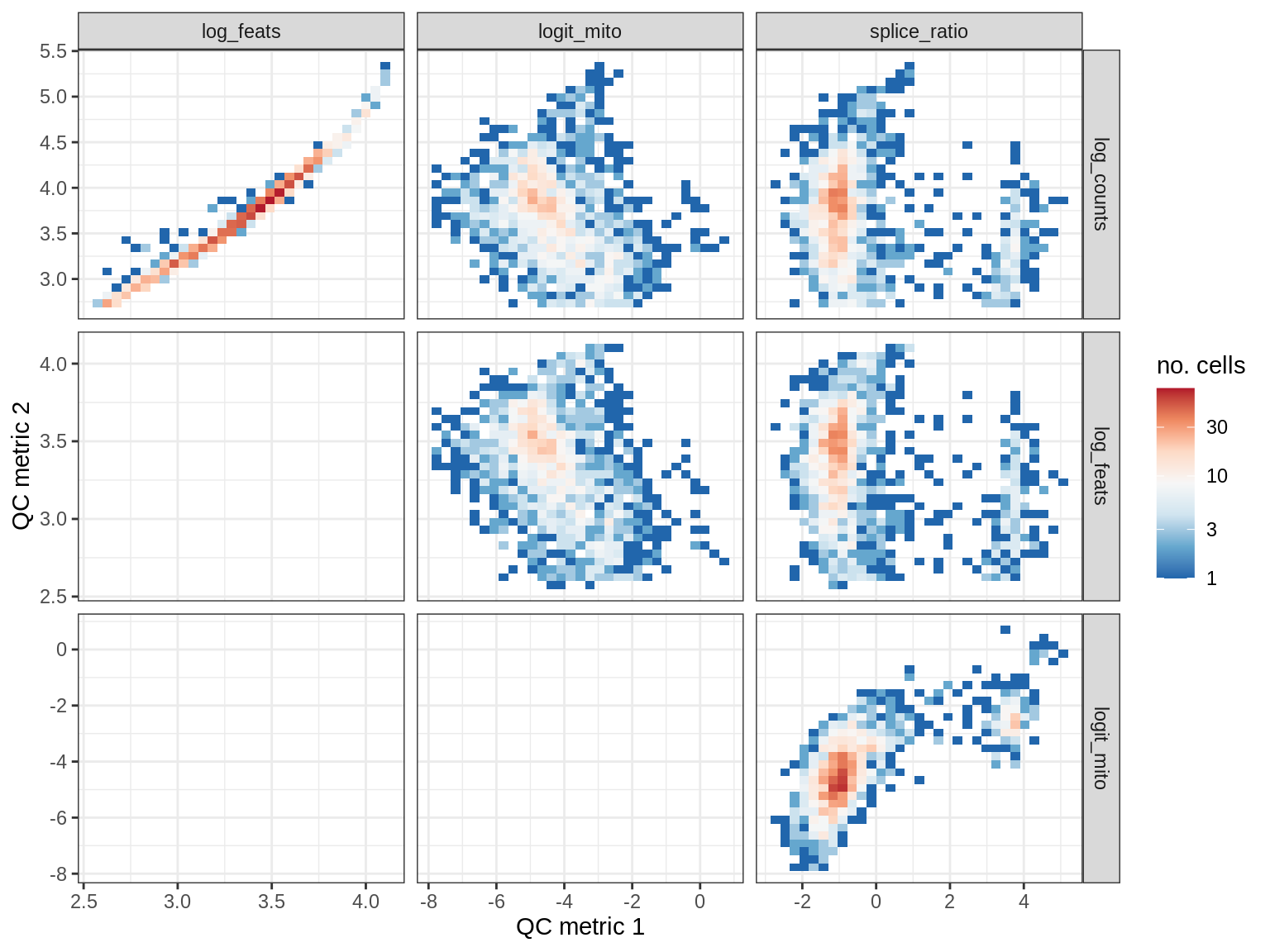
WM102
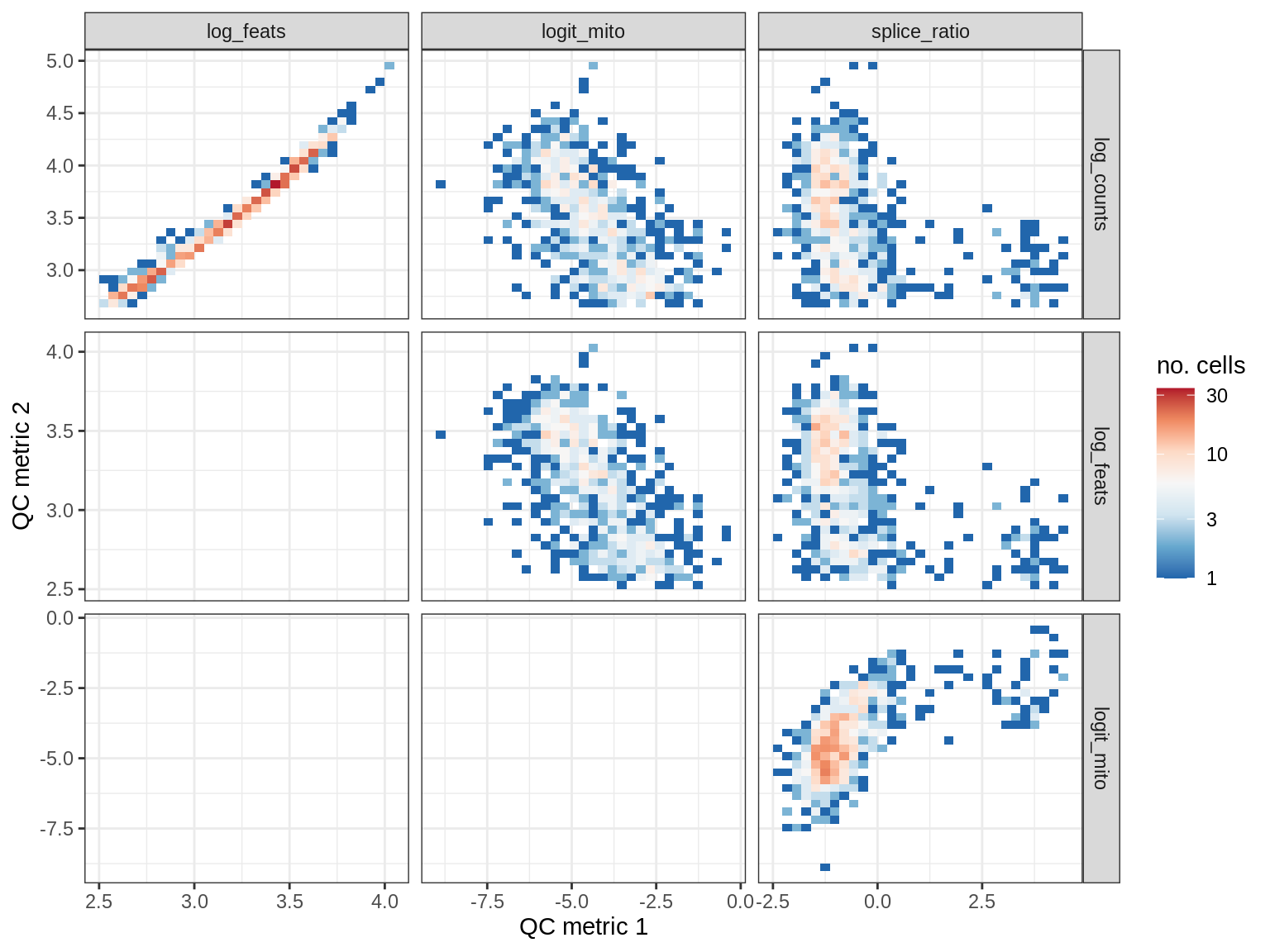
WM103
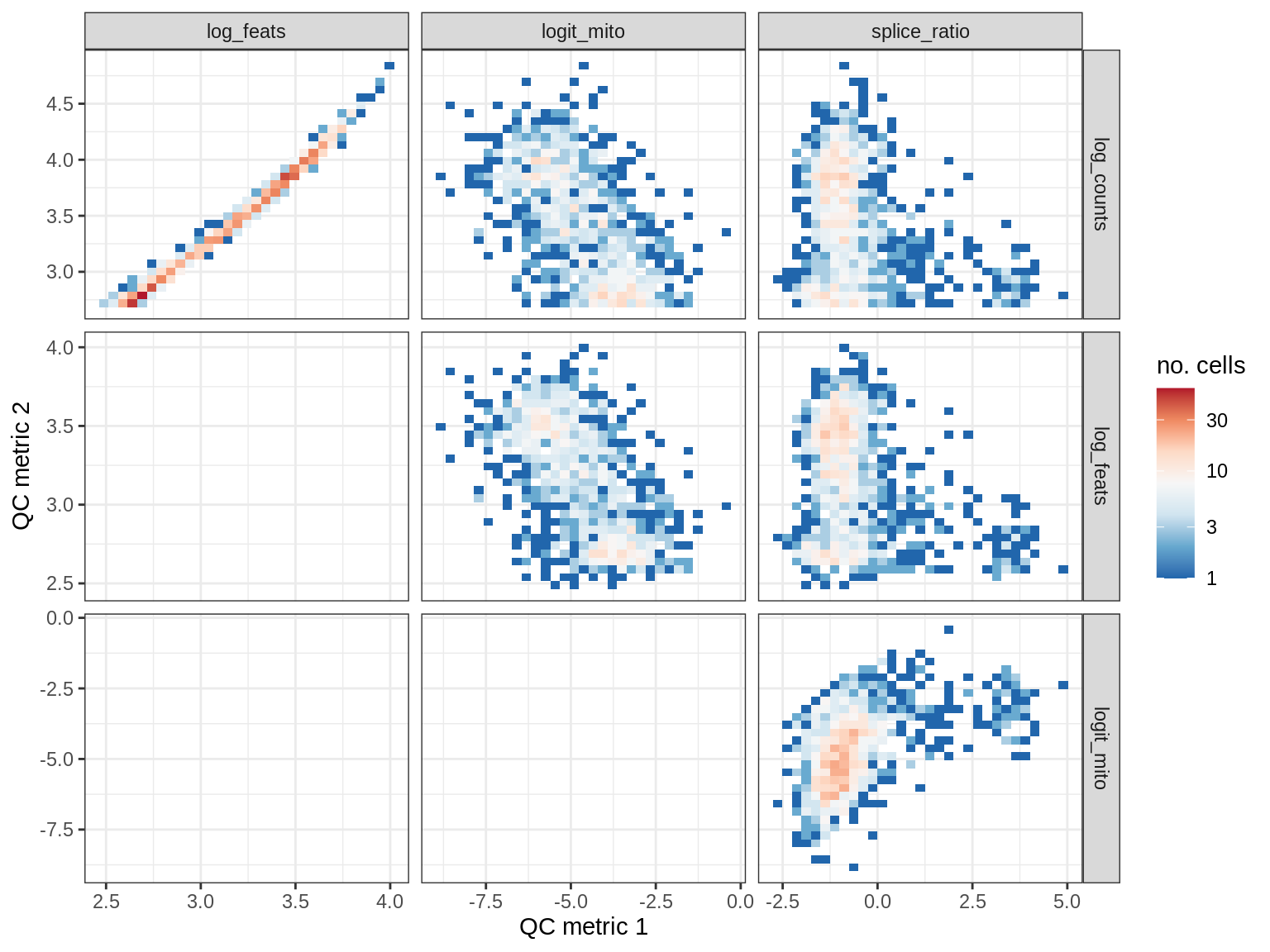
WM104
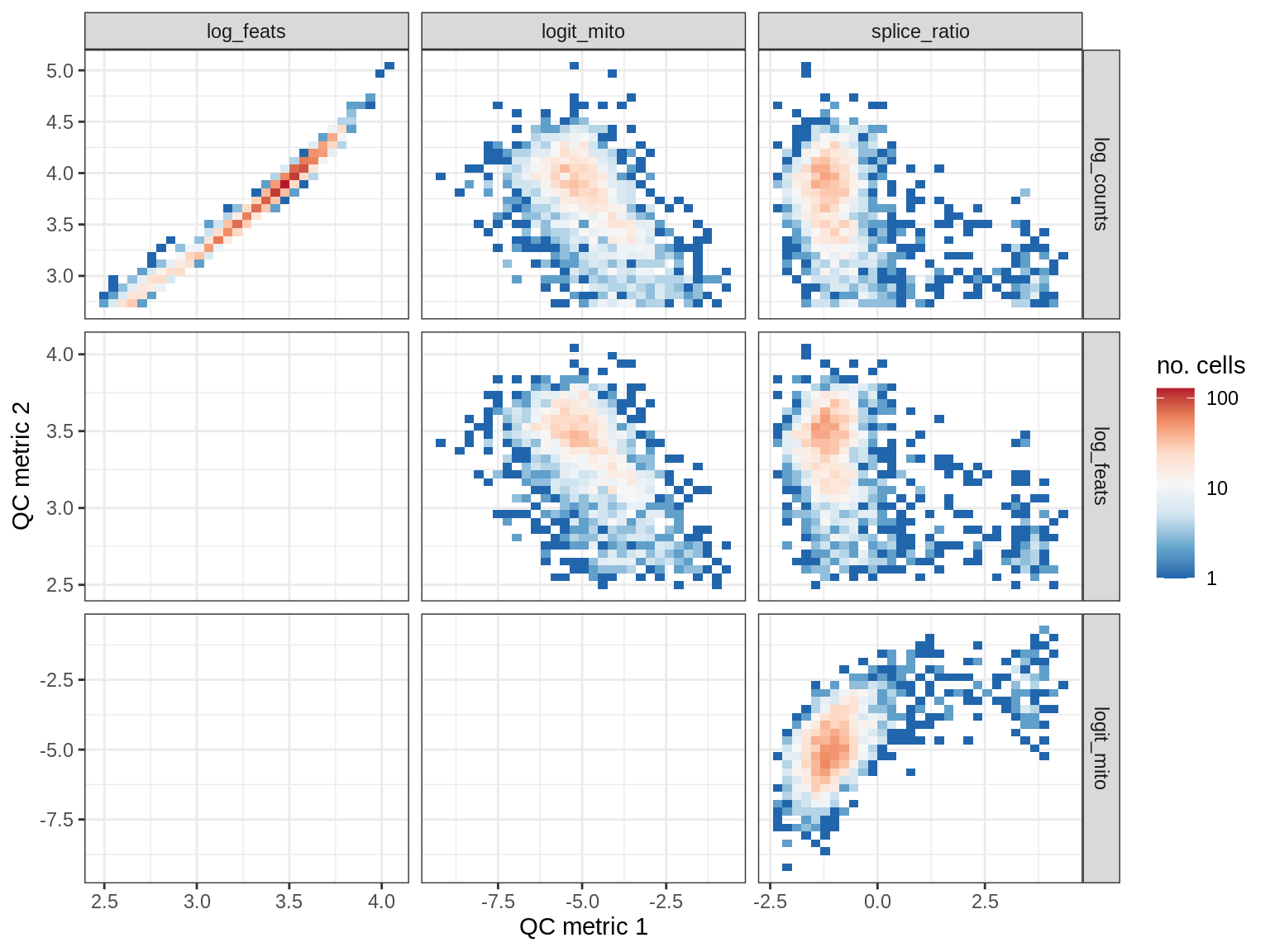
WM105
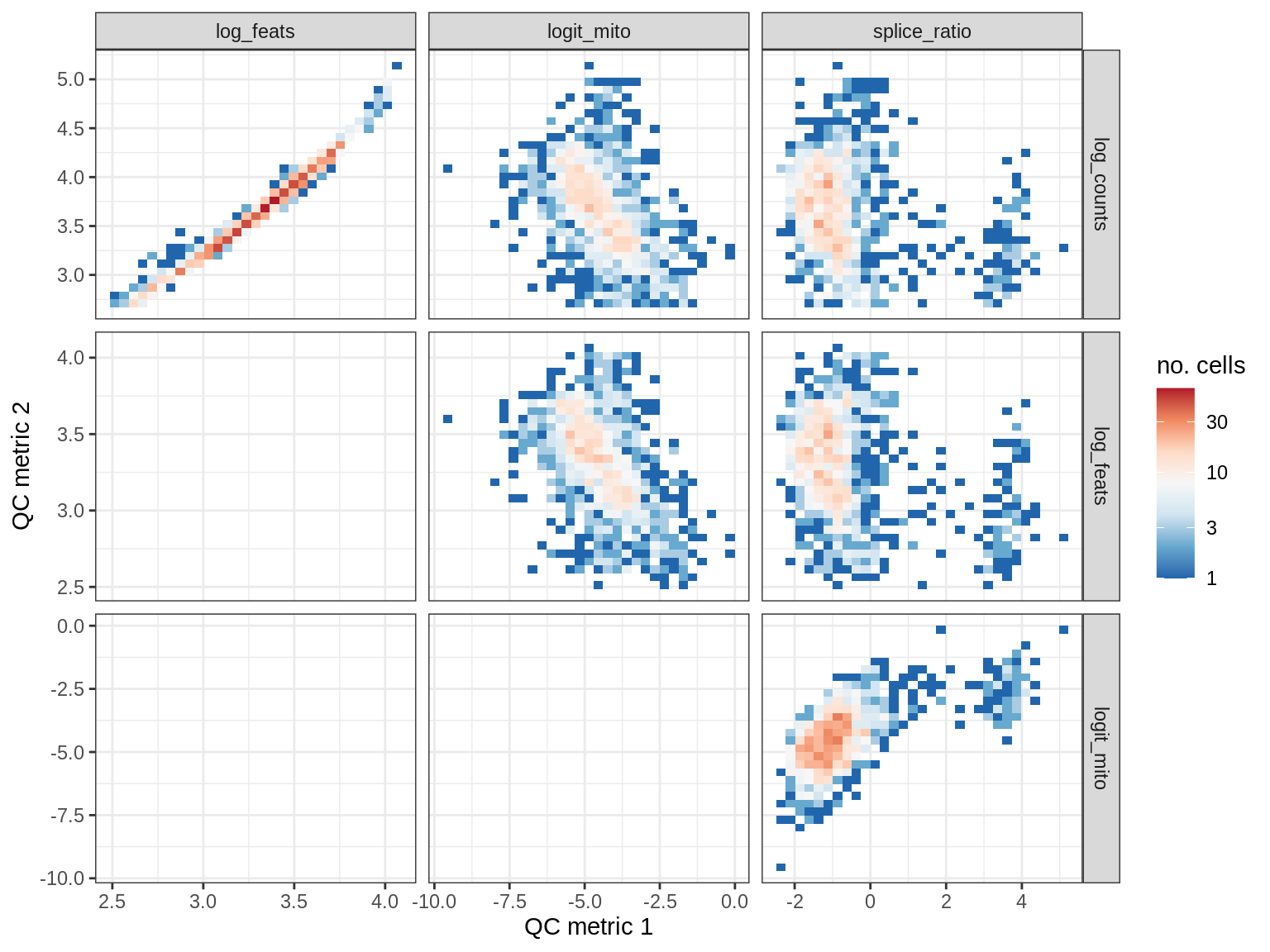
WM106
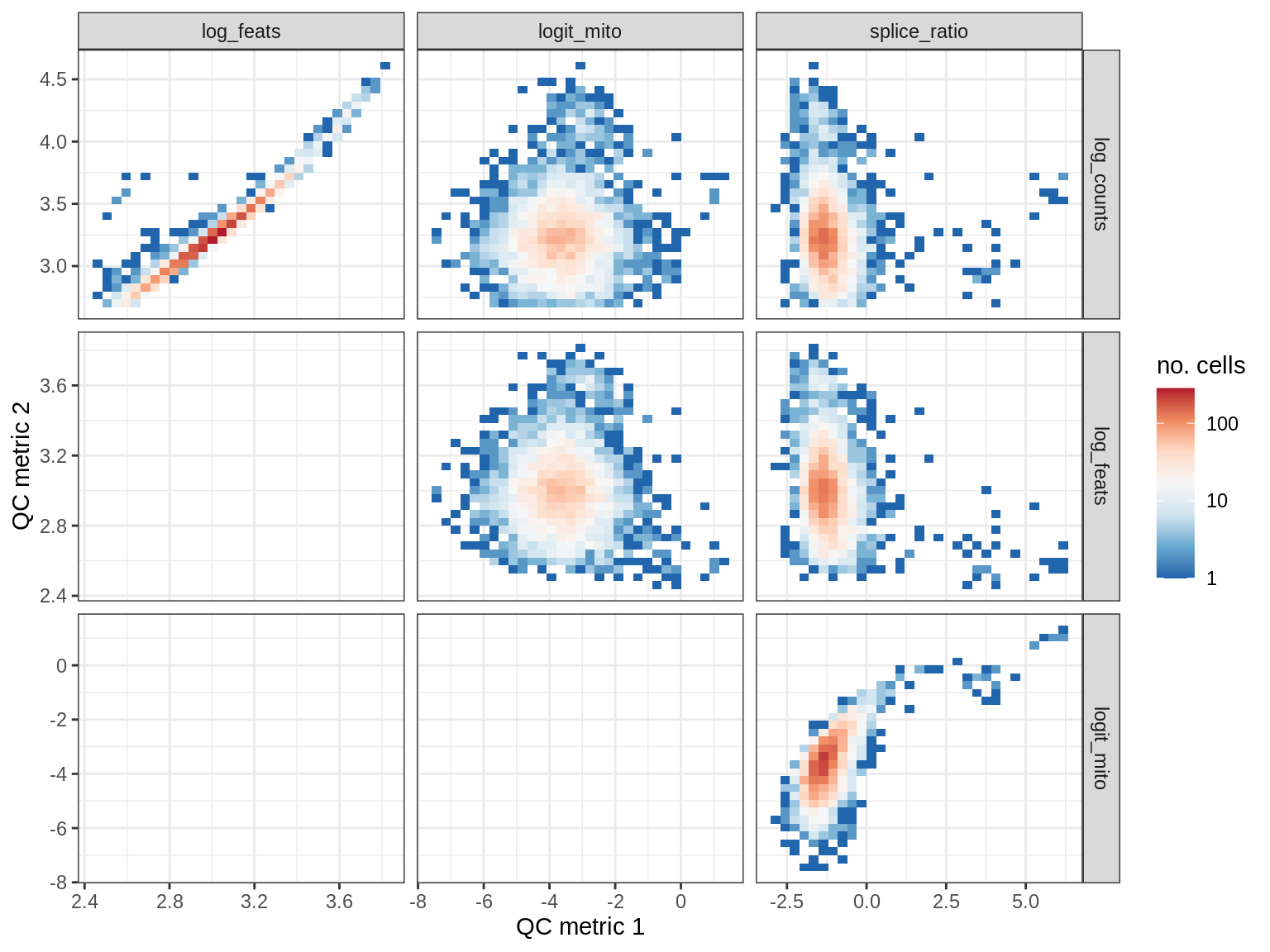
WM107
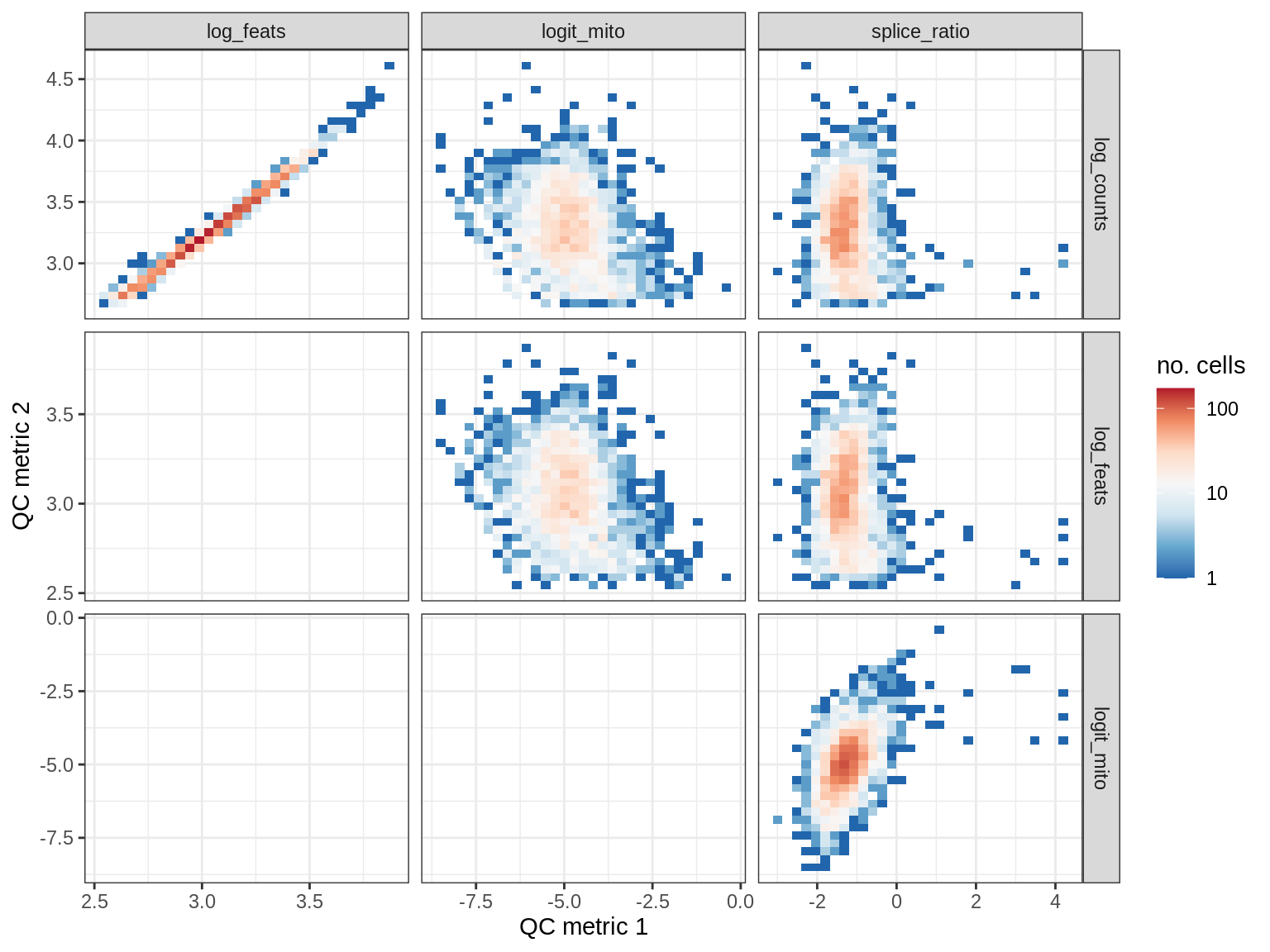
WM108
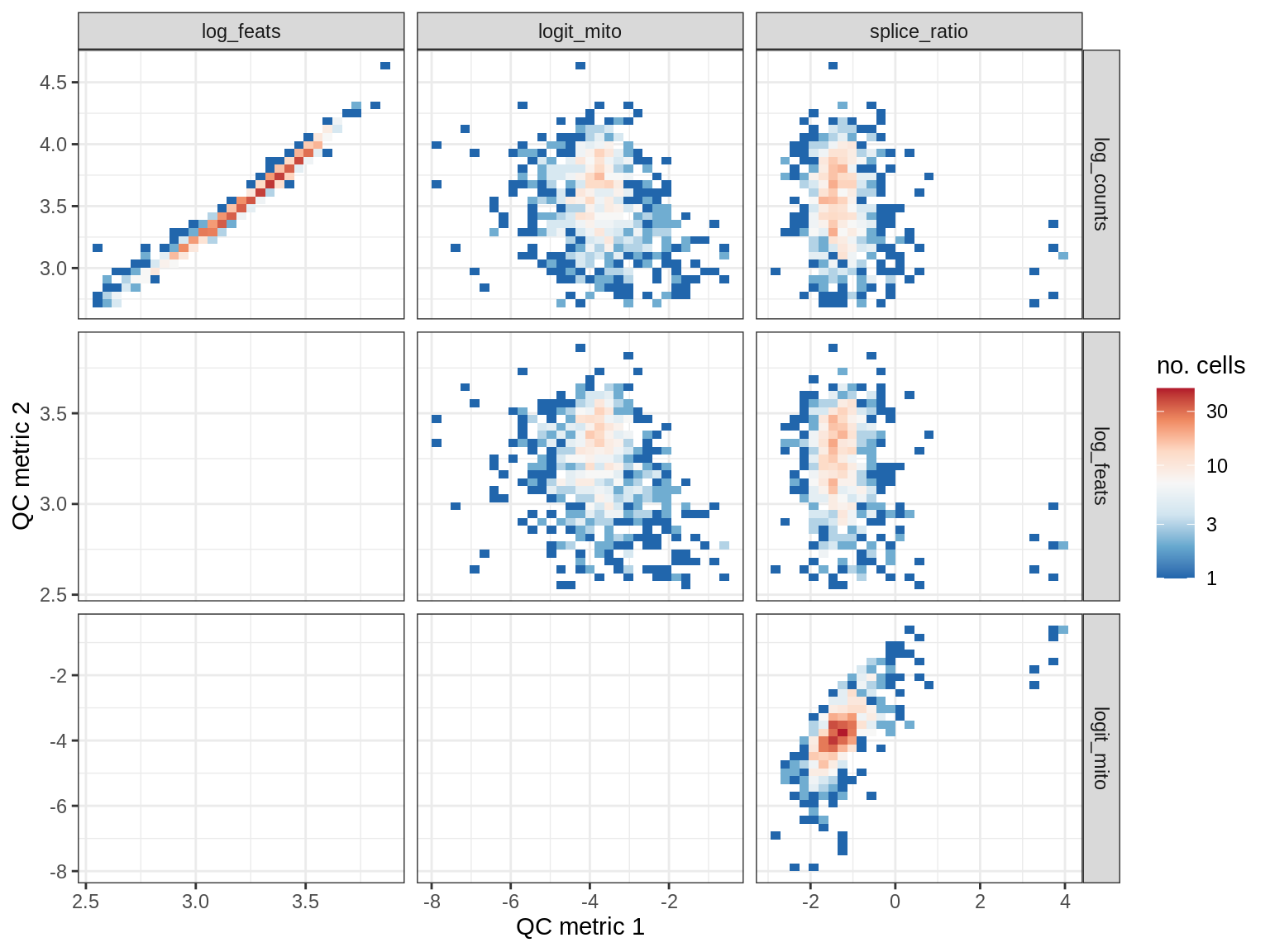
WM109
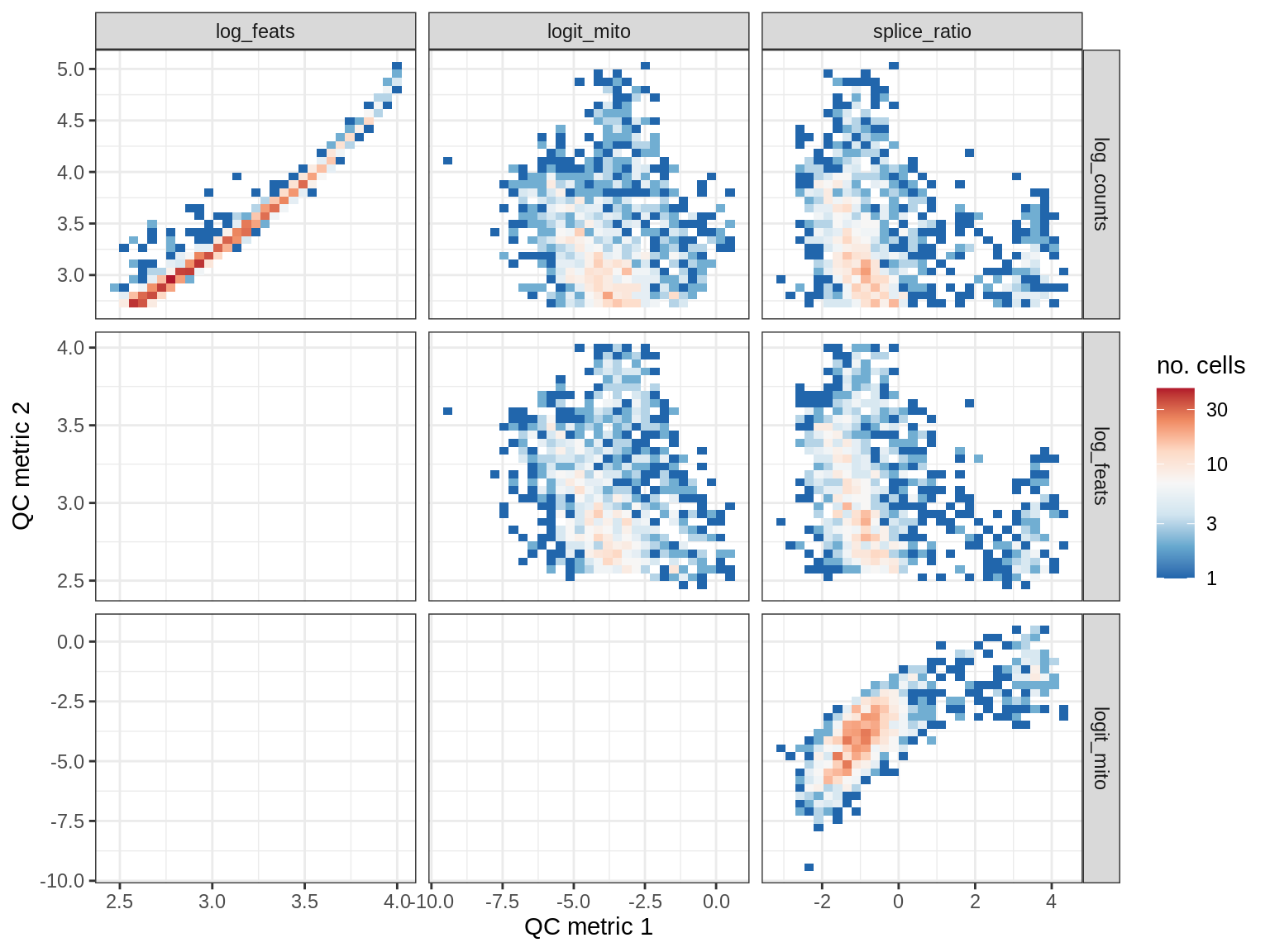
WM110
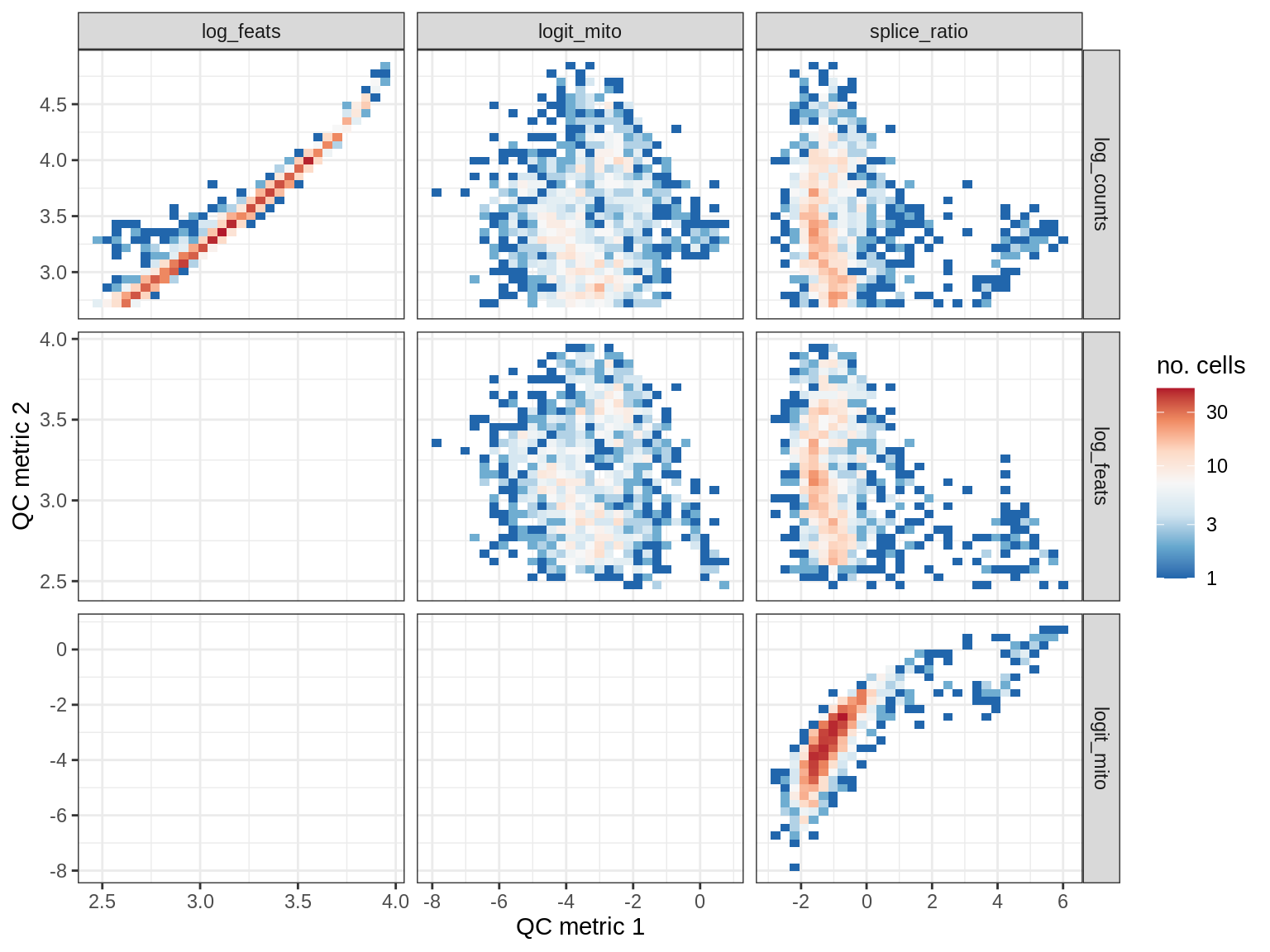
WM112
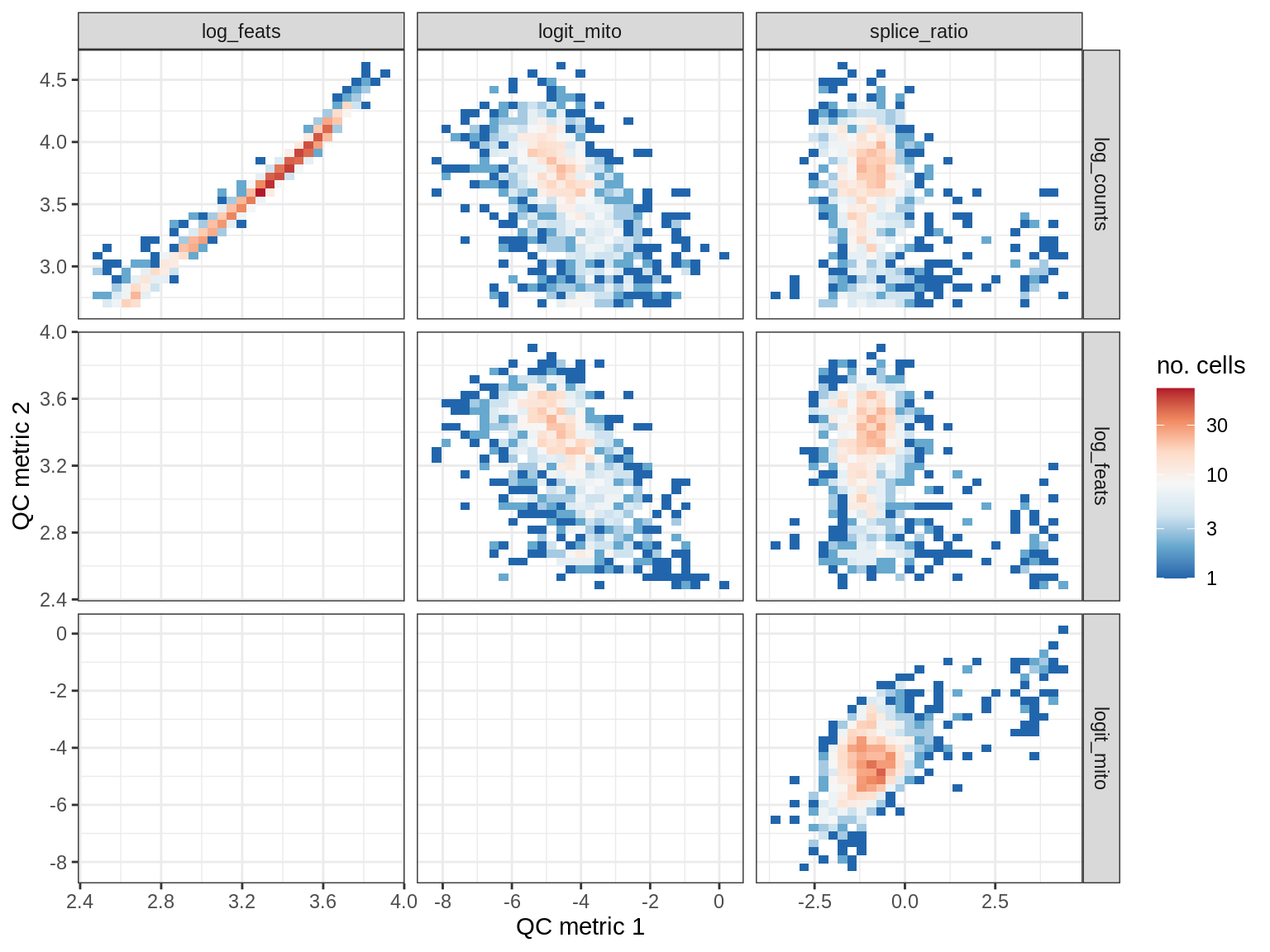
WM113
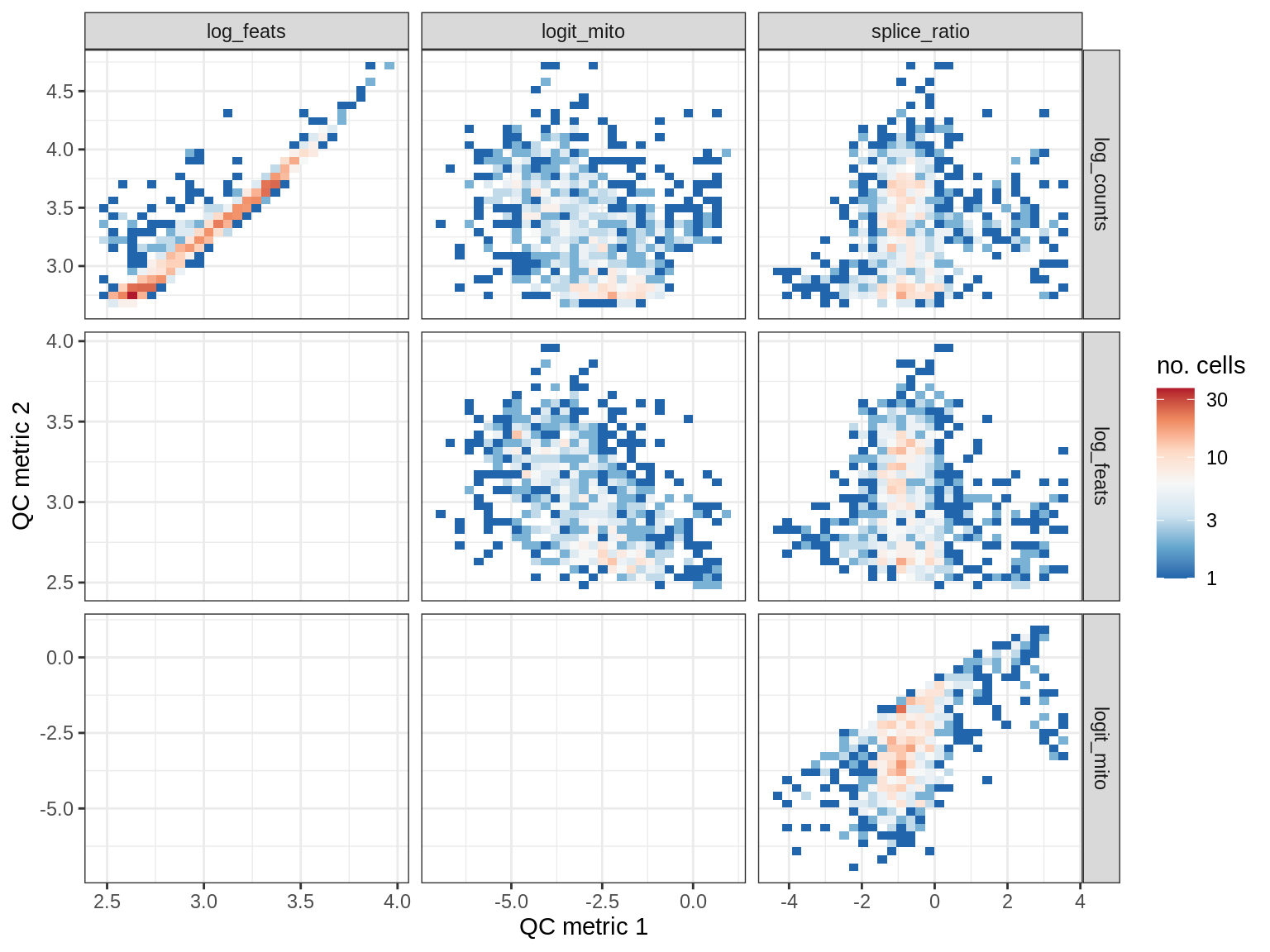
WM116
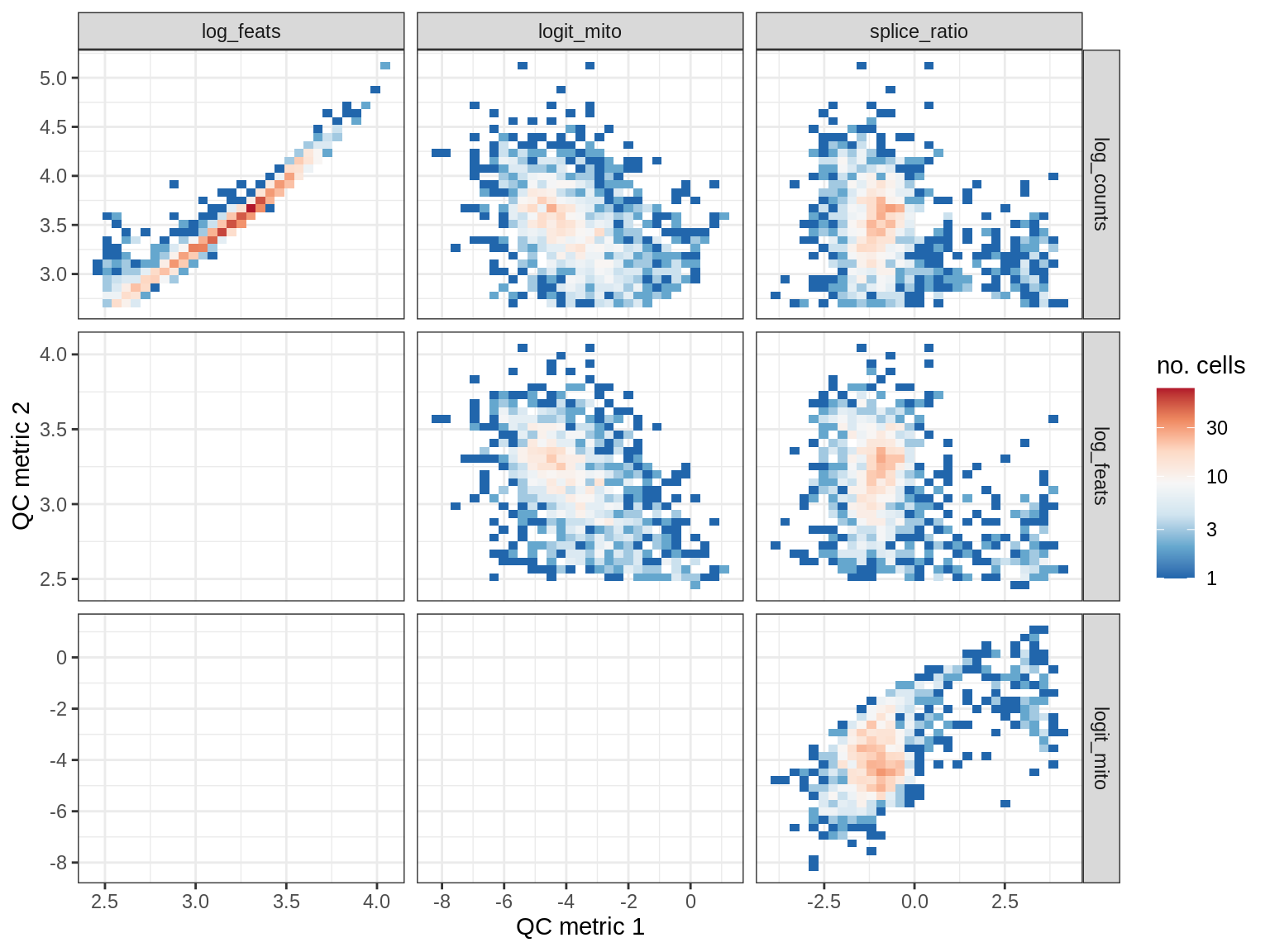
WM117
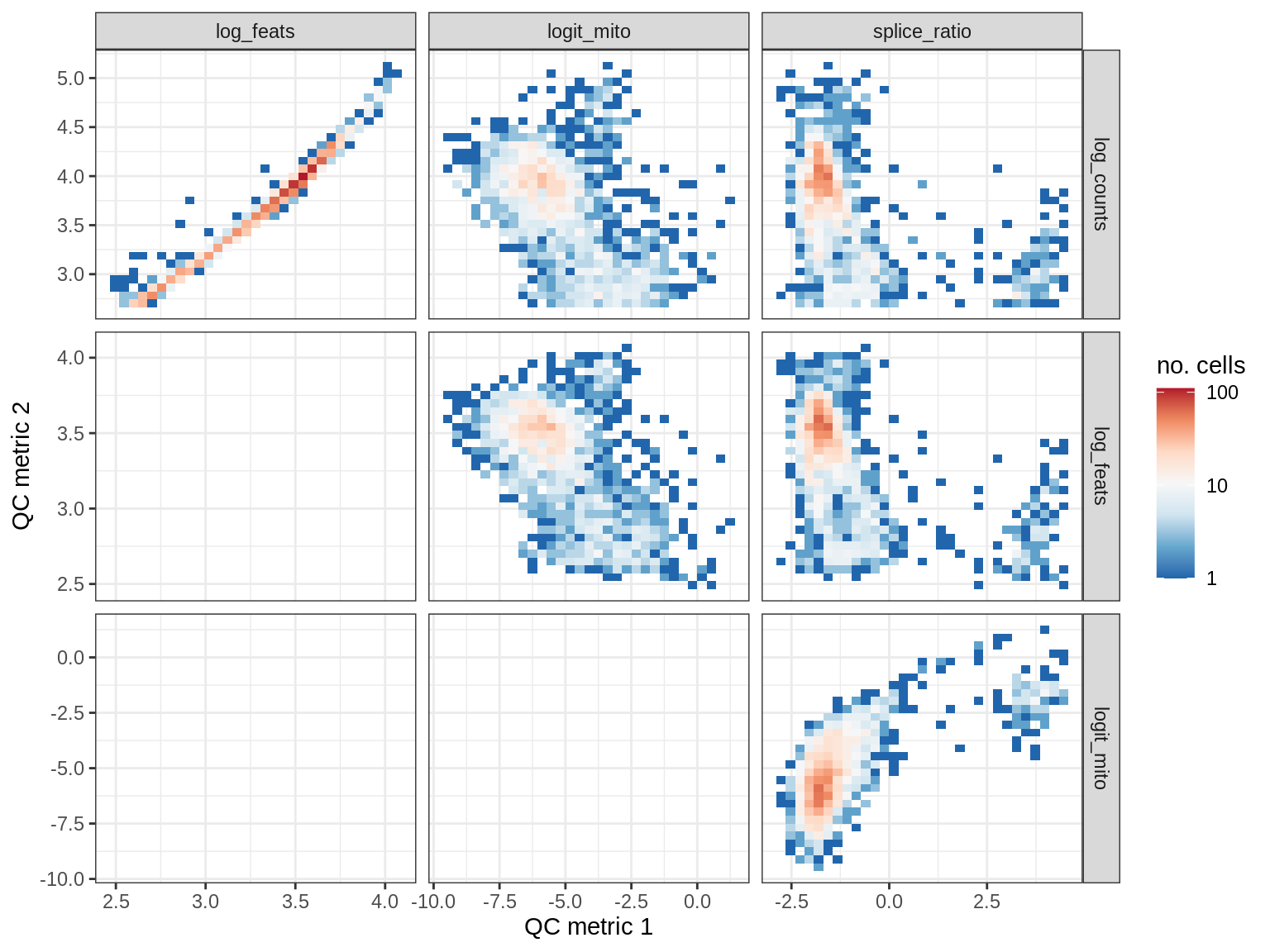
WM118
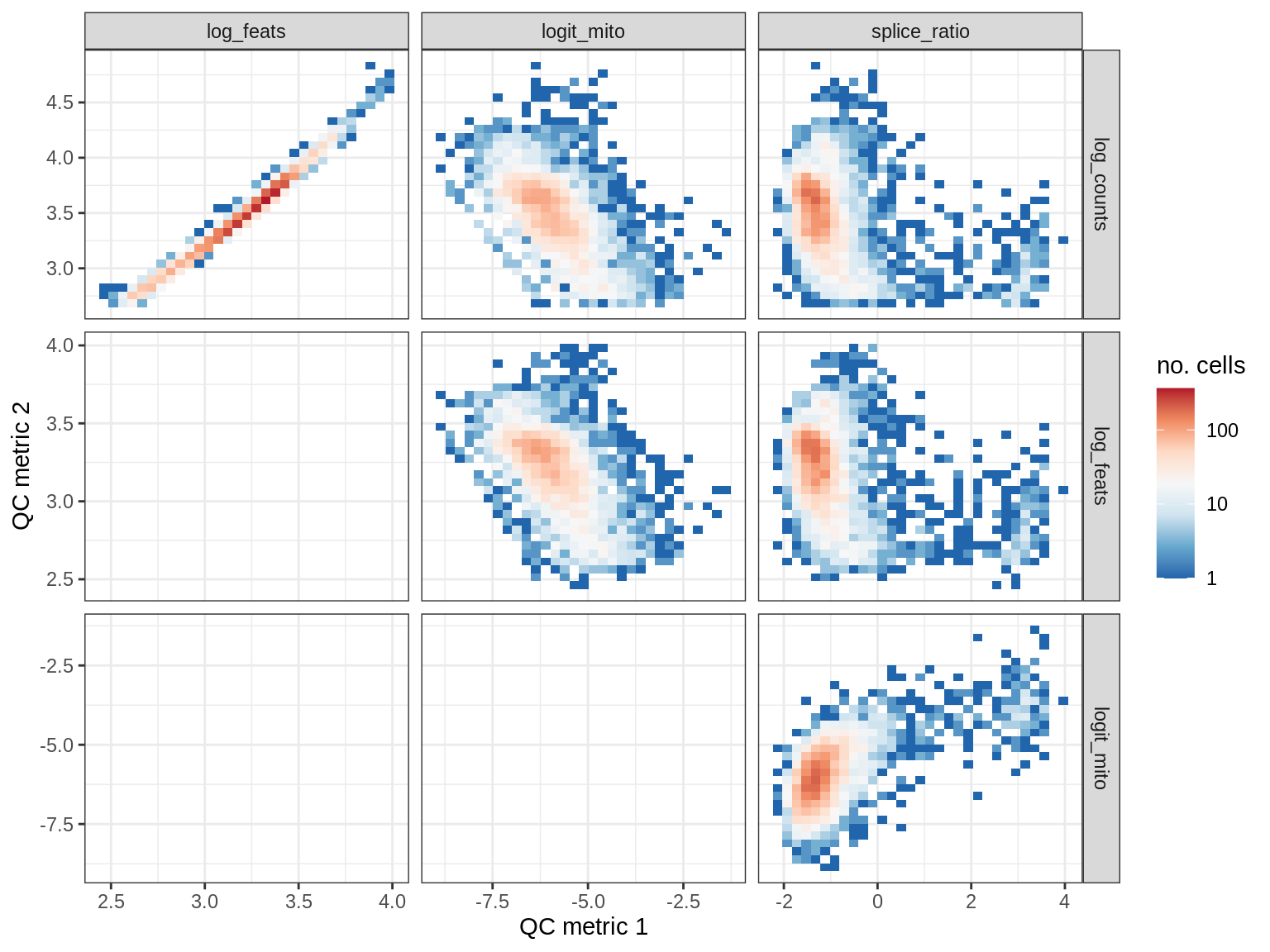
WM119
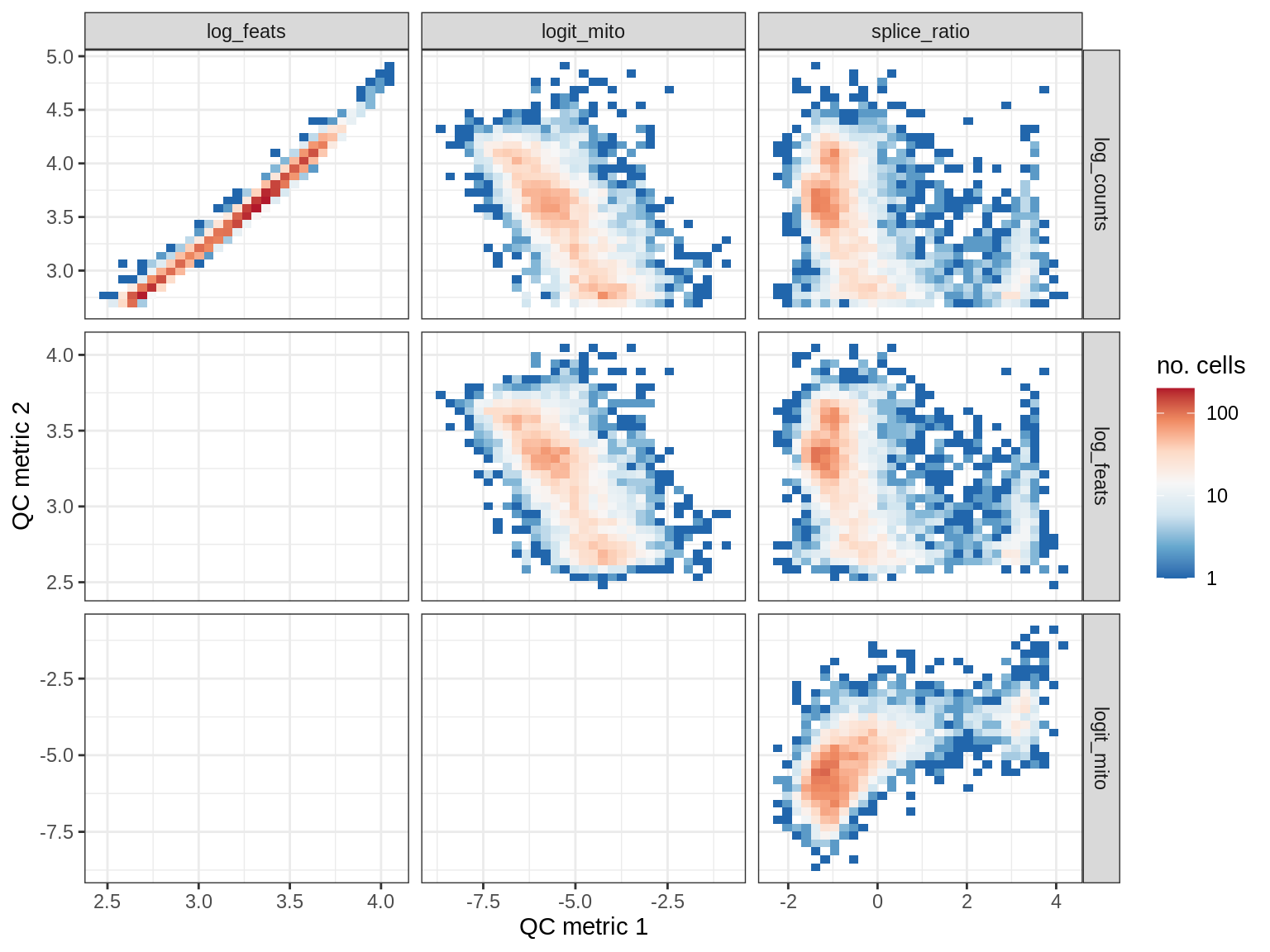
WM120
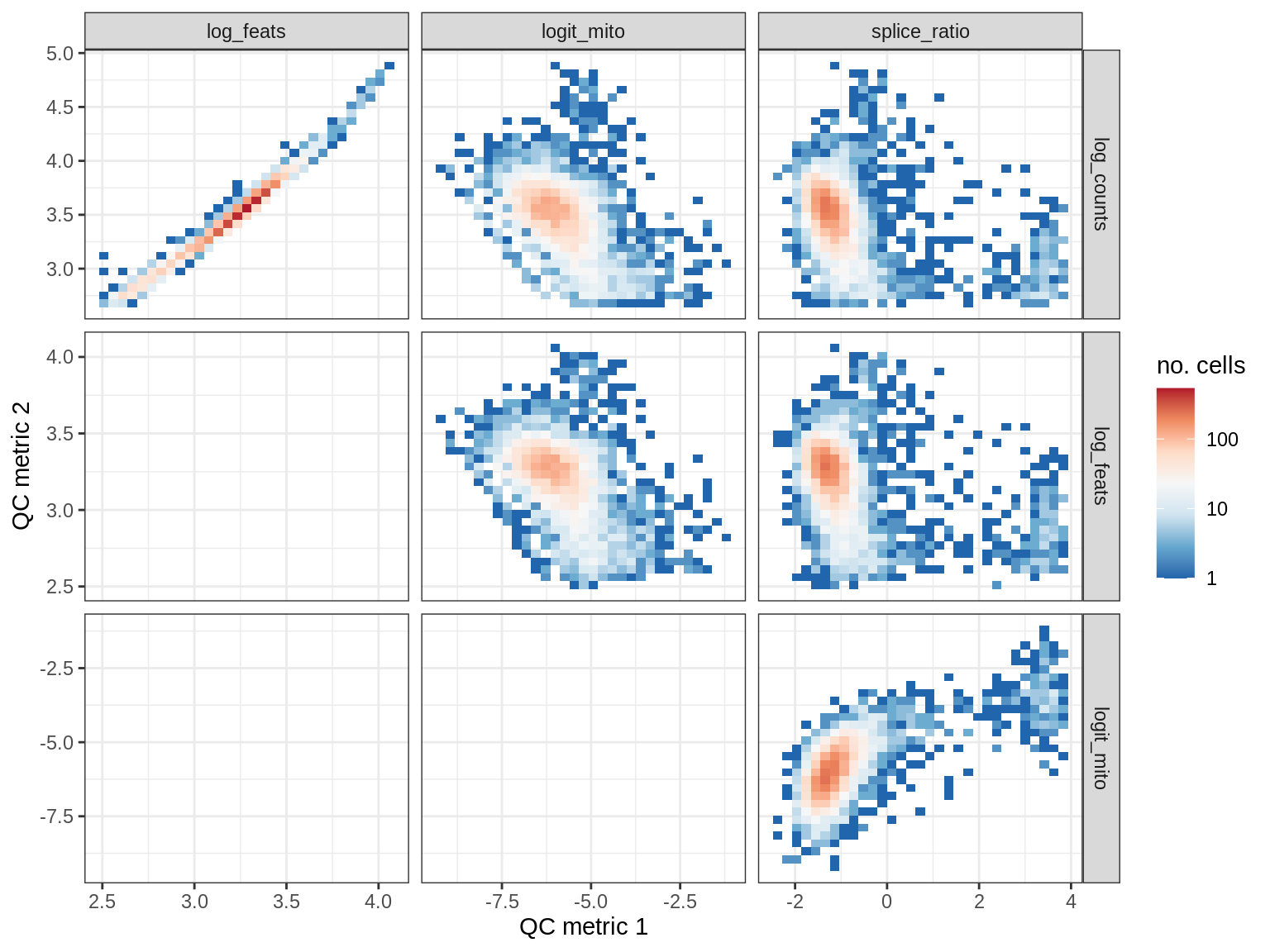
WM121
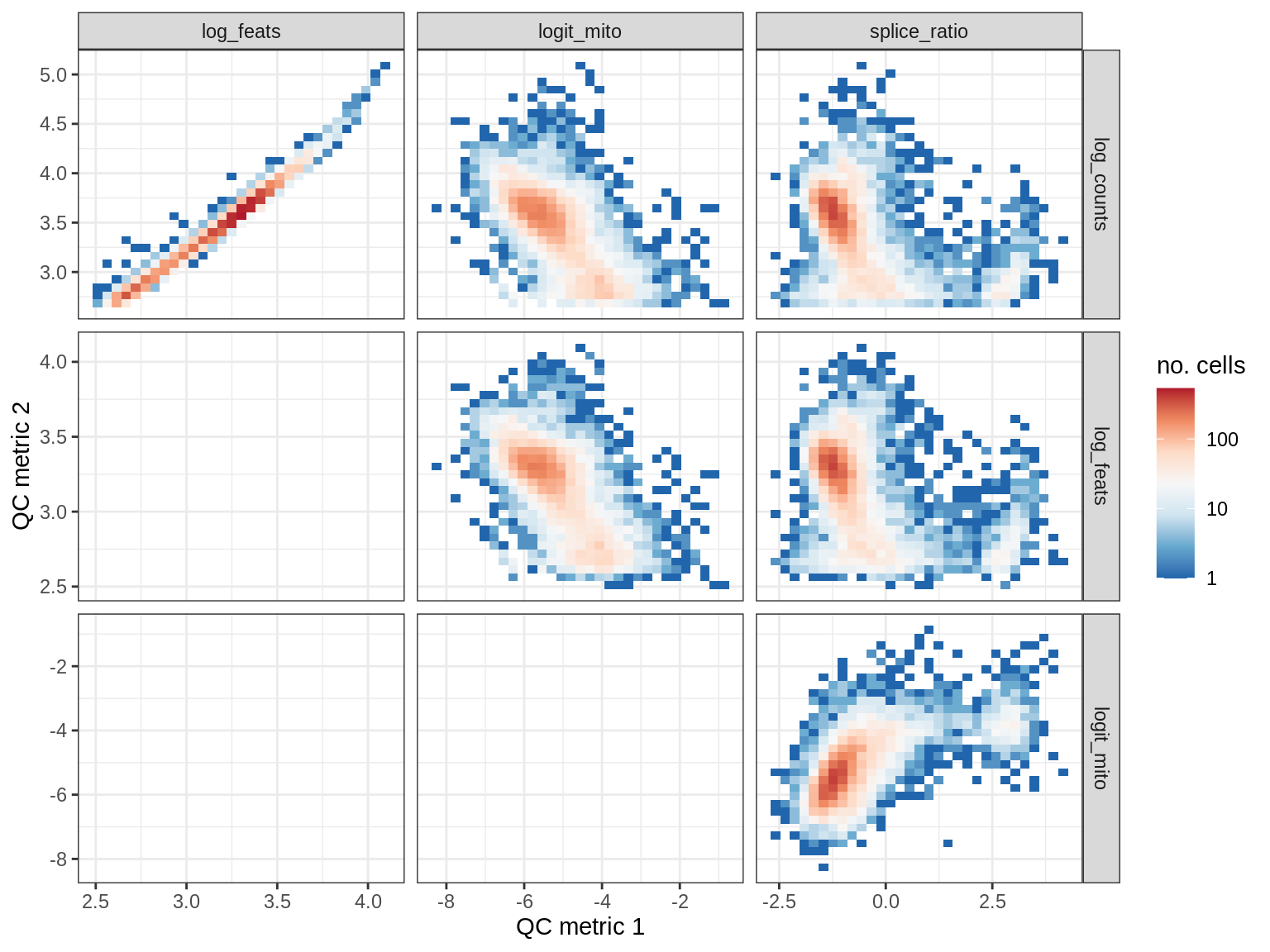
WM122
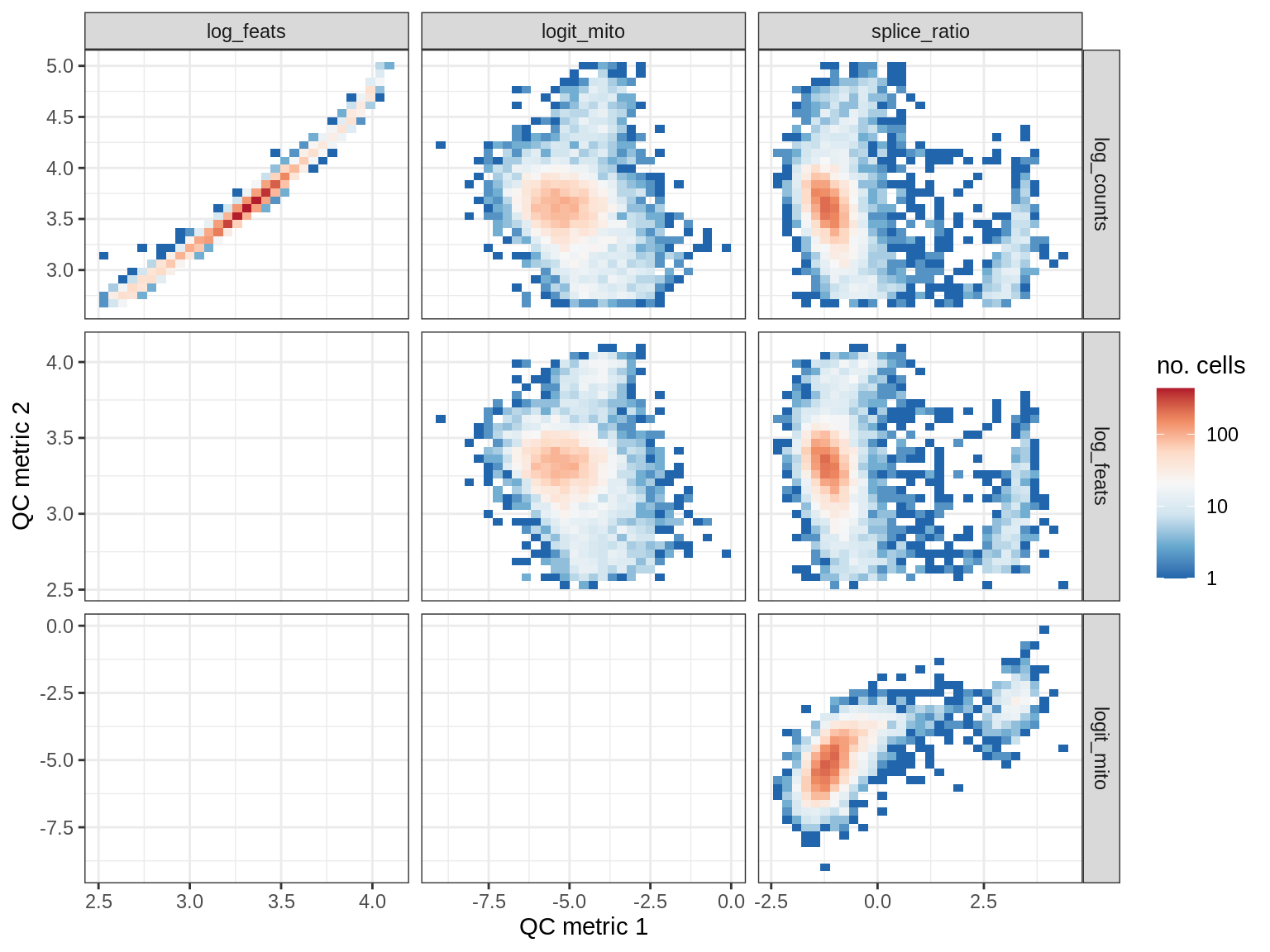
WM123
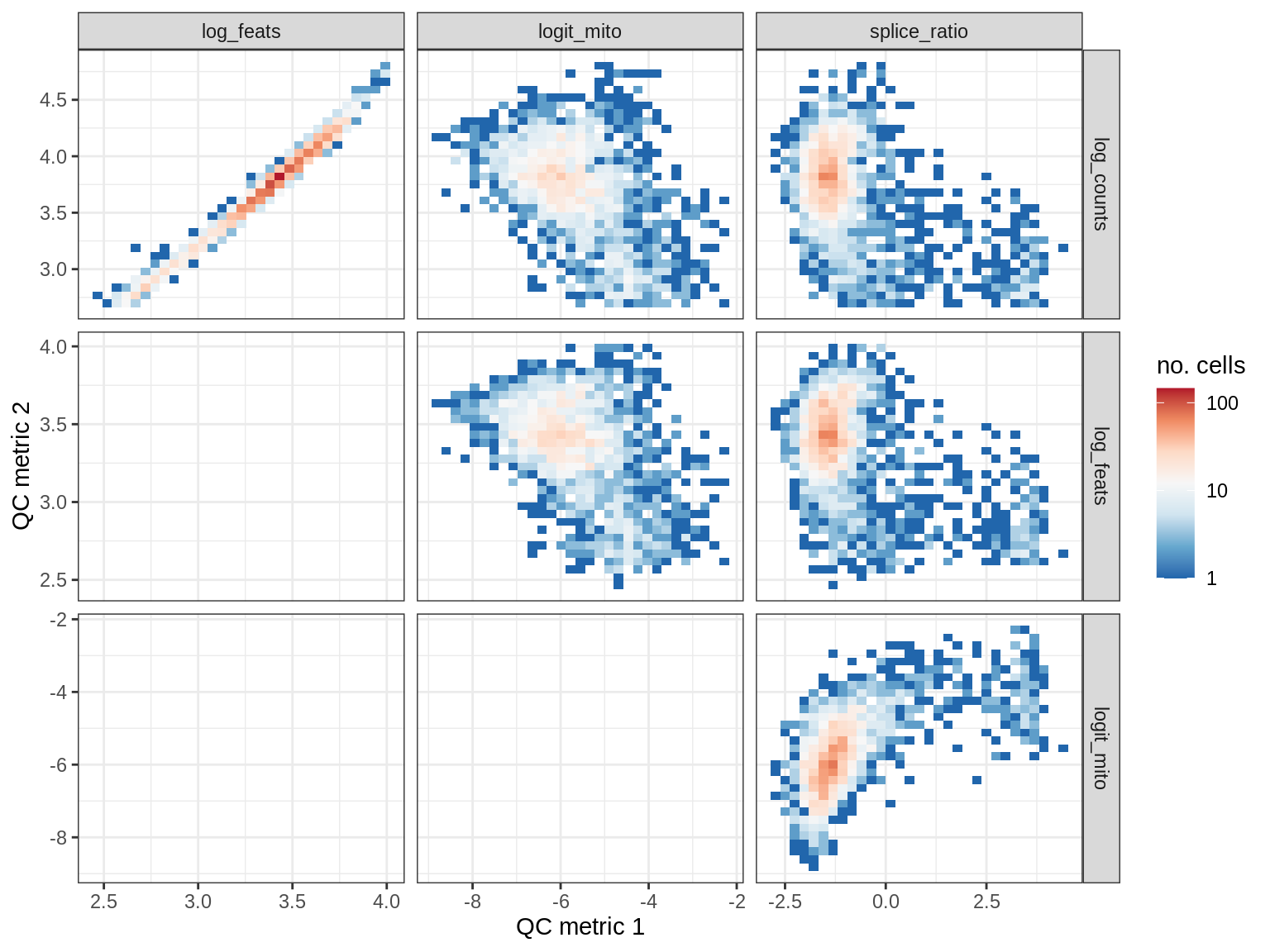
WM124
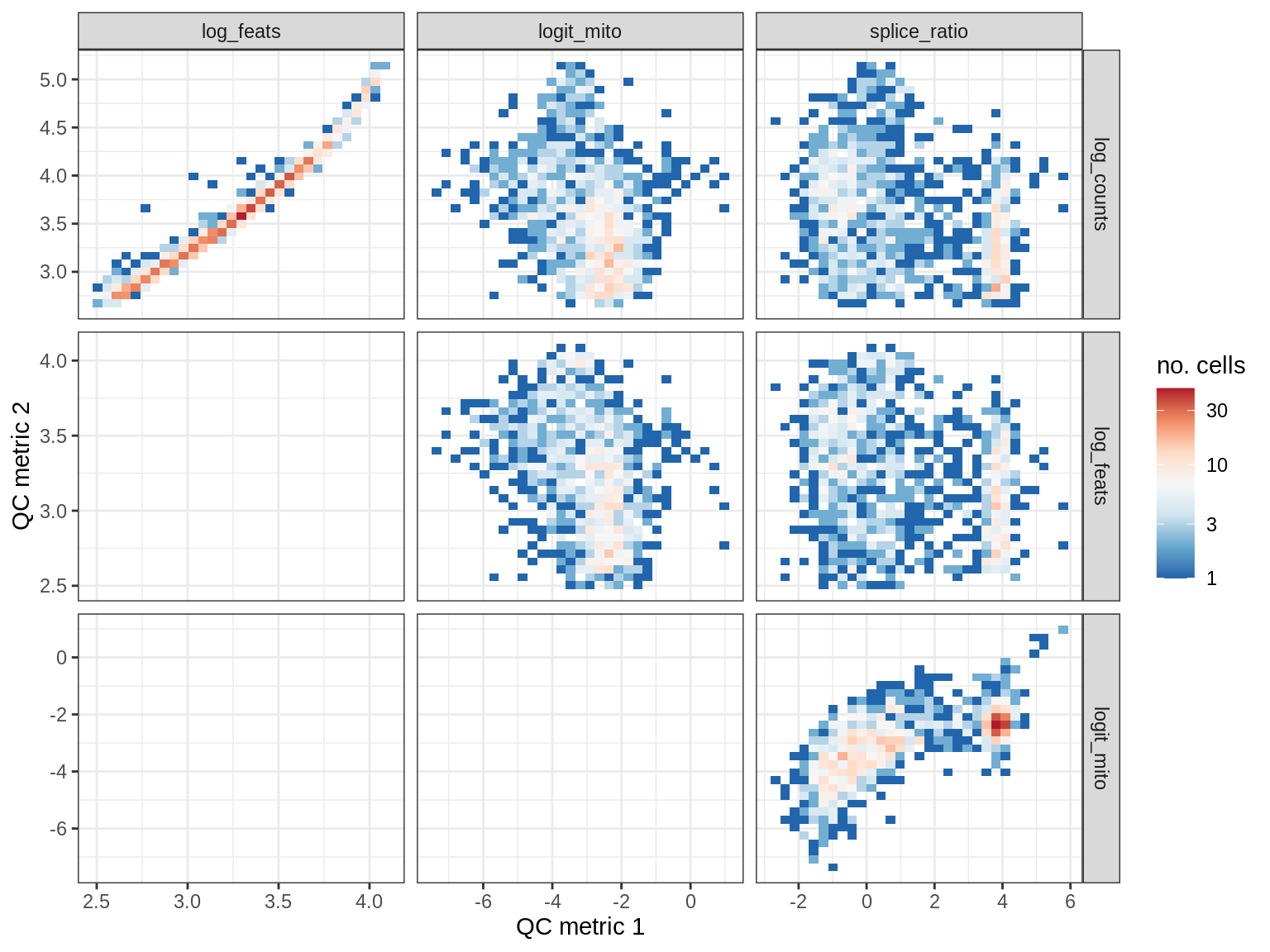
WM125
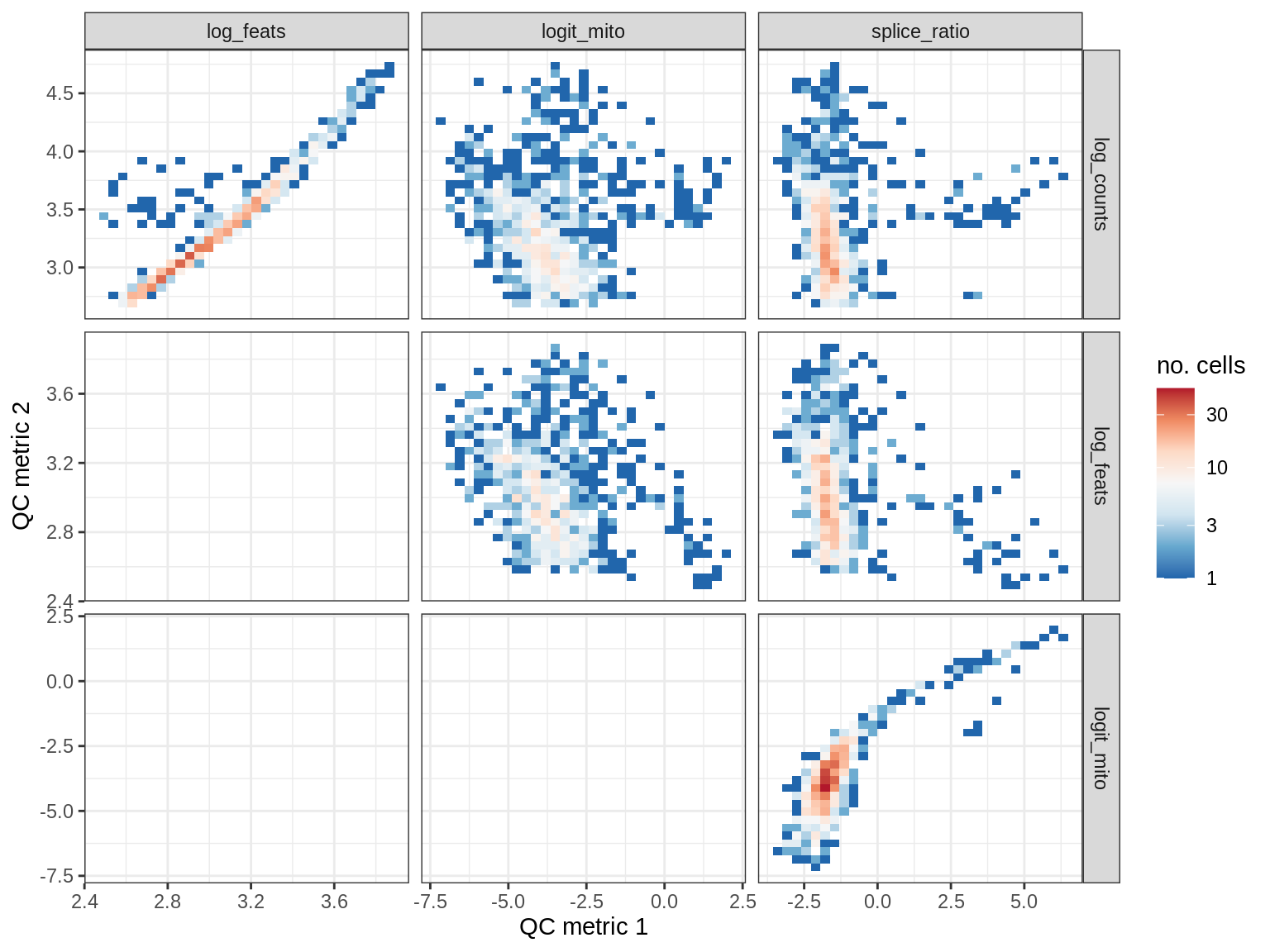
WM126
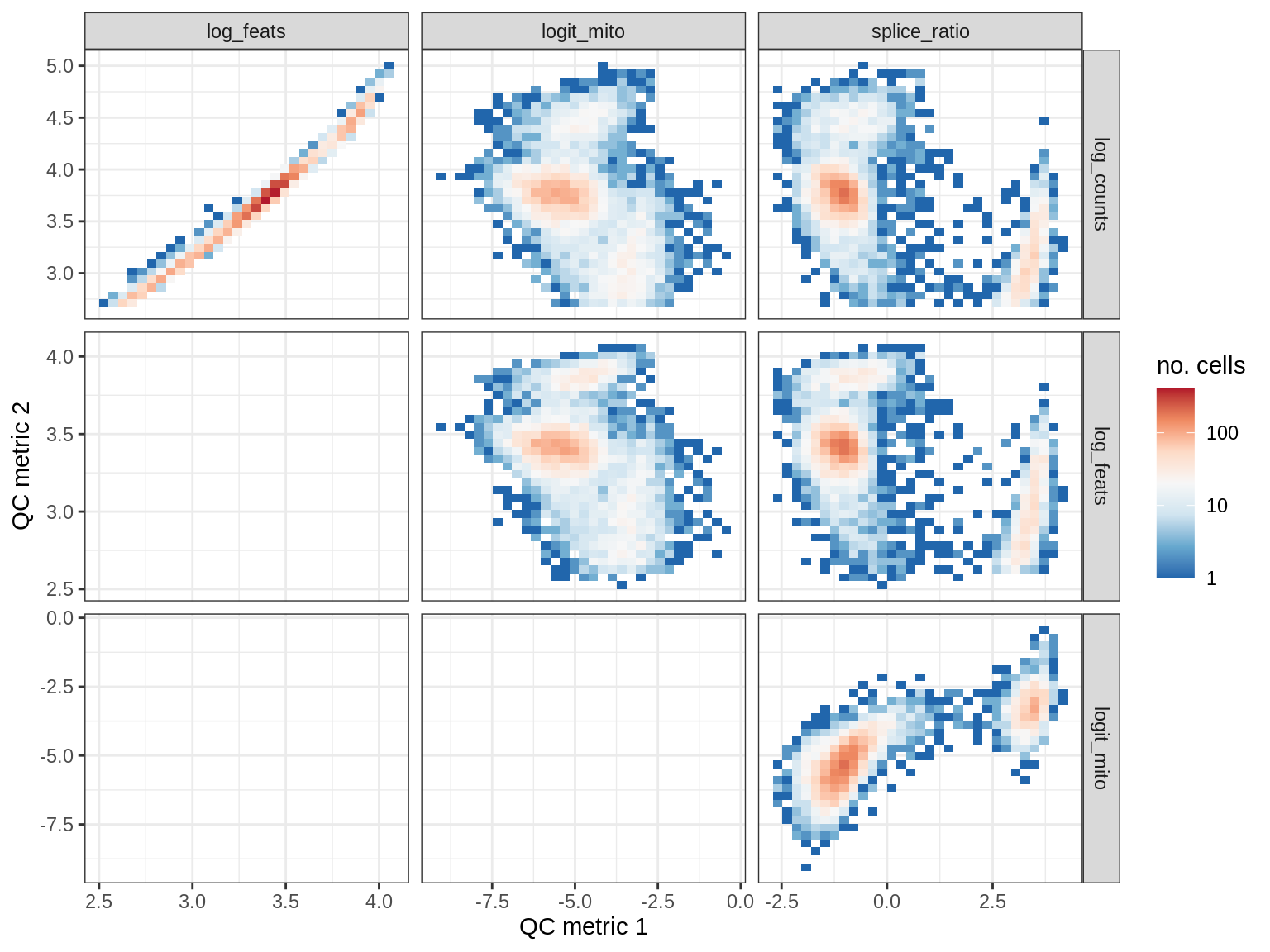
WM127
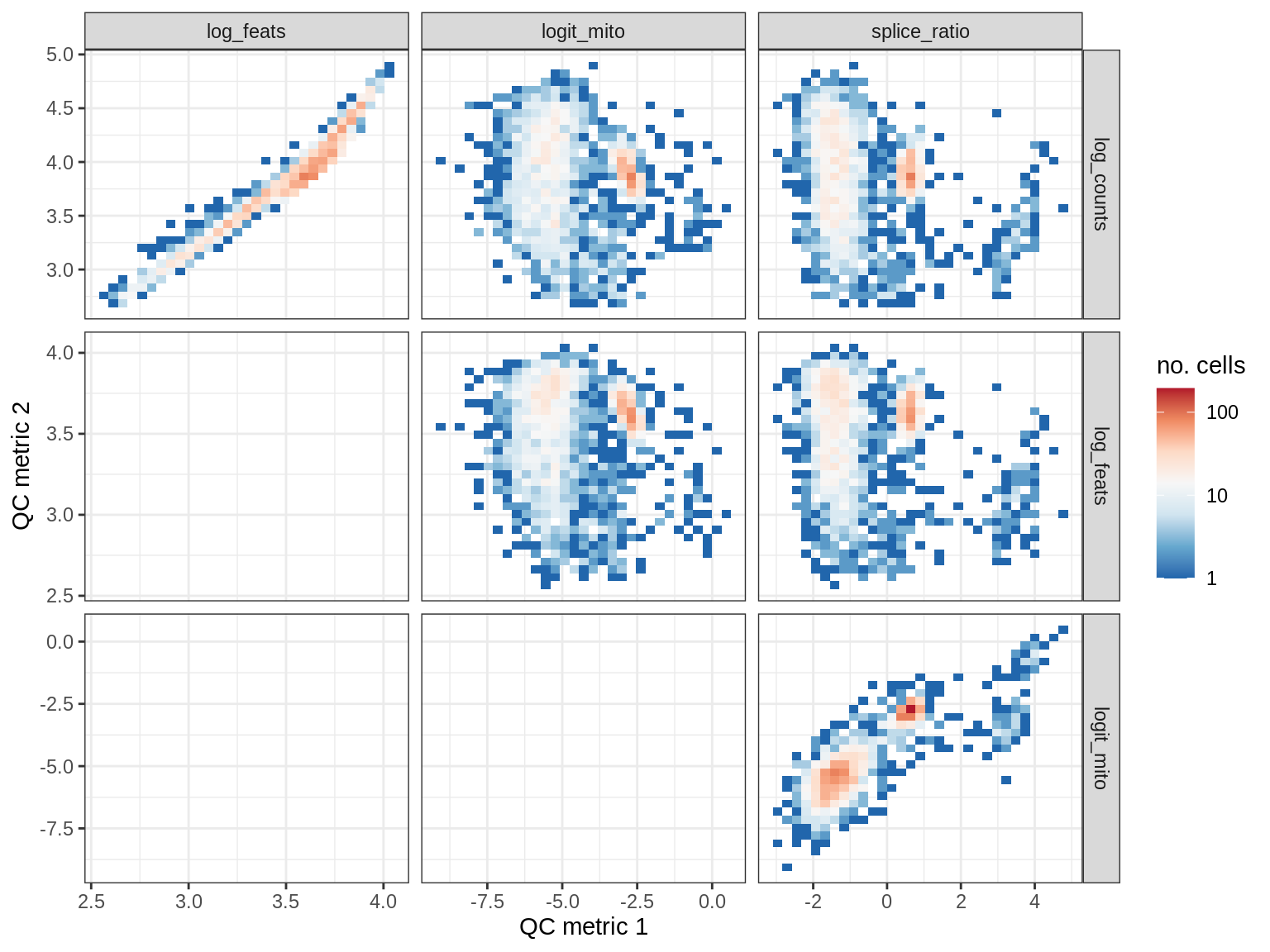
WM128
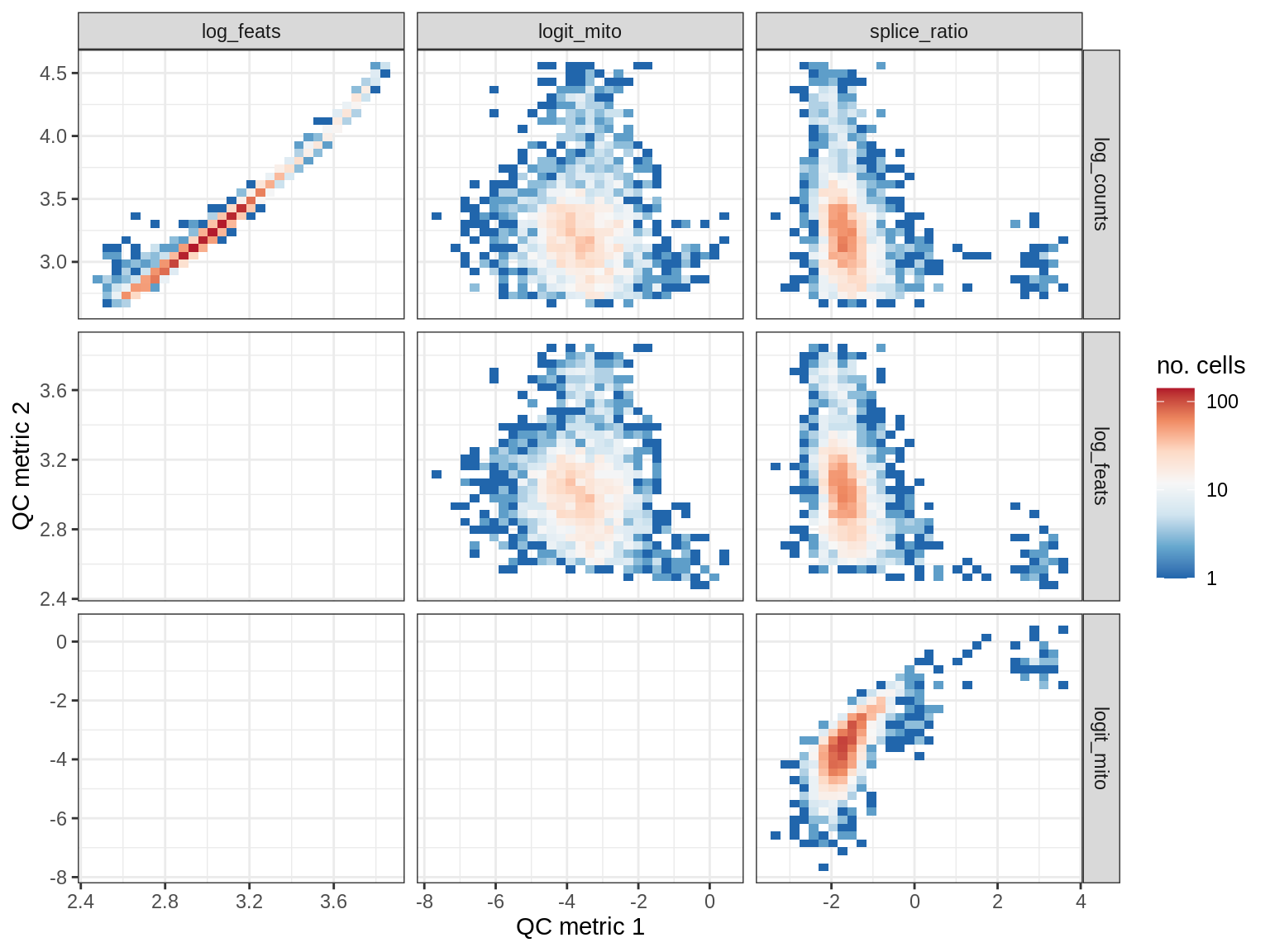
WM129
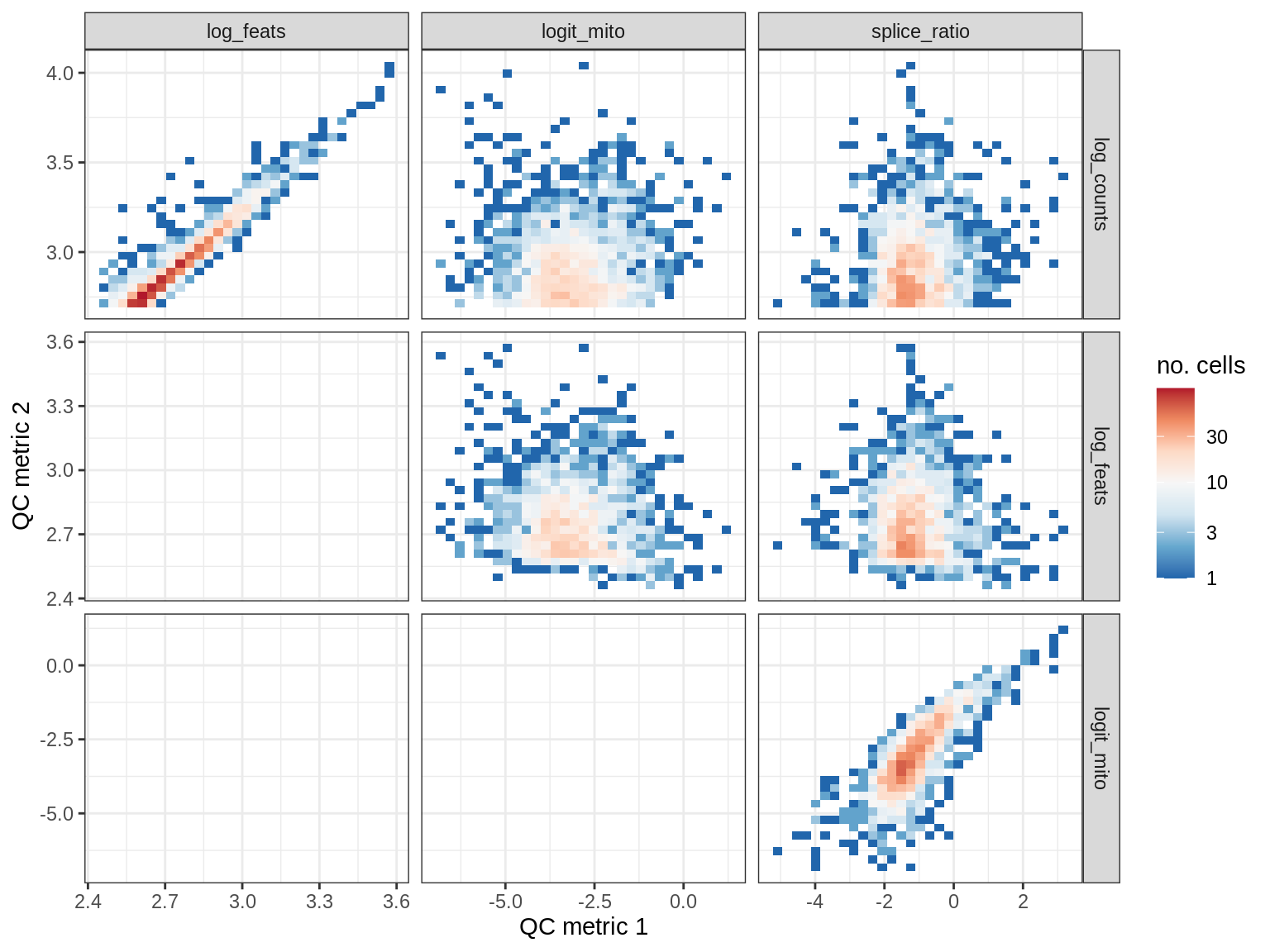
WM130
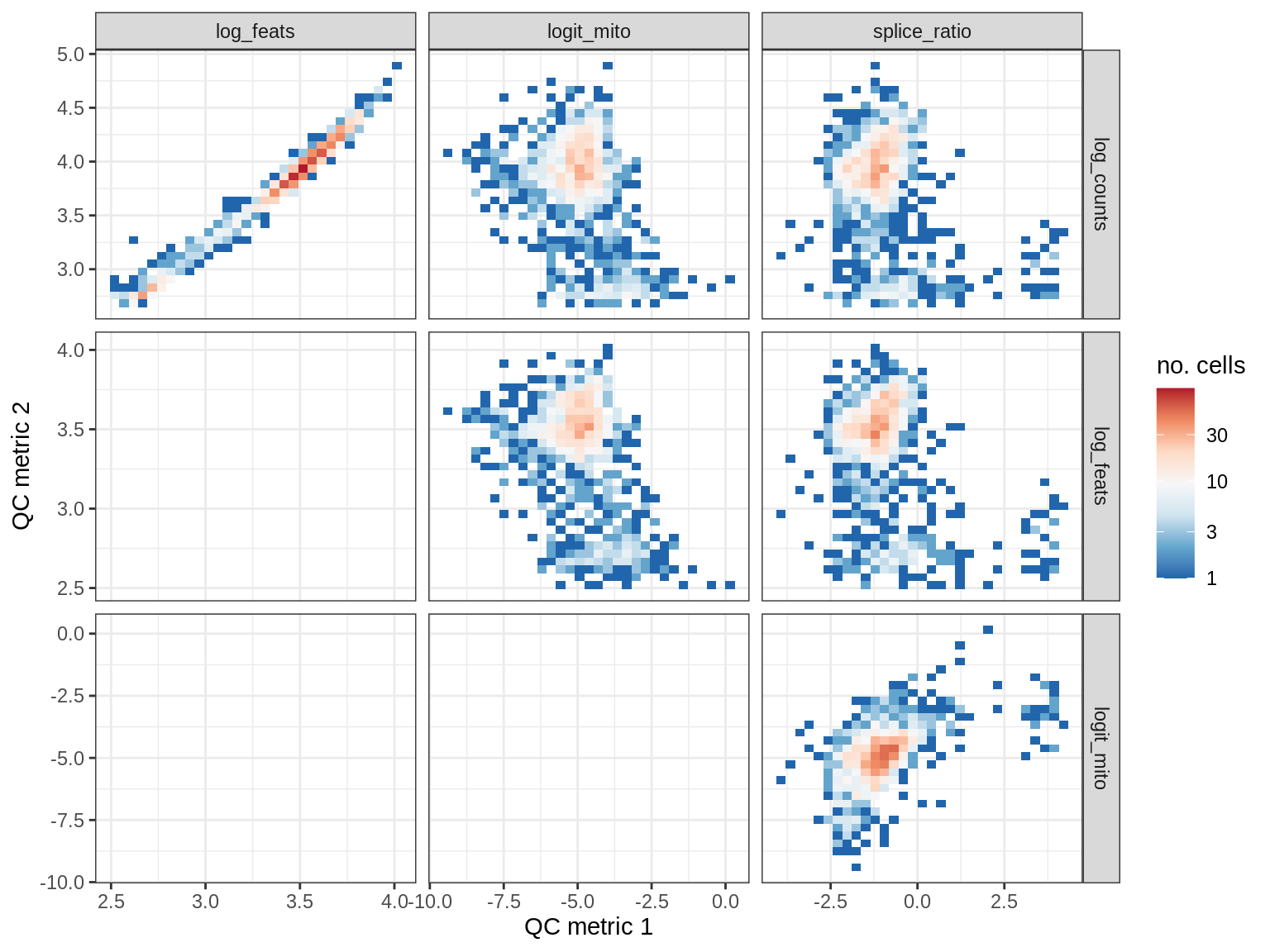
WM131
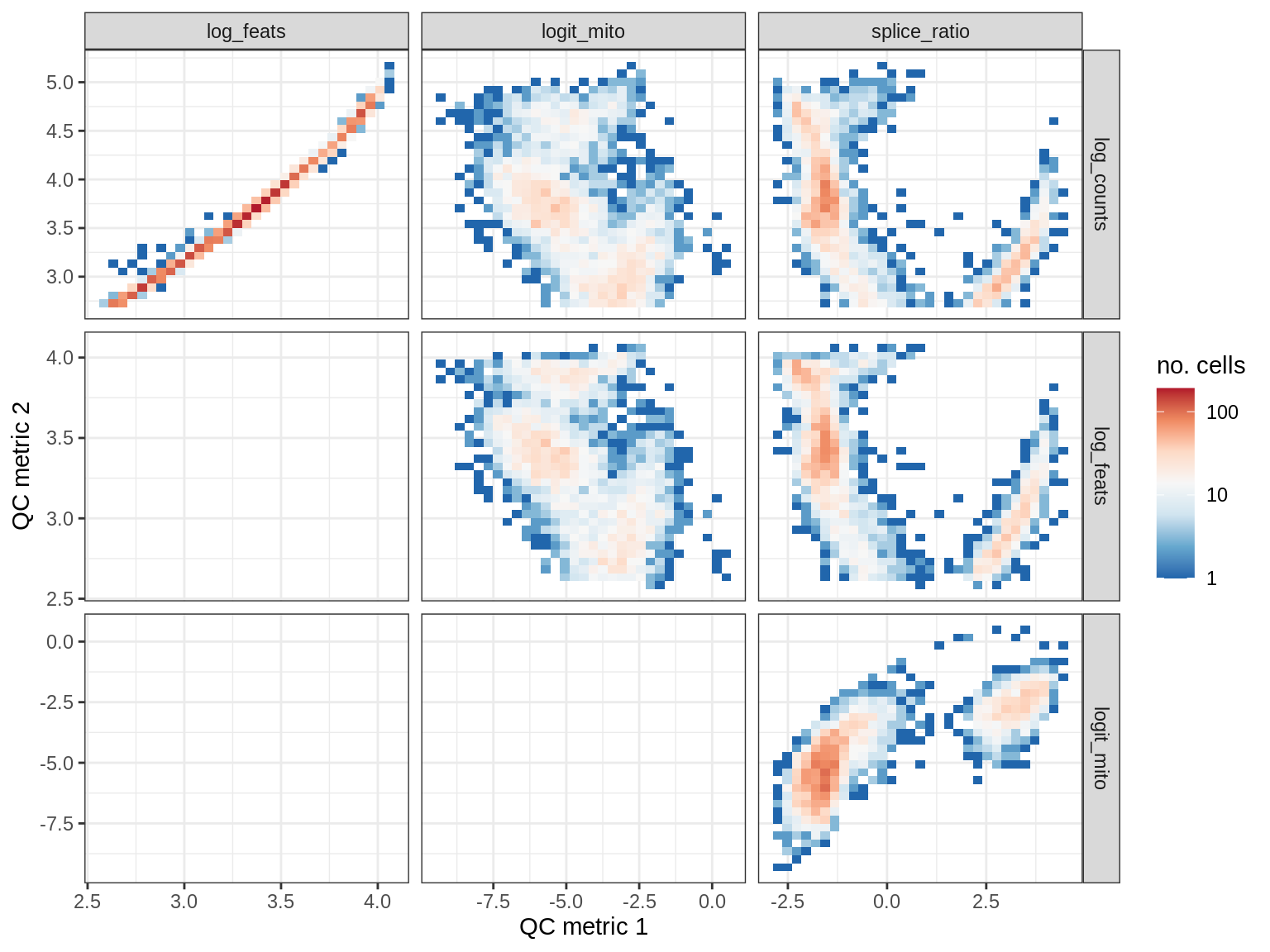
WM132
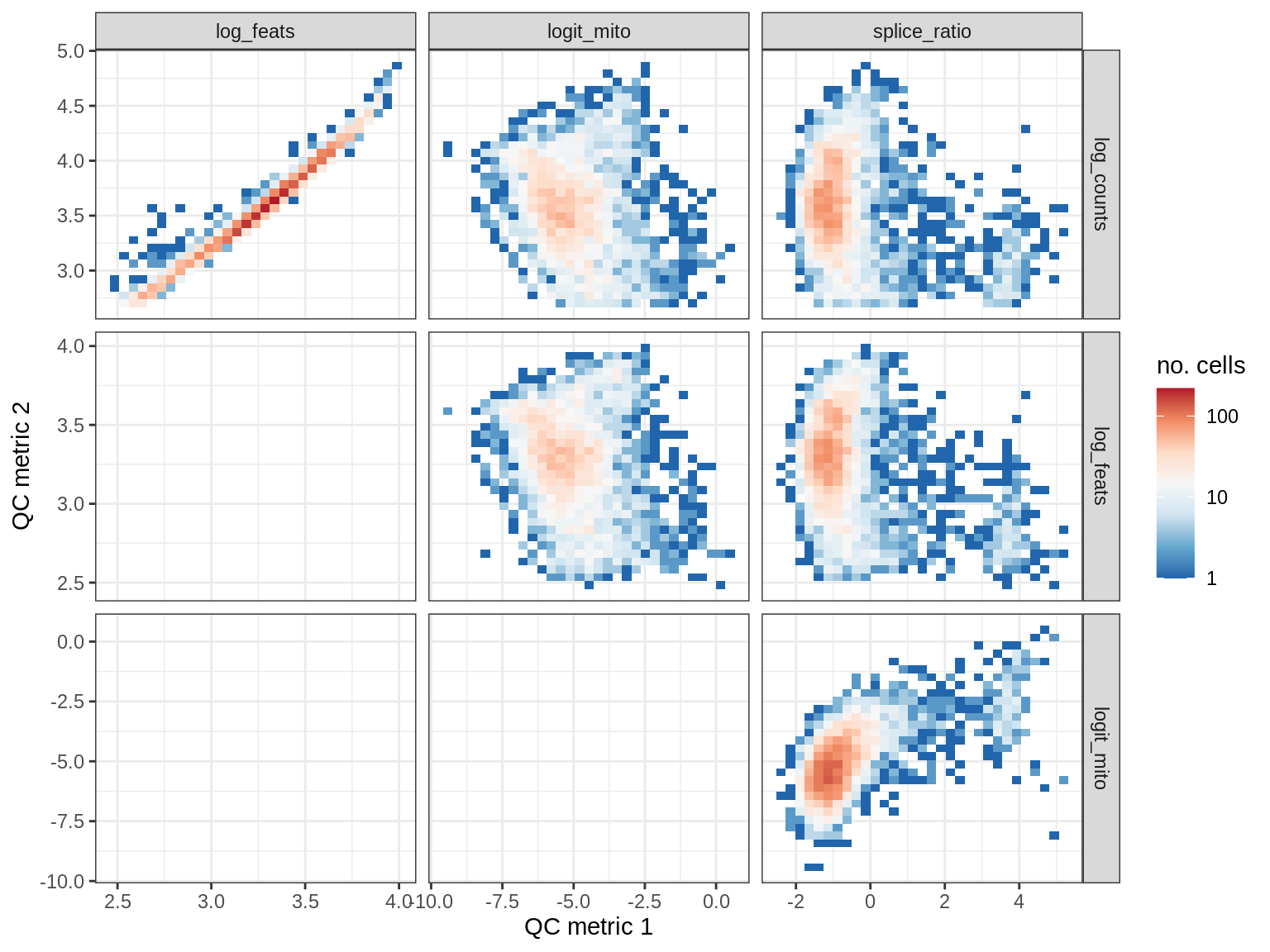
WM133
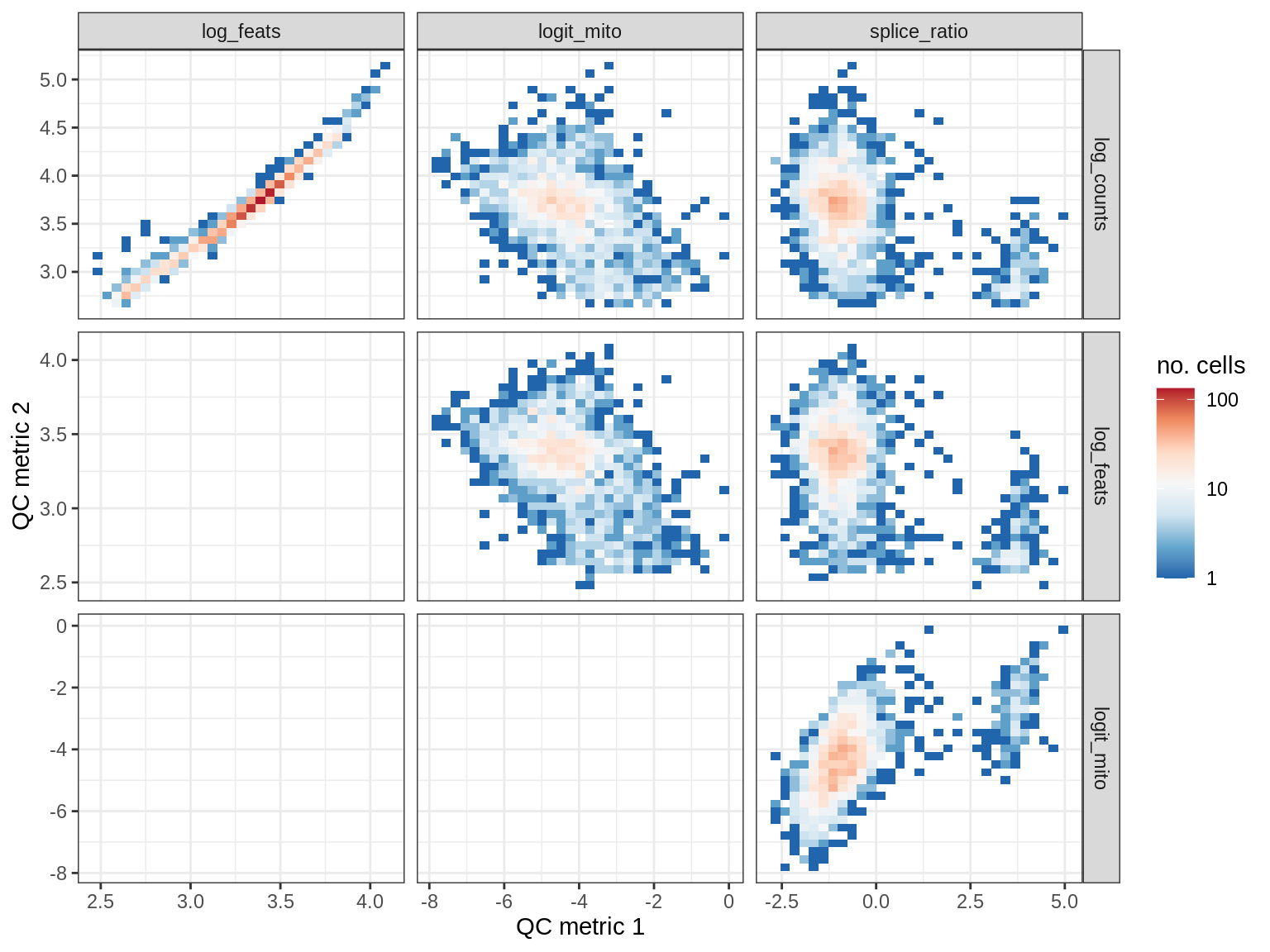
WM134
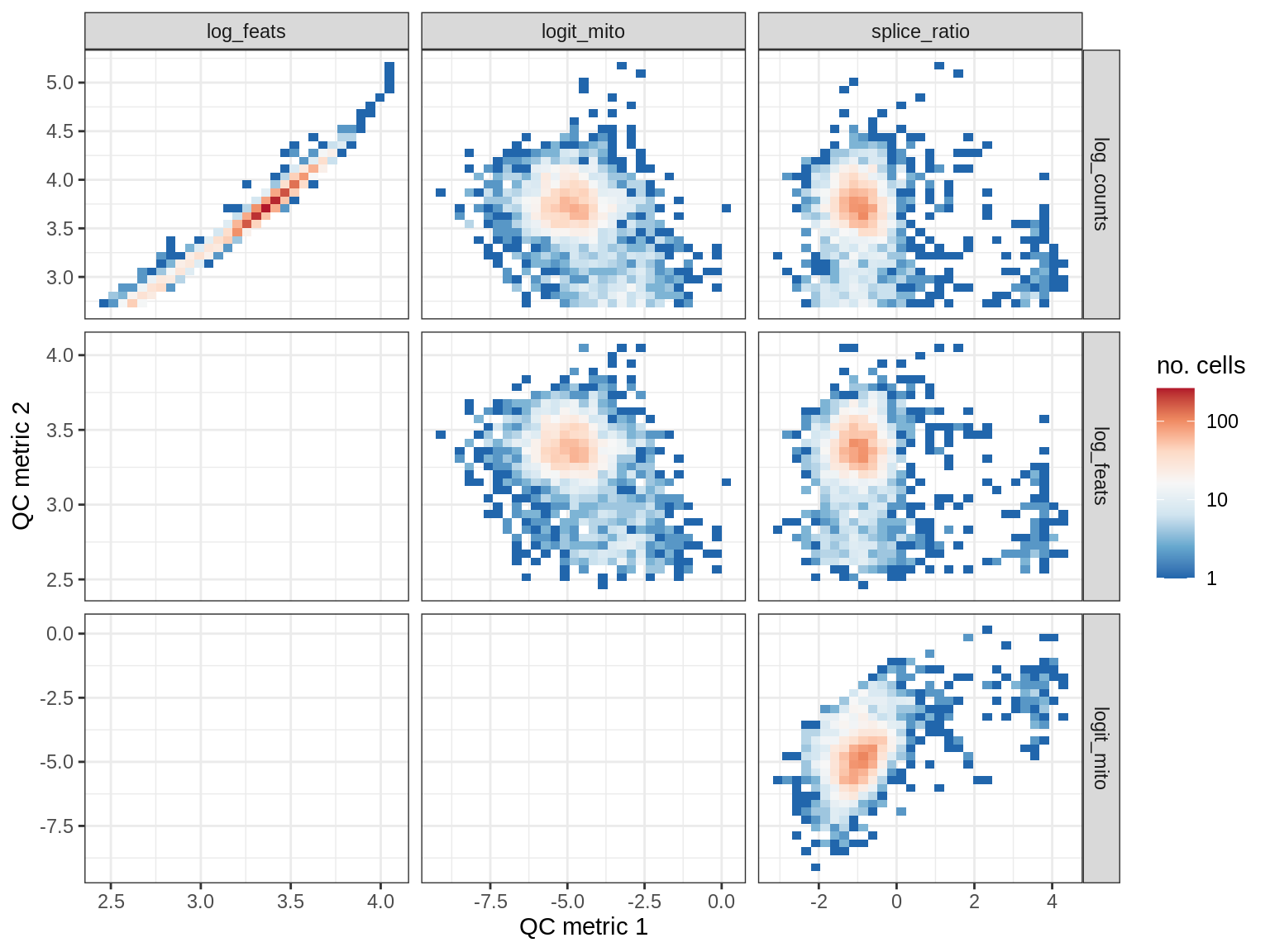
WM135
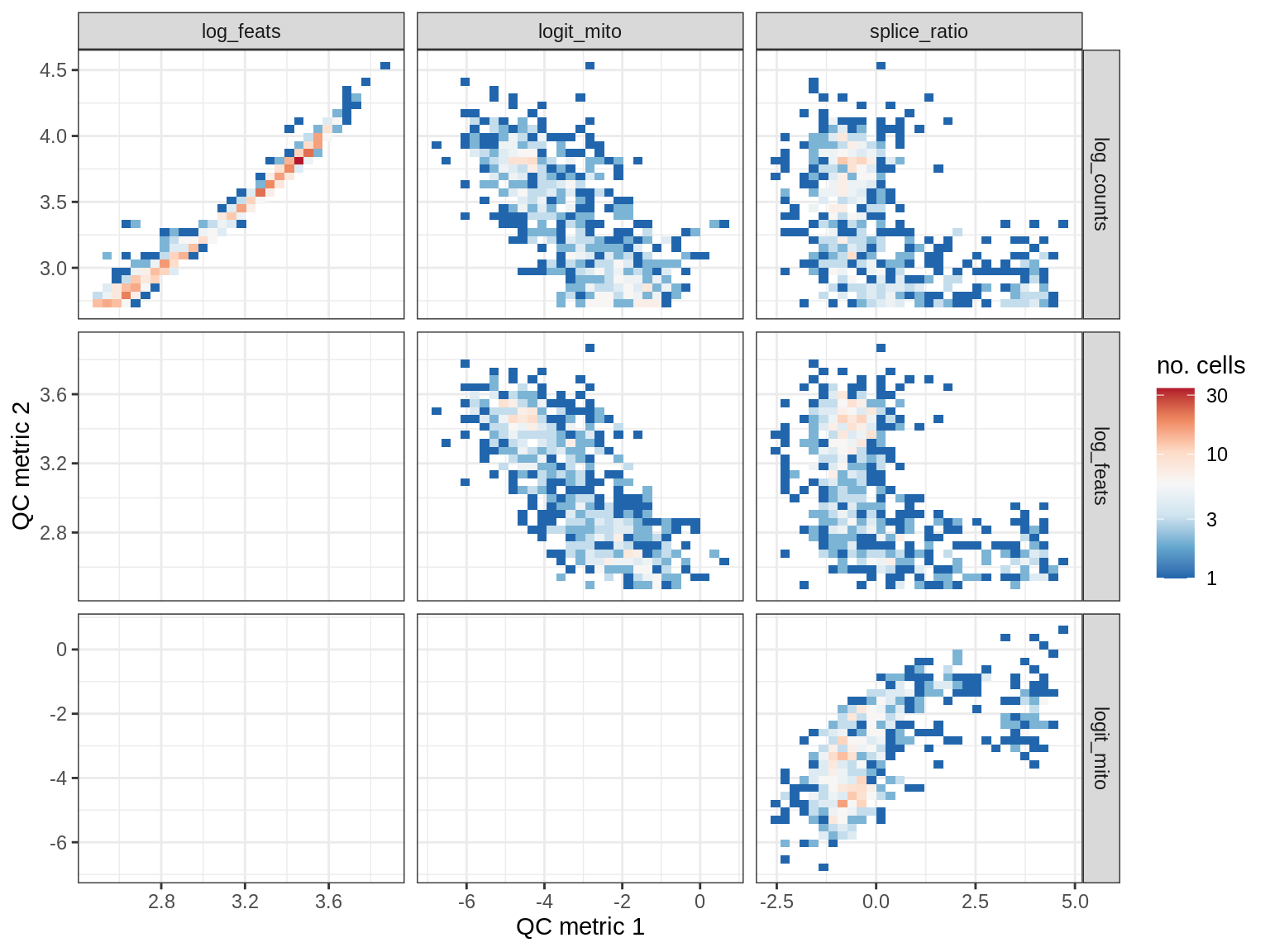
WM136
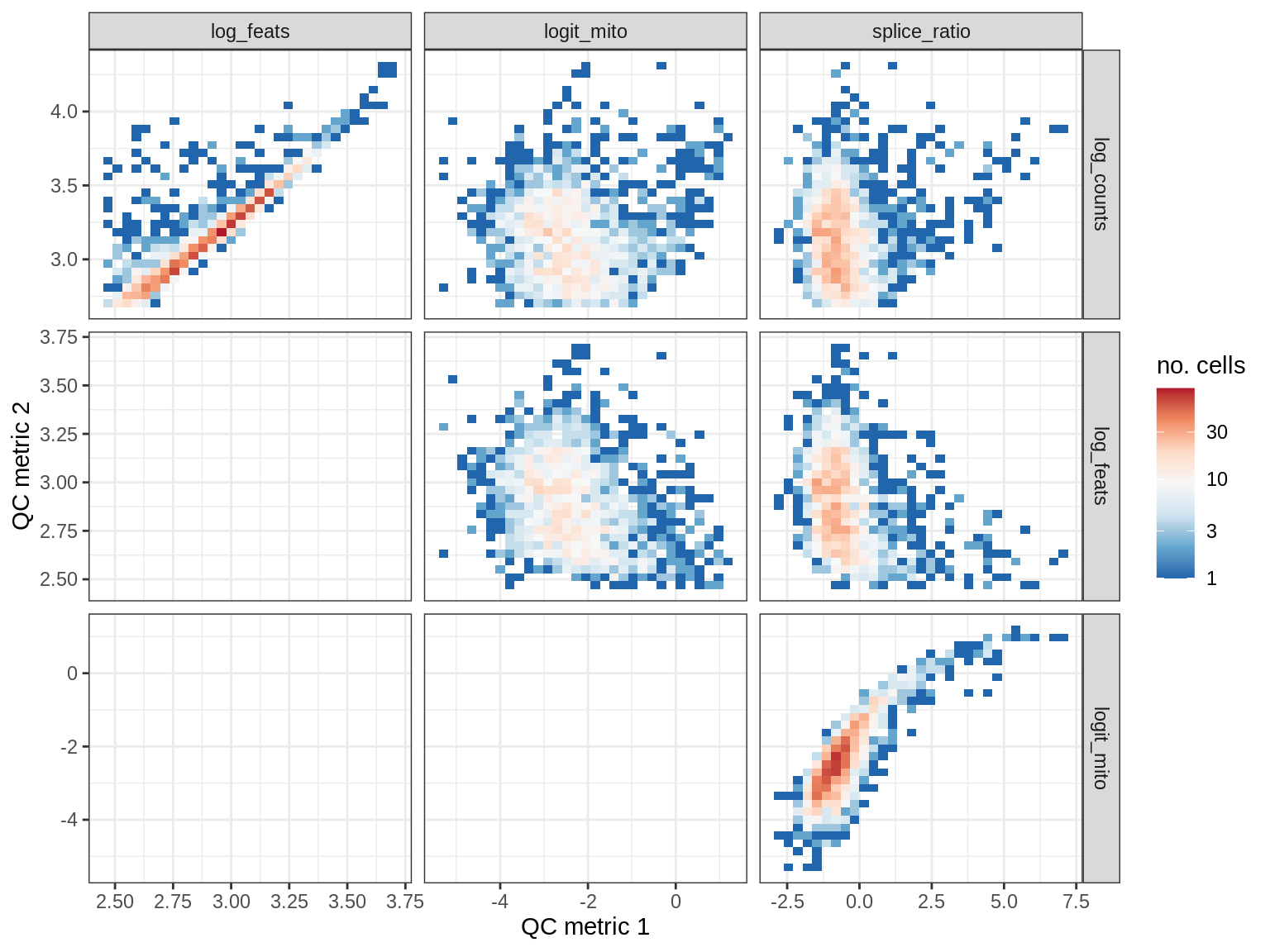
WM137
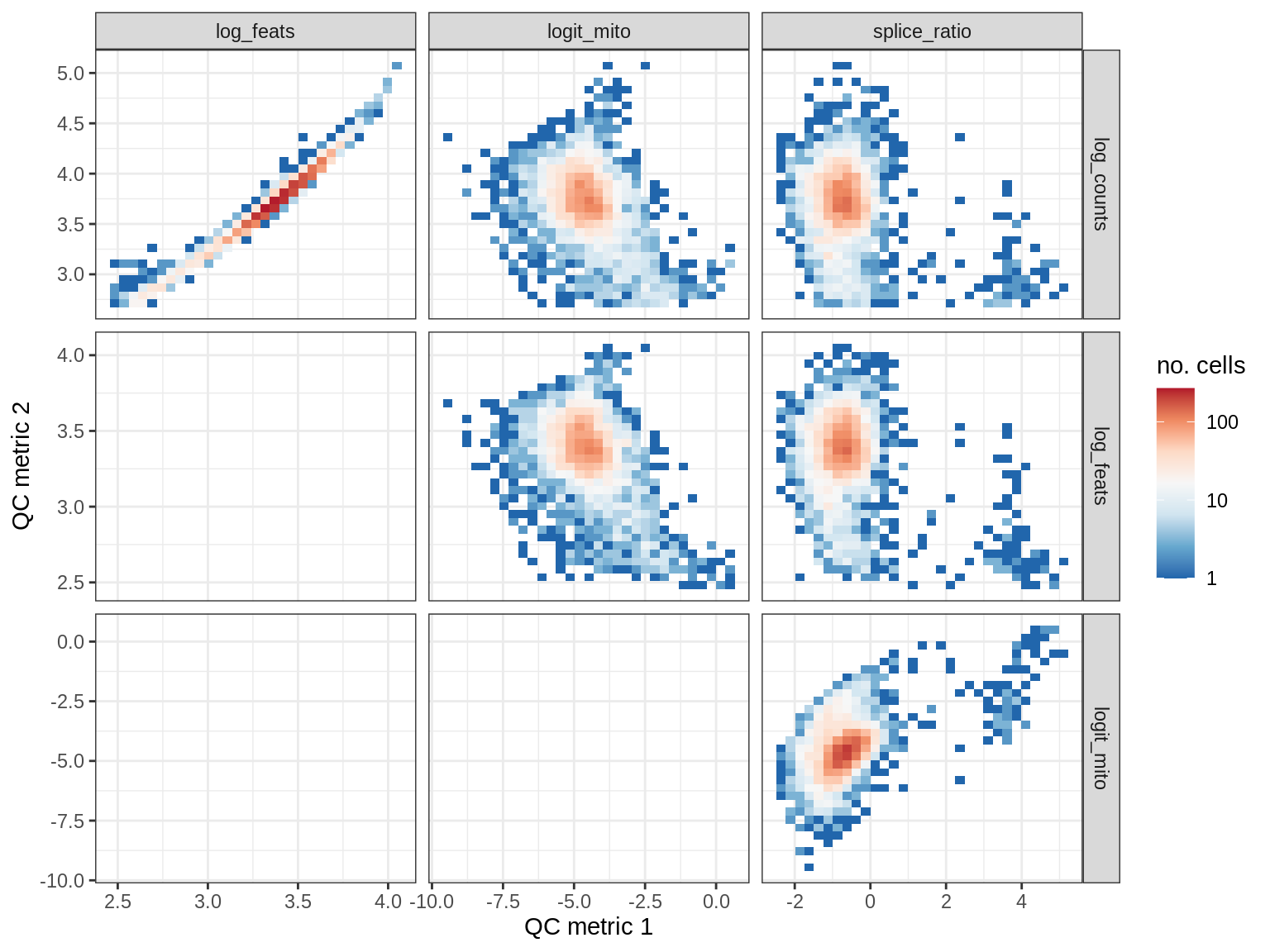
WM138
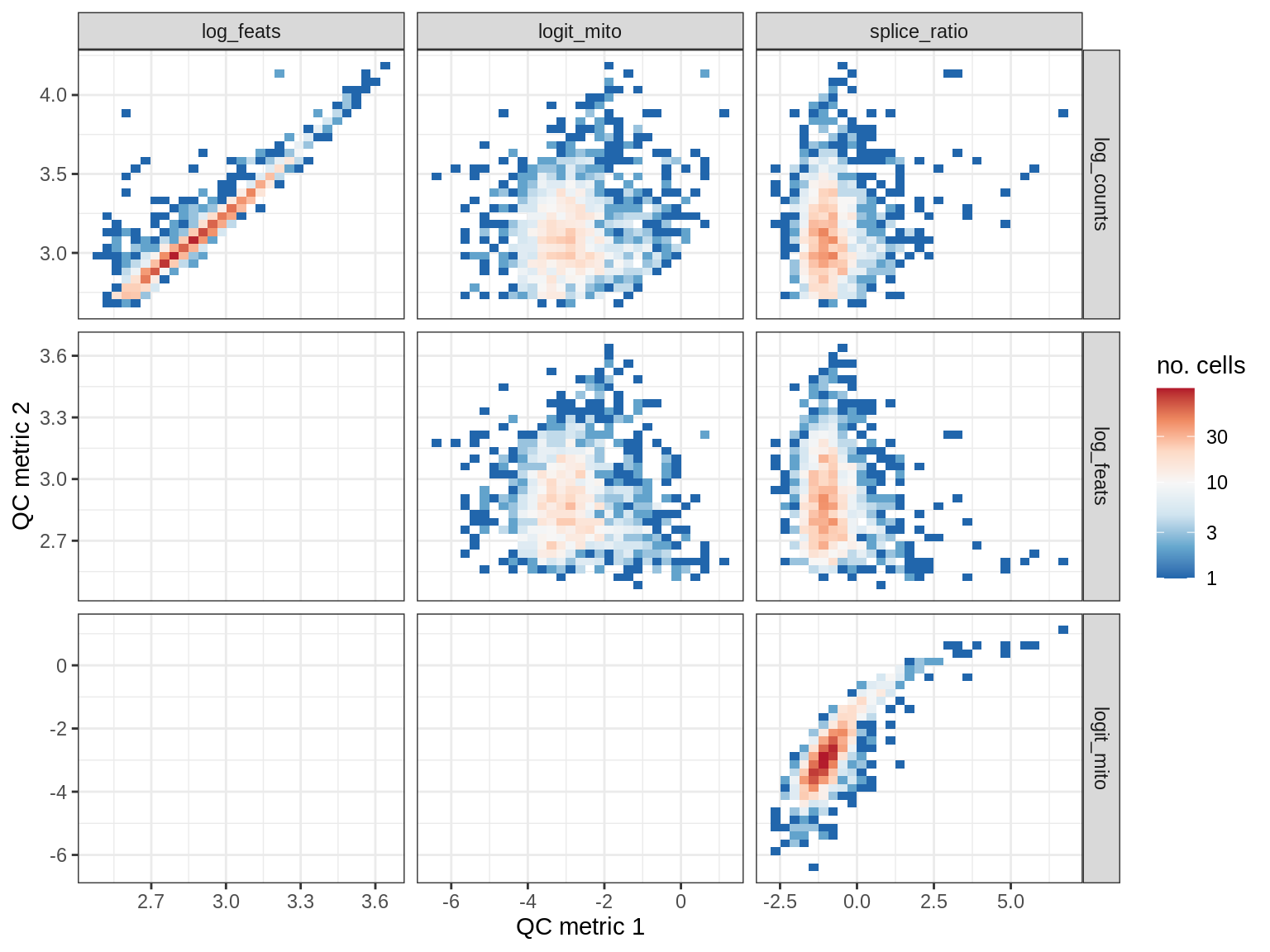
WM139
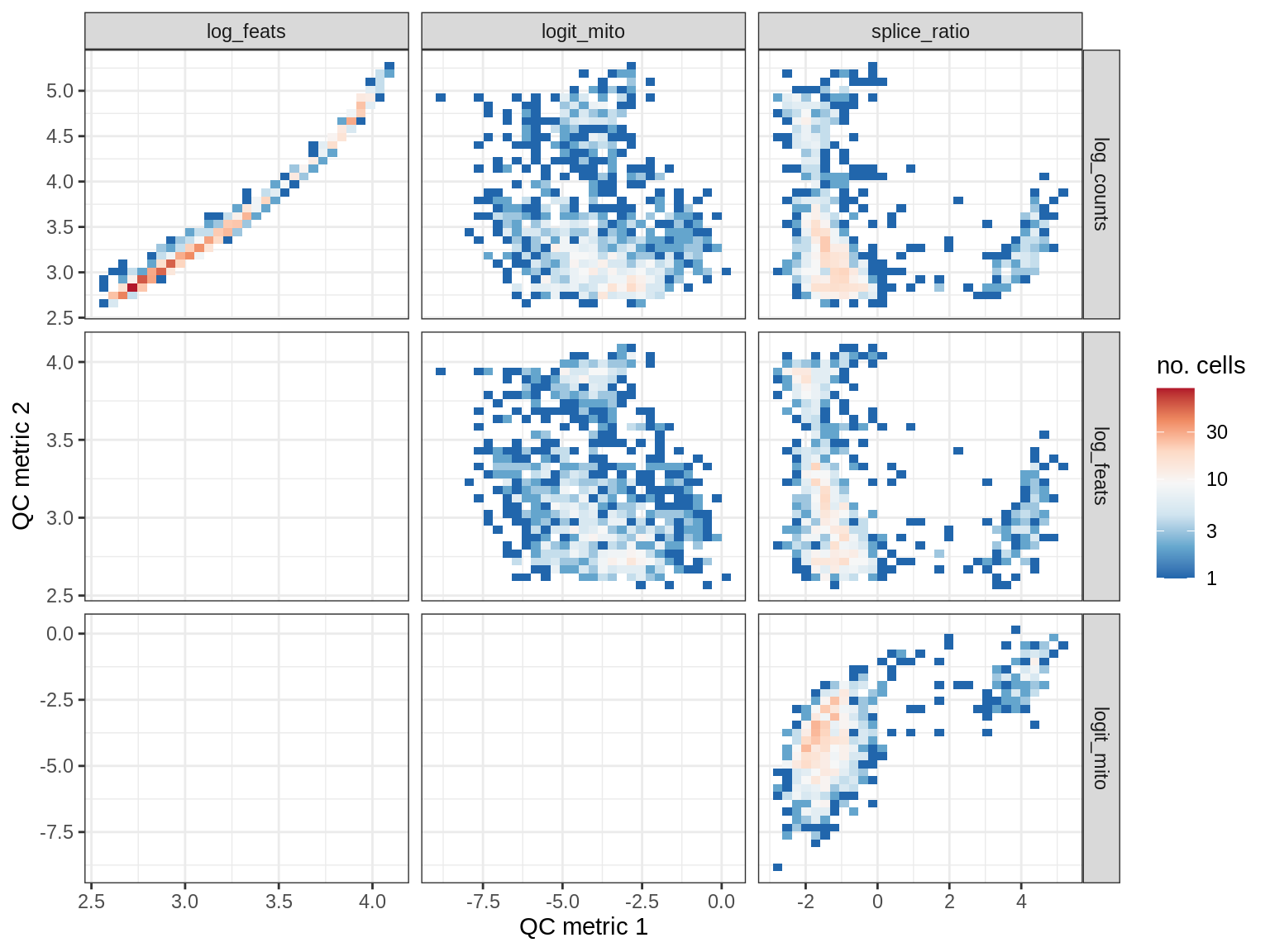
WM140
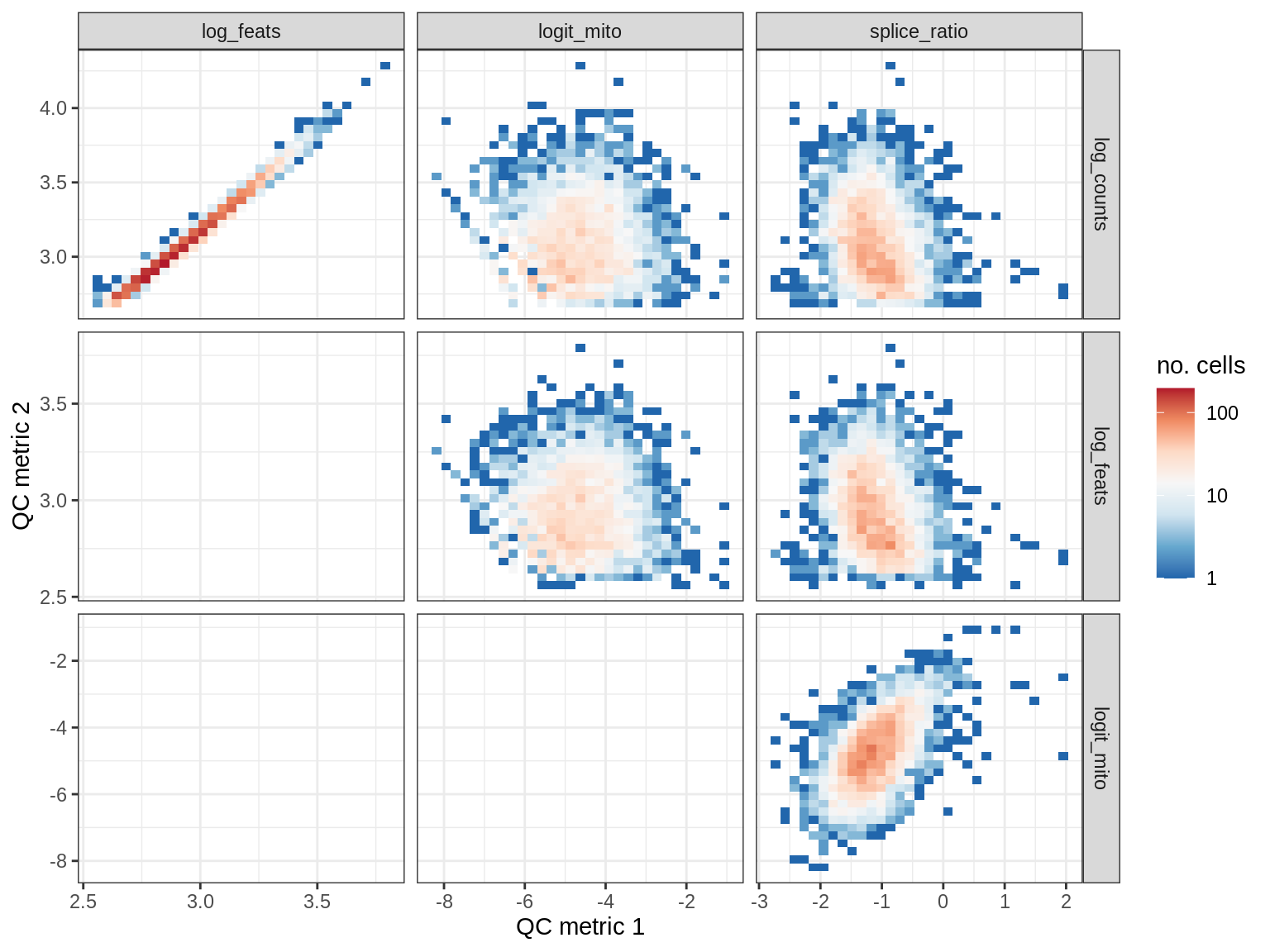
WM141
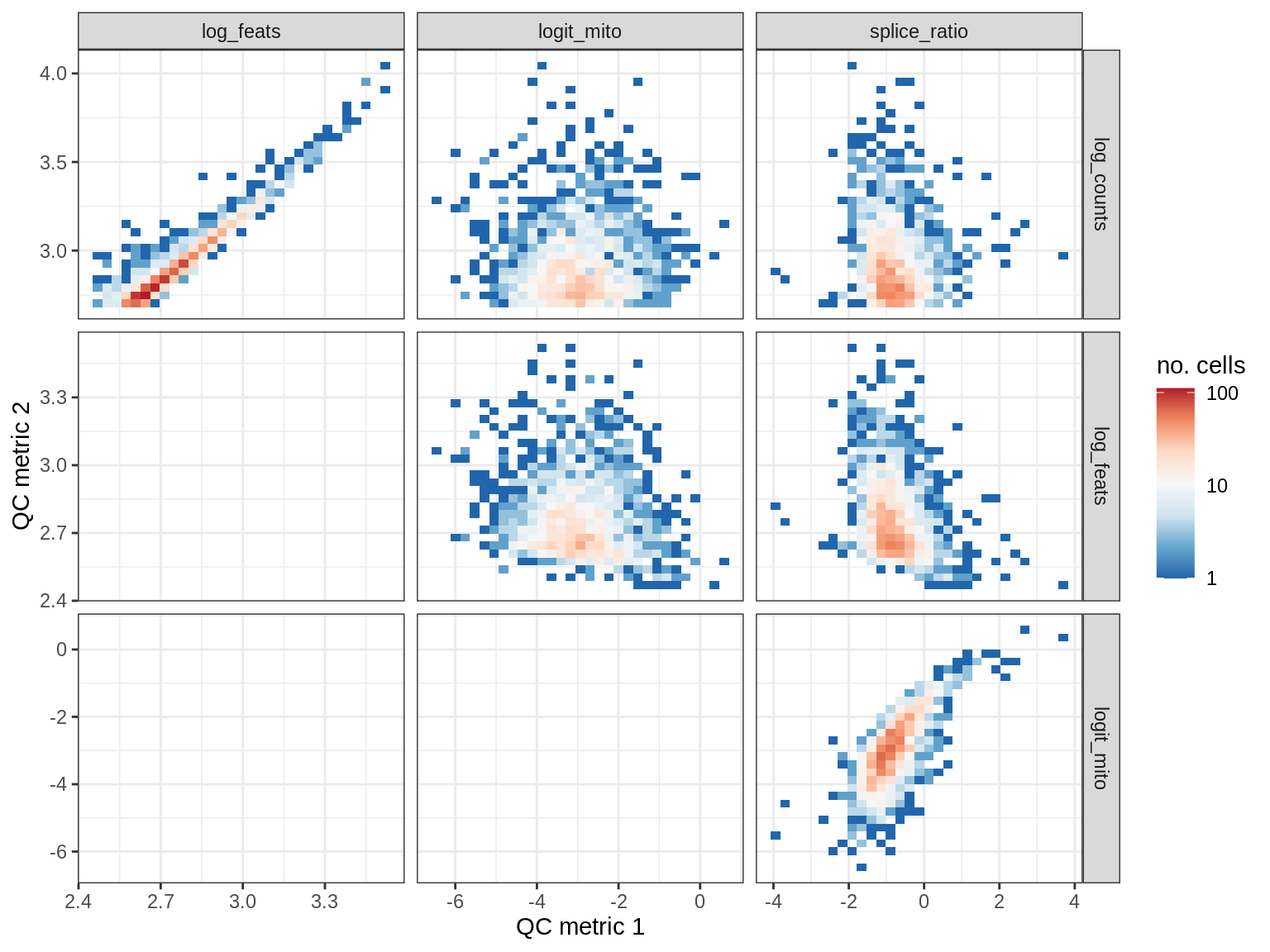
WM142
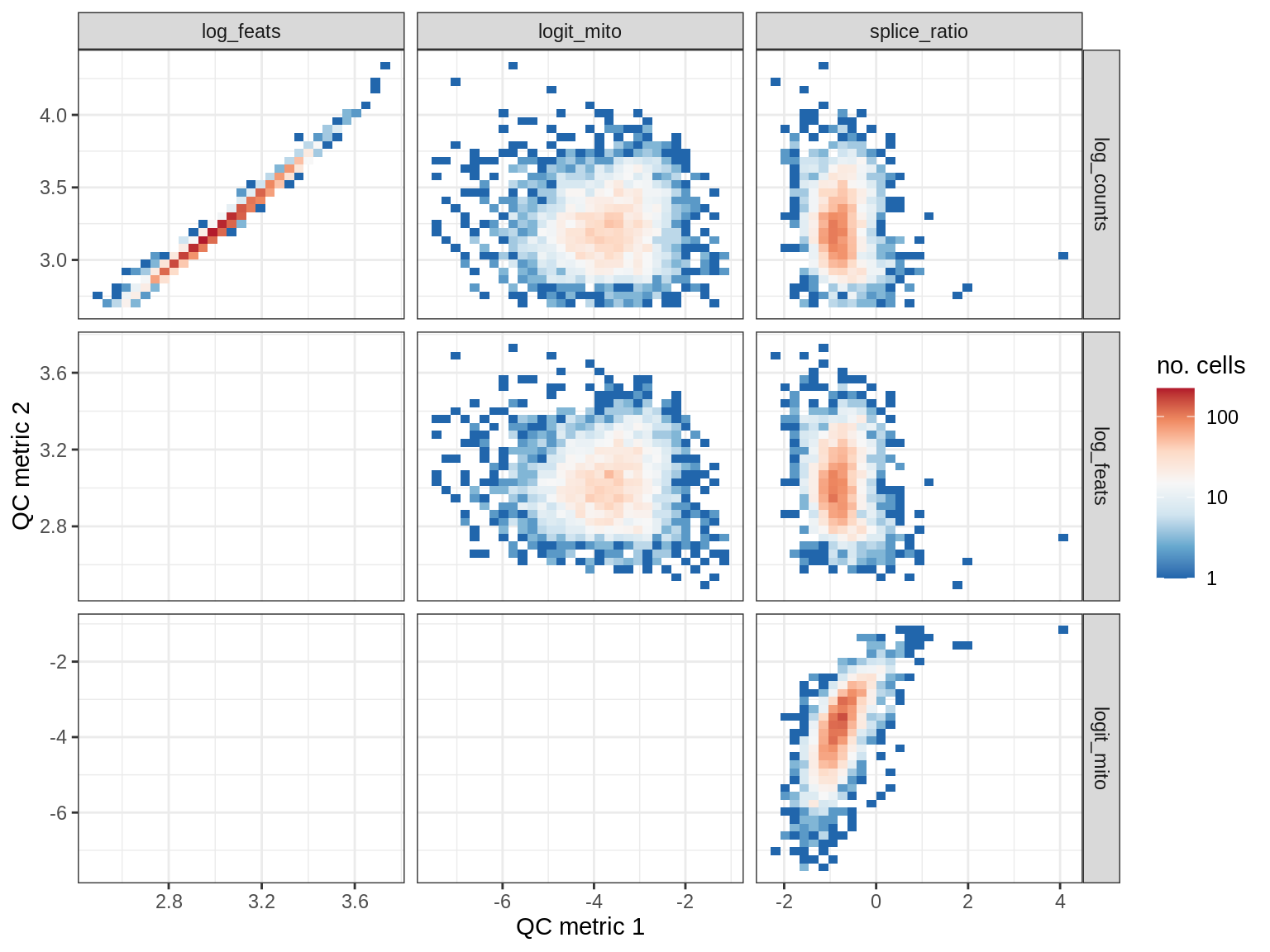
WM143
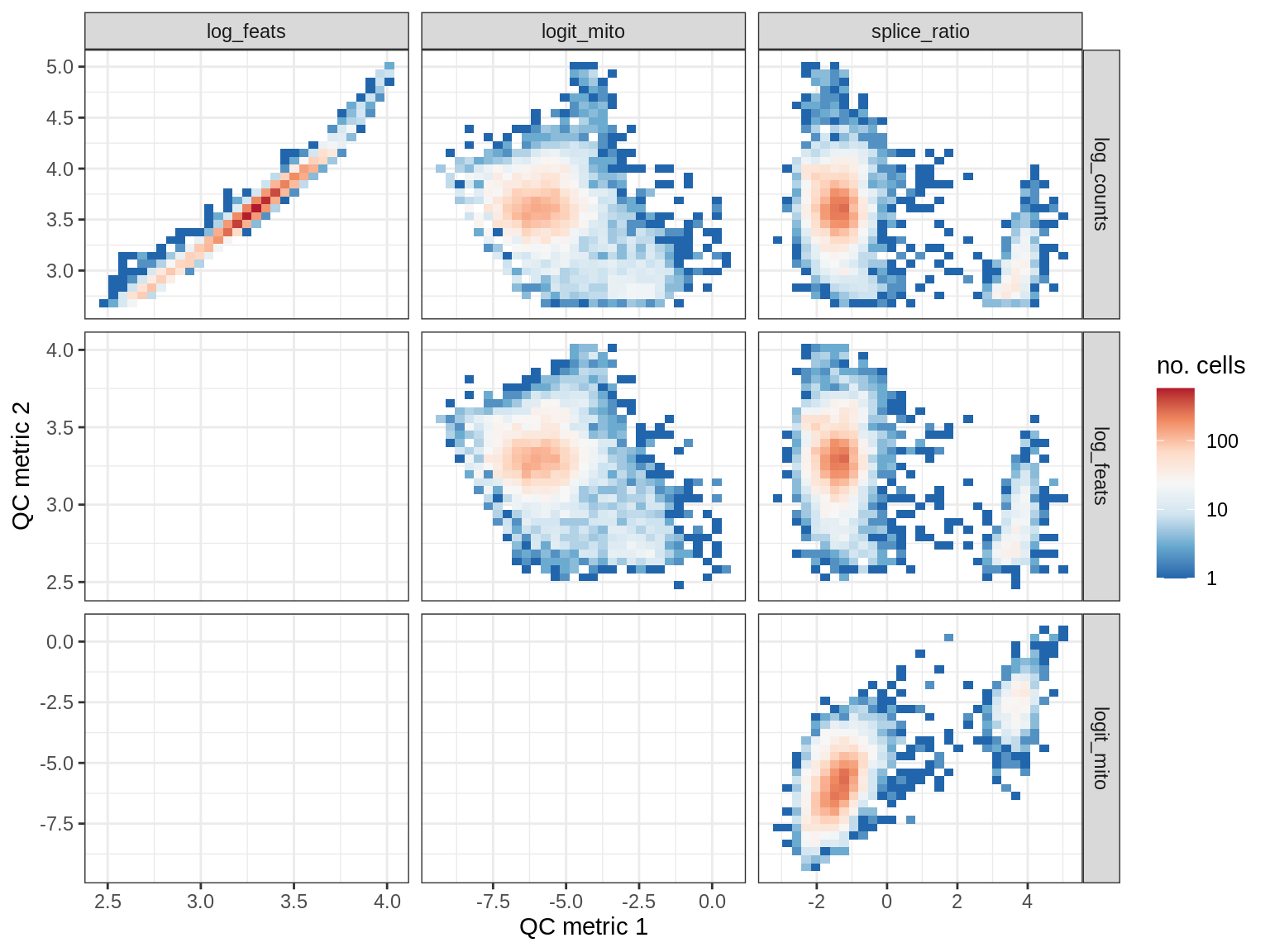
WM145
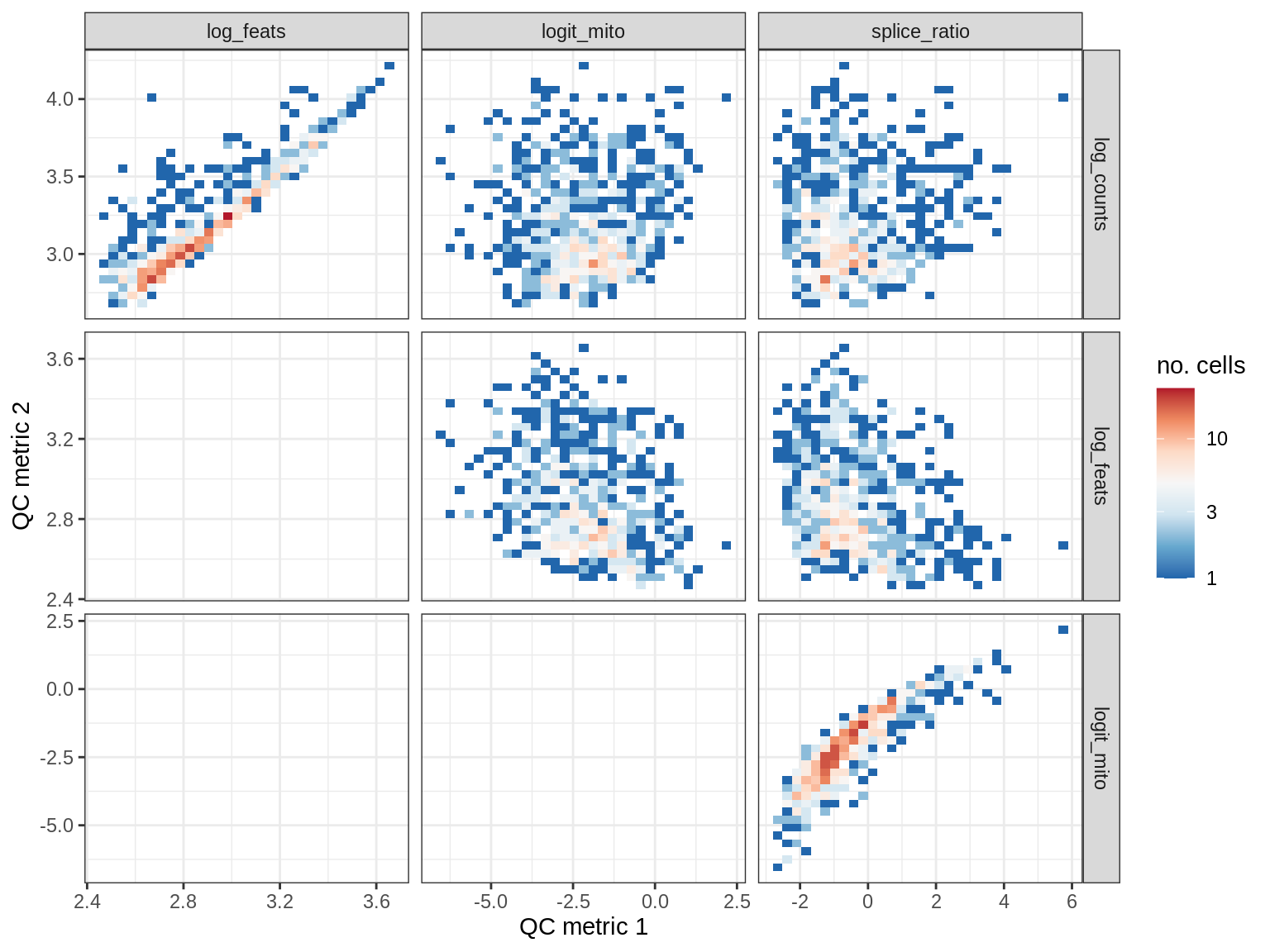
WM146
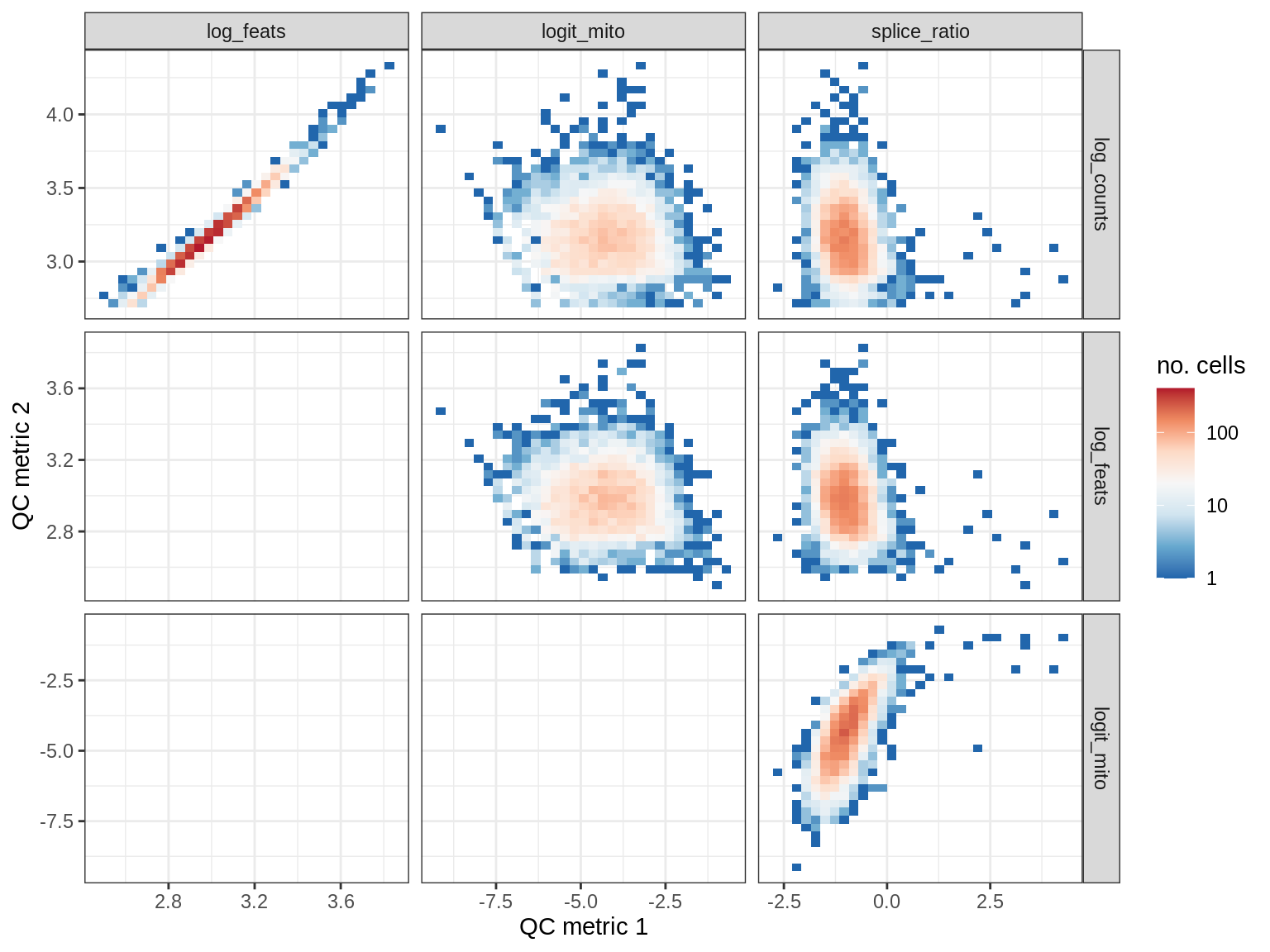
WM147
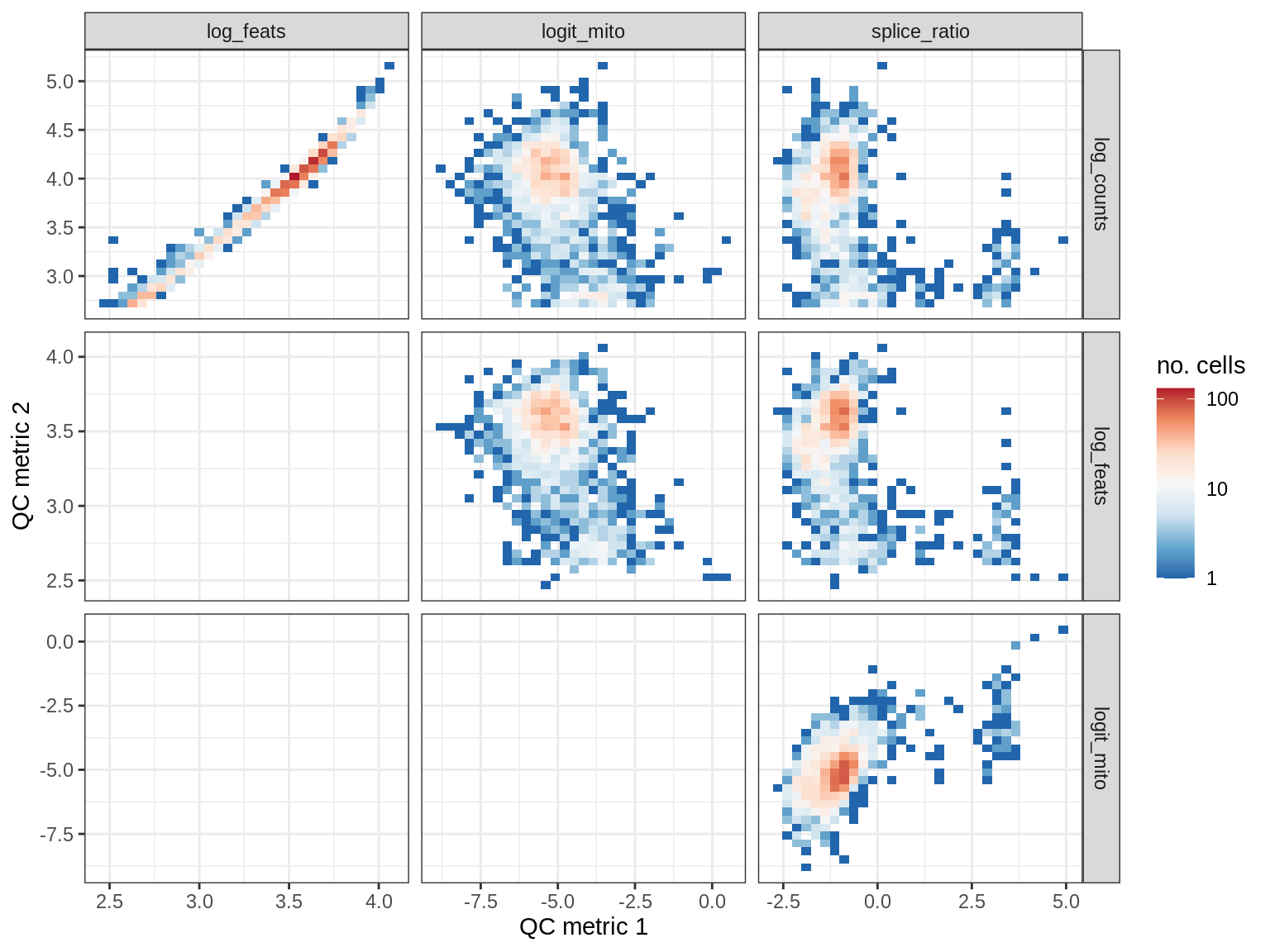
WM148
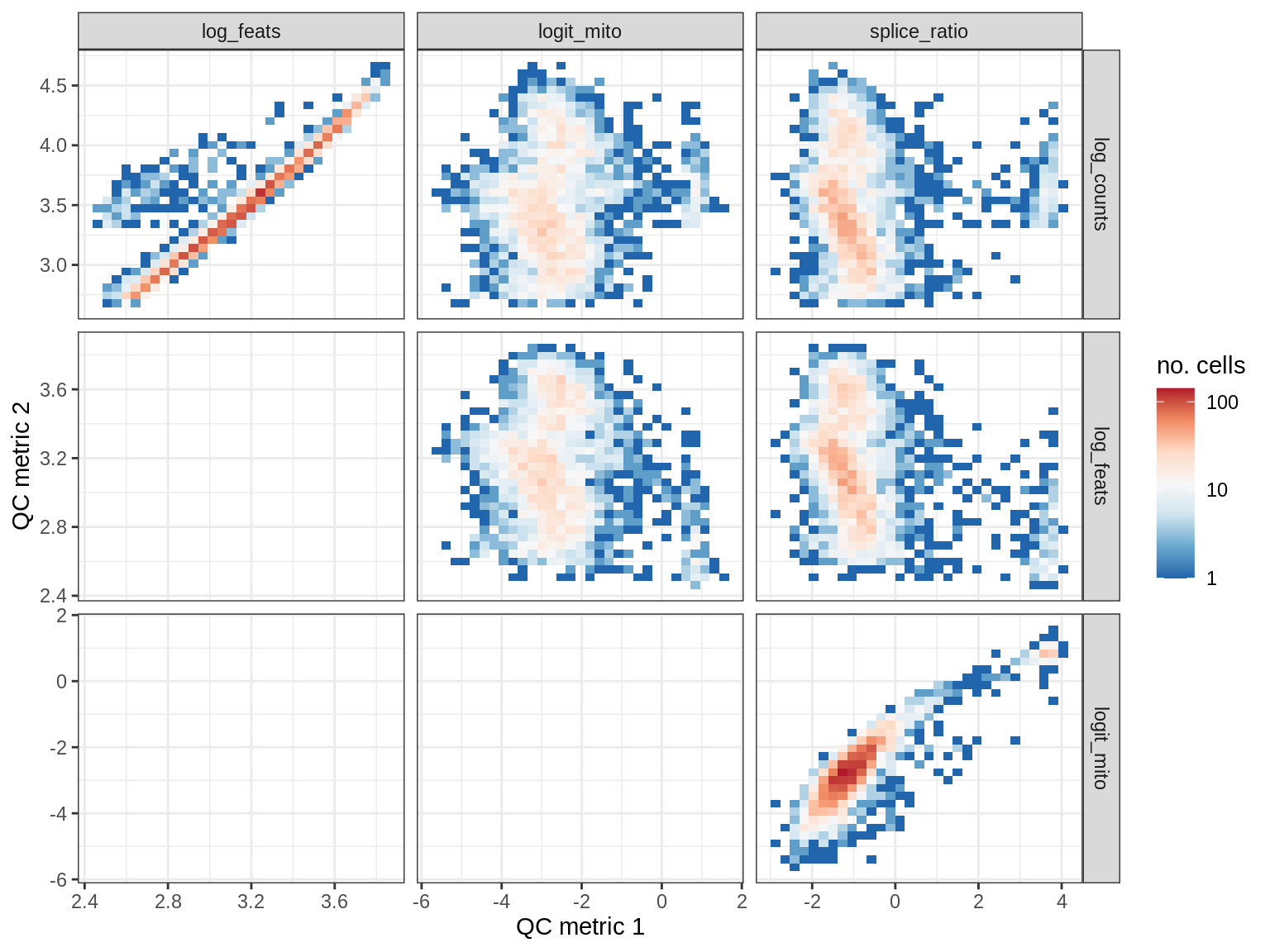
WM149
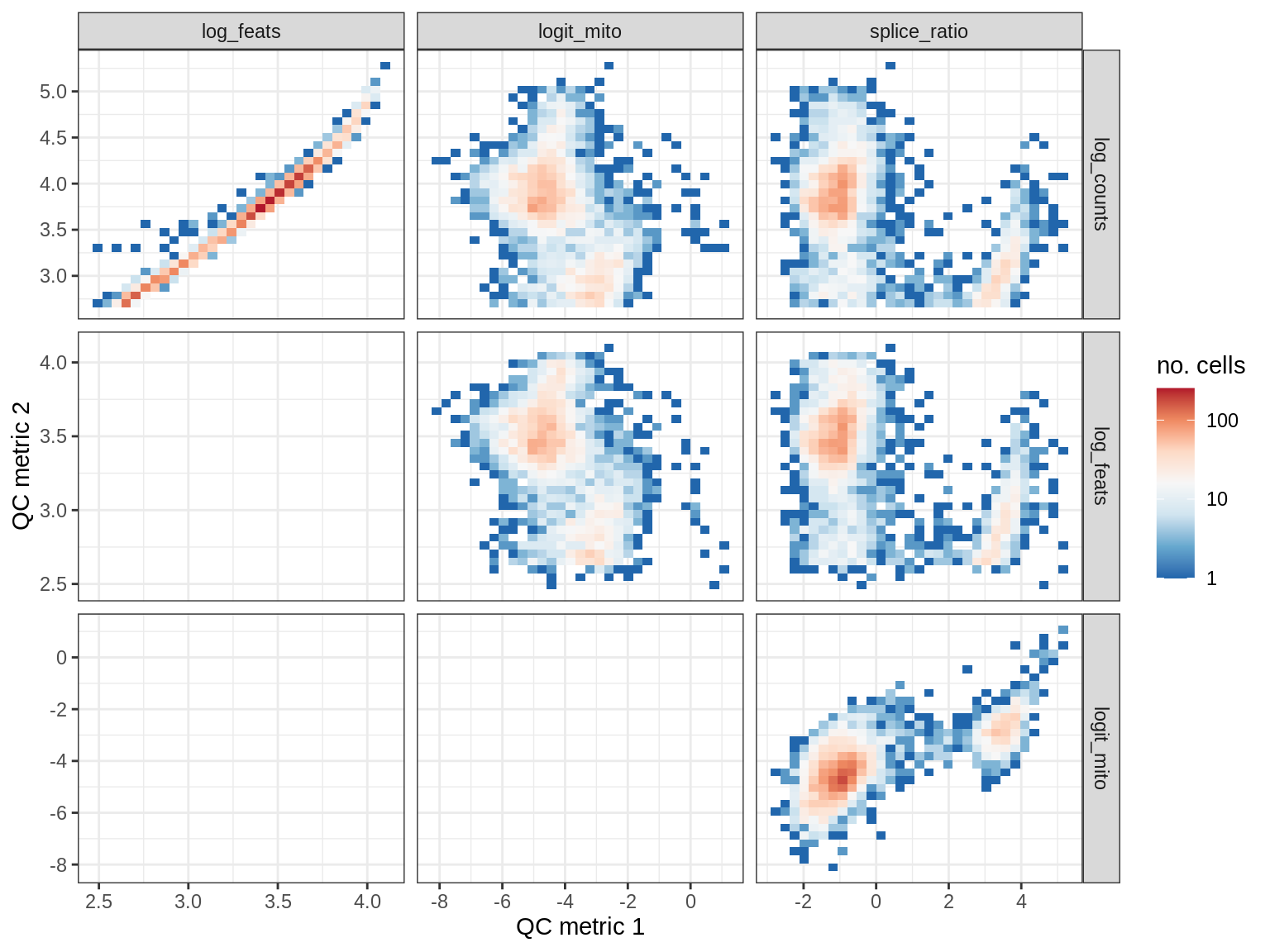
WM150
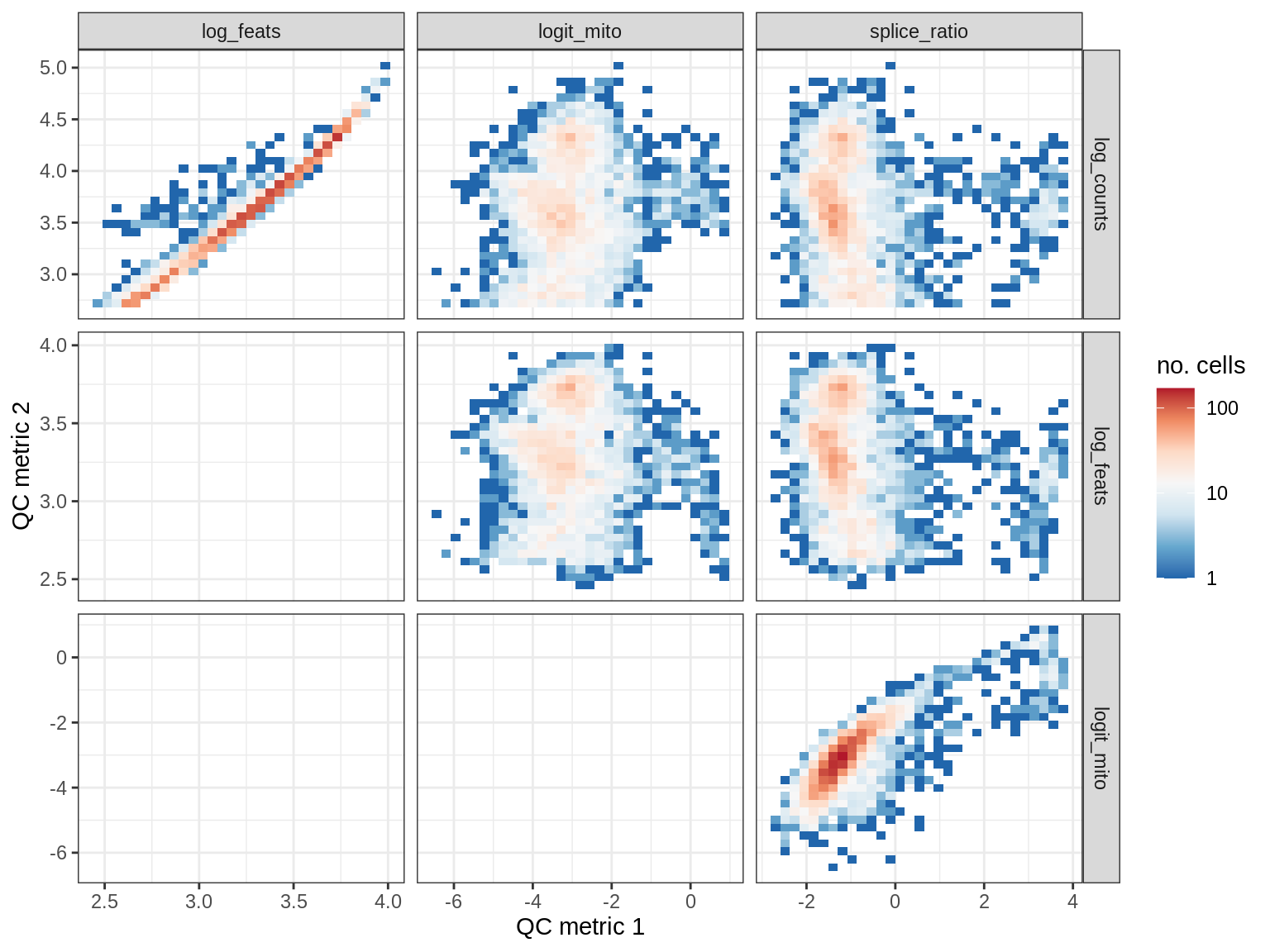
WM151
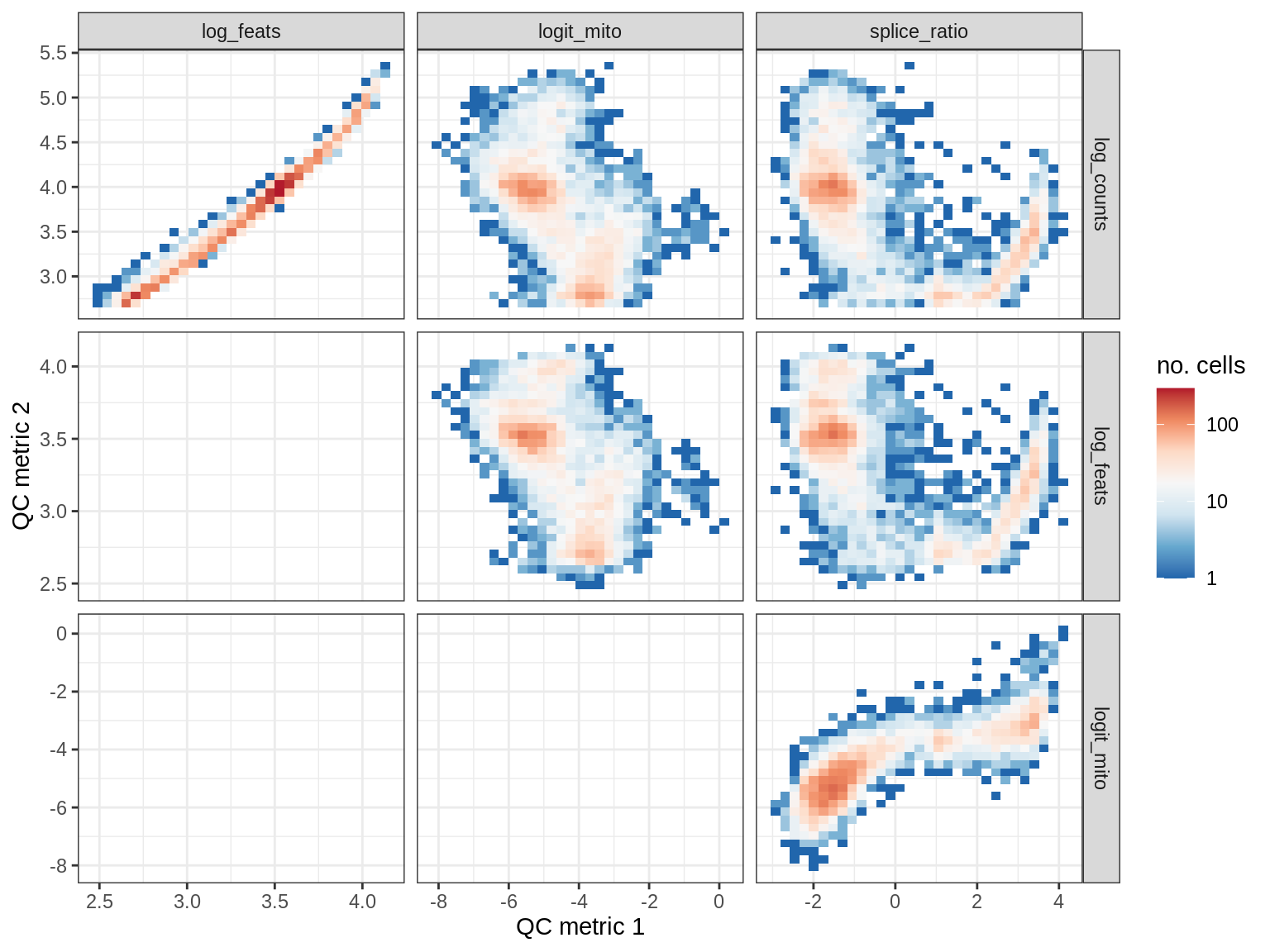
WM152
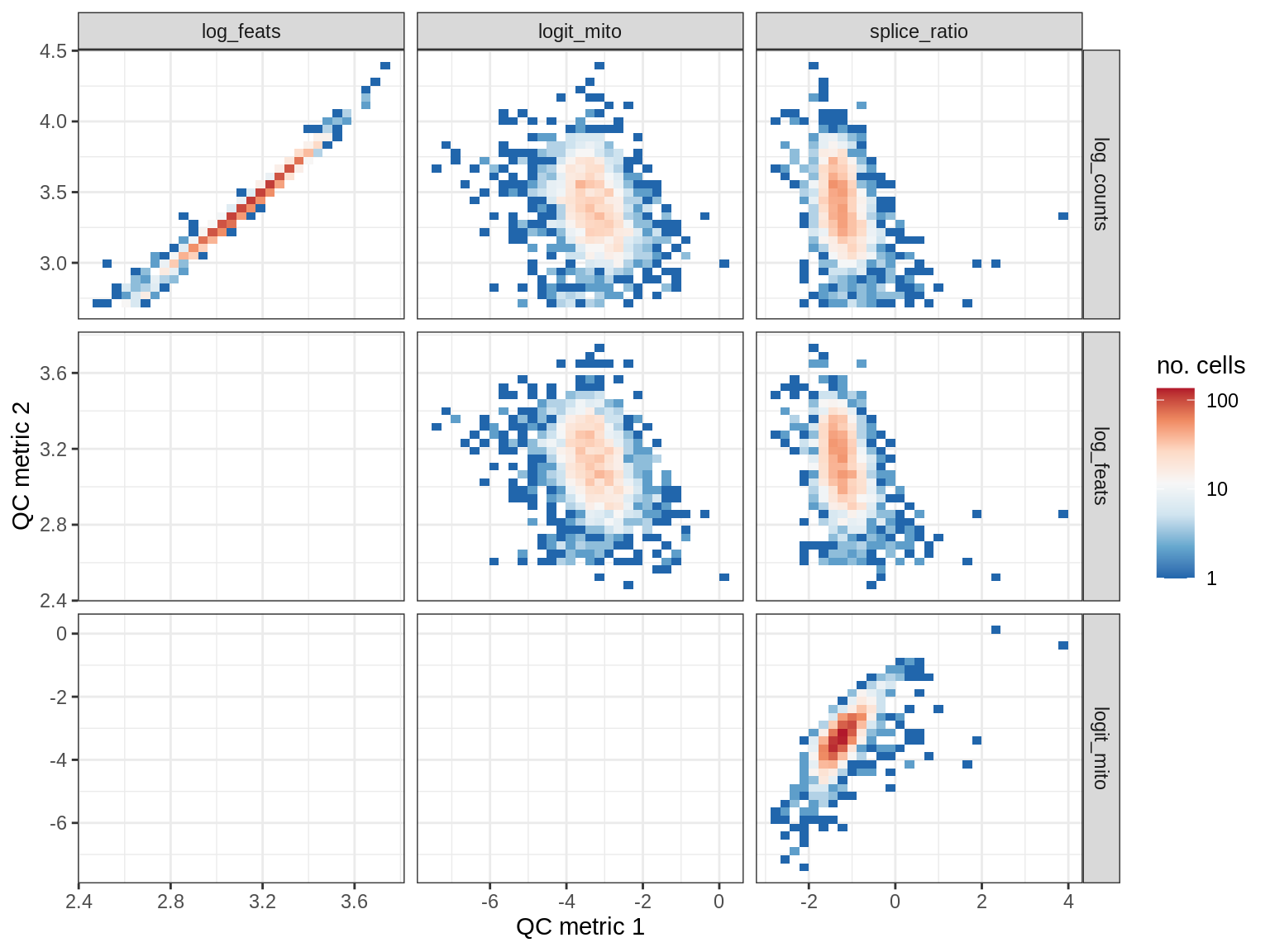
WM153
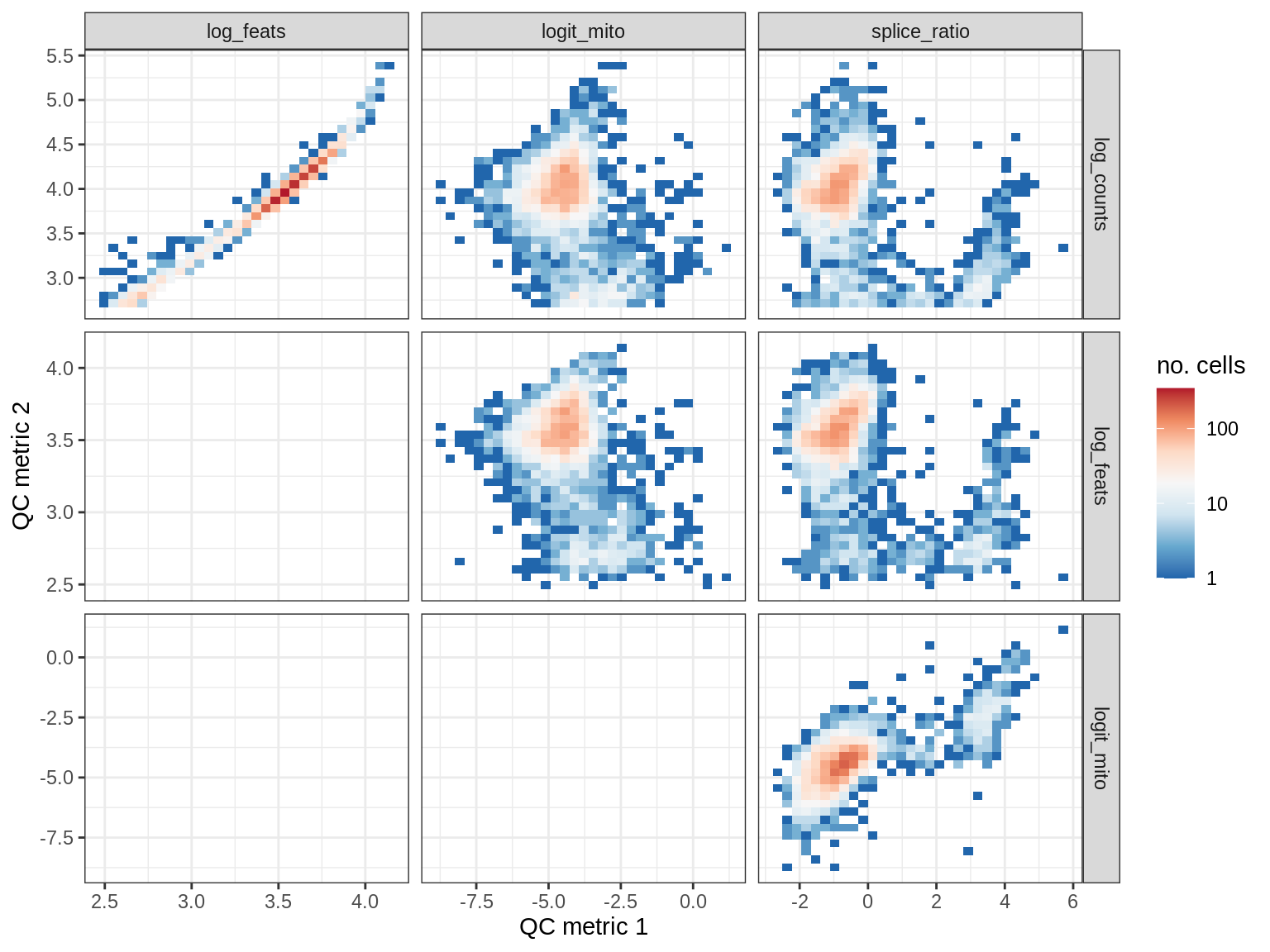
WM154
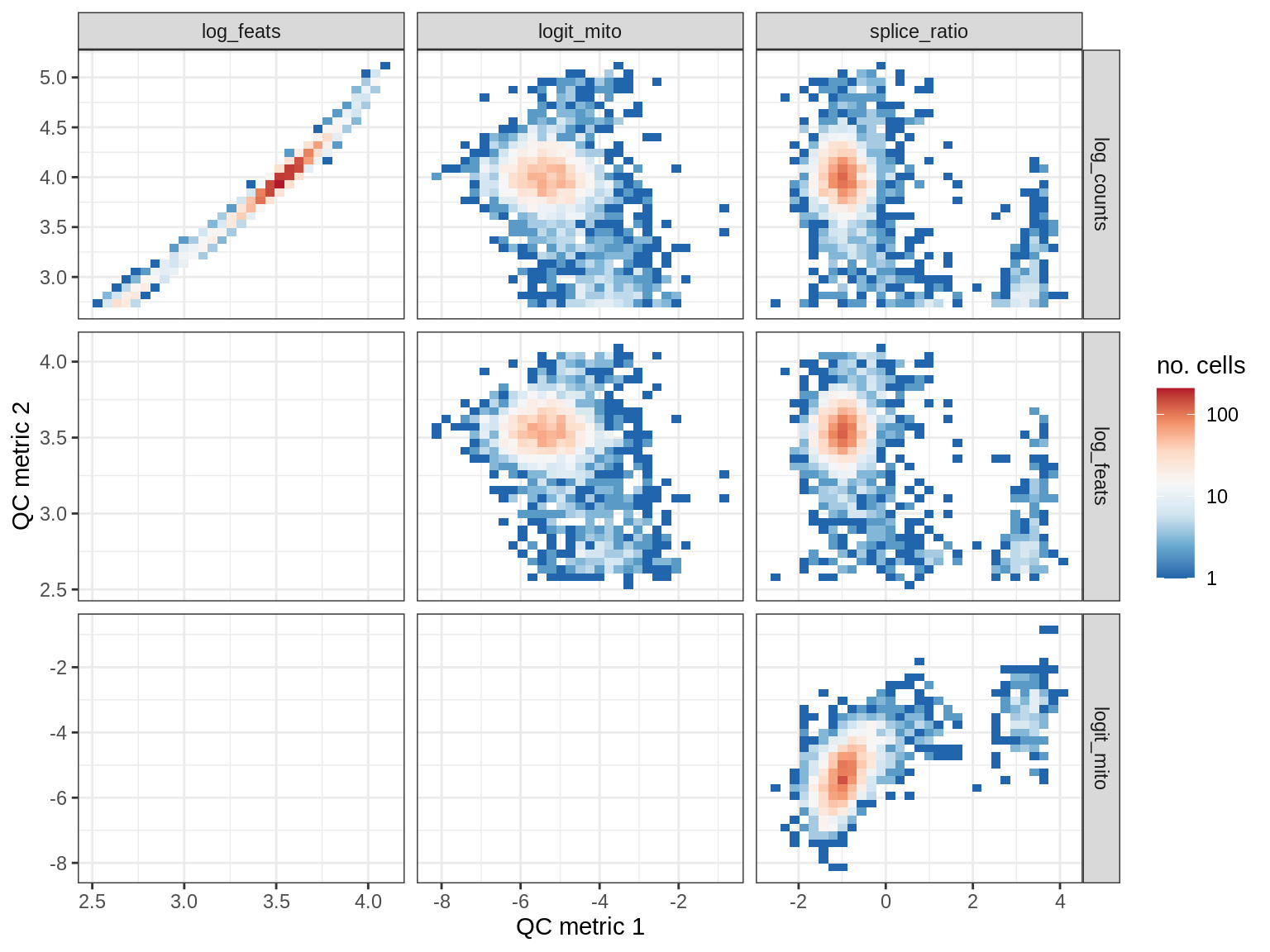
WM155
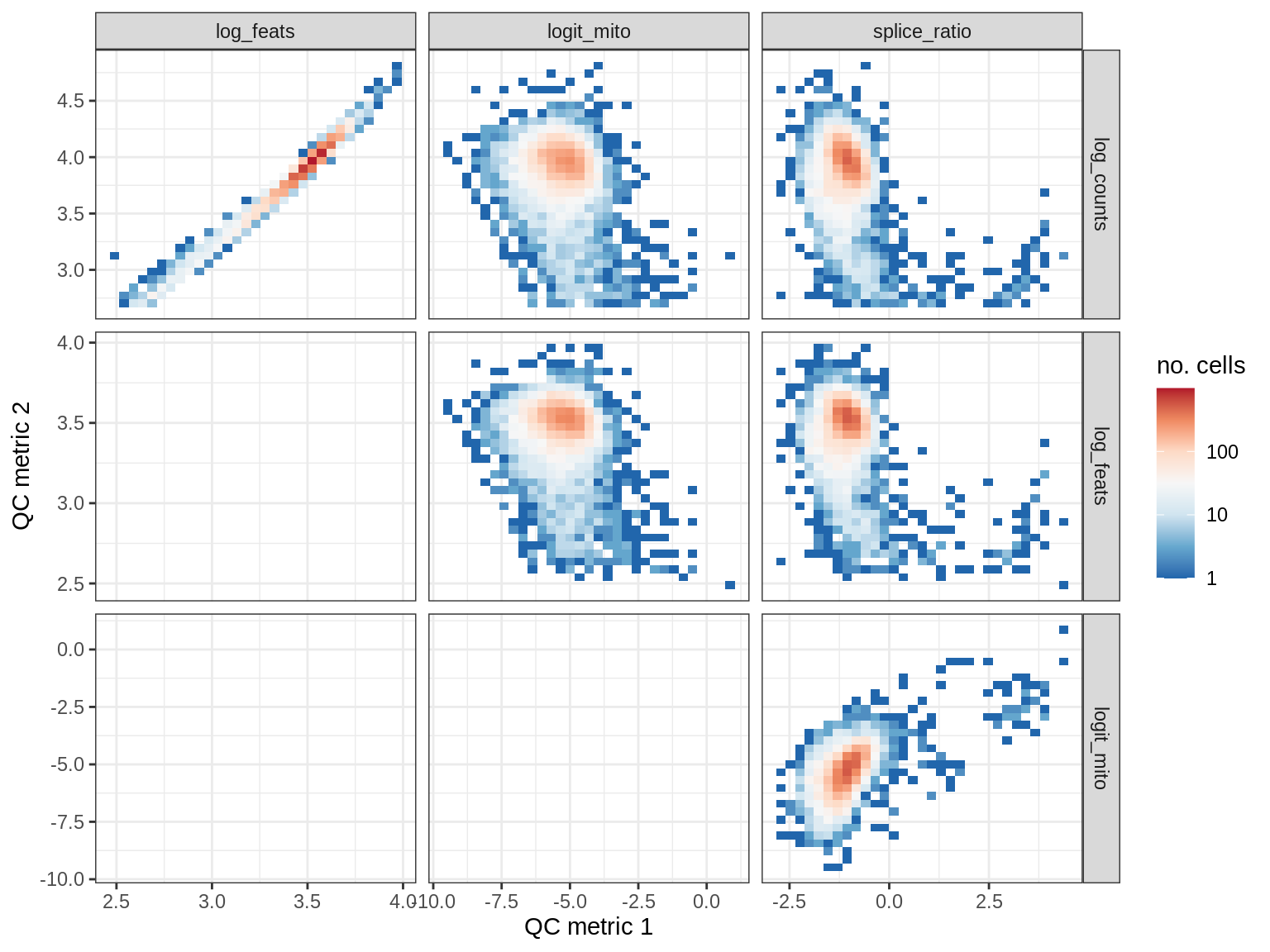
WM156
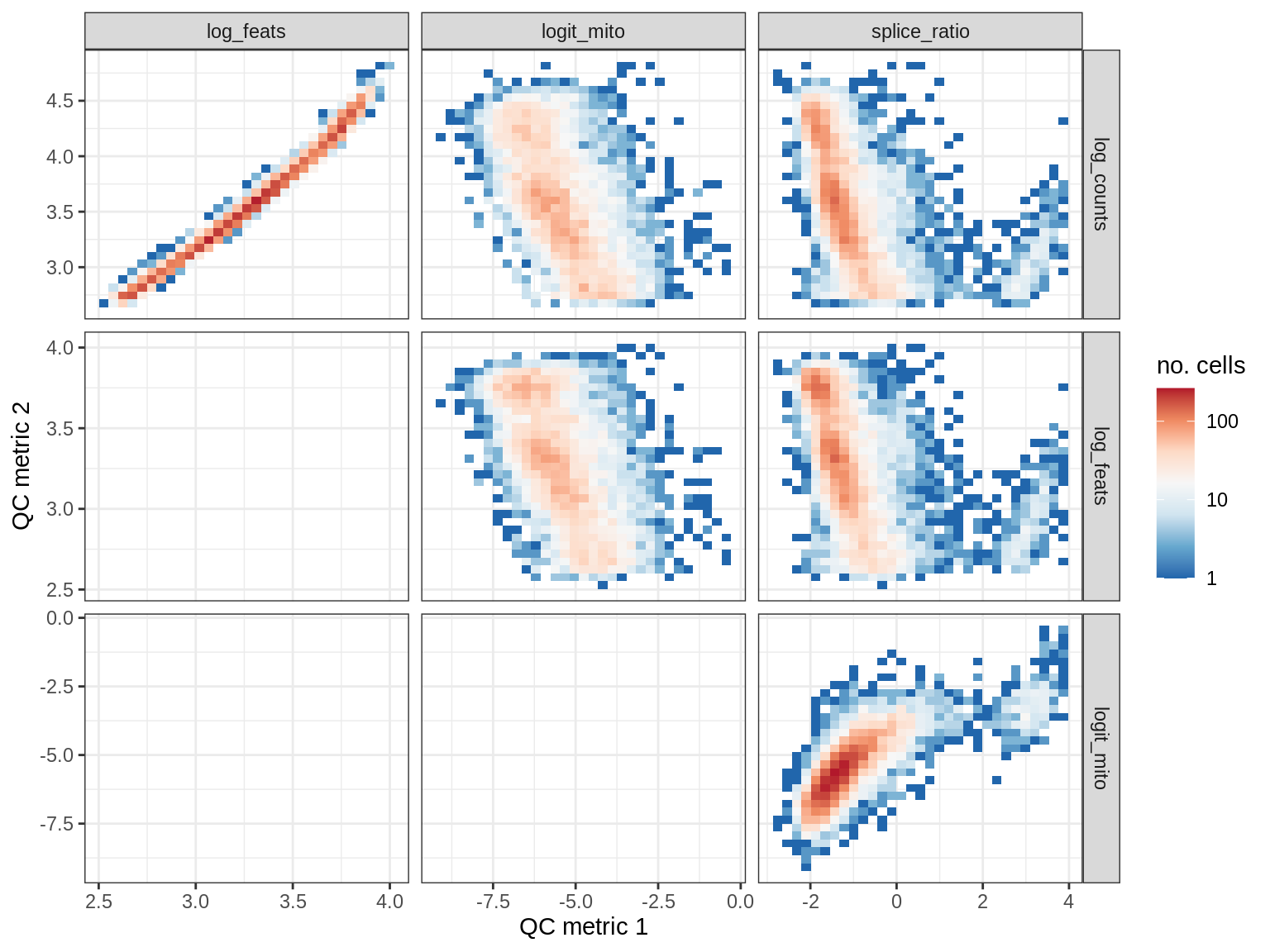
WM157
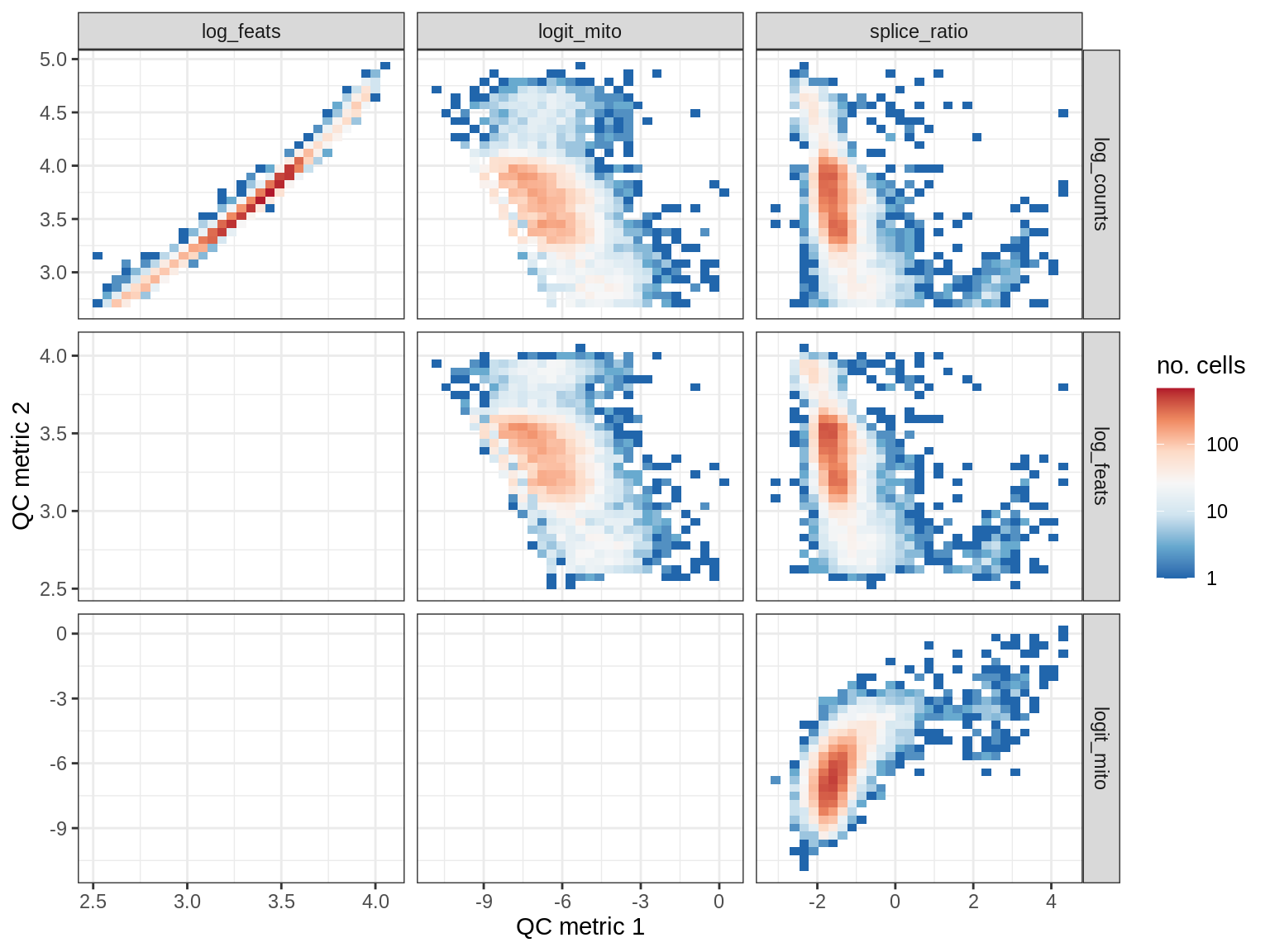
WM158
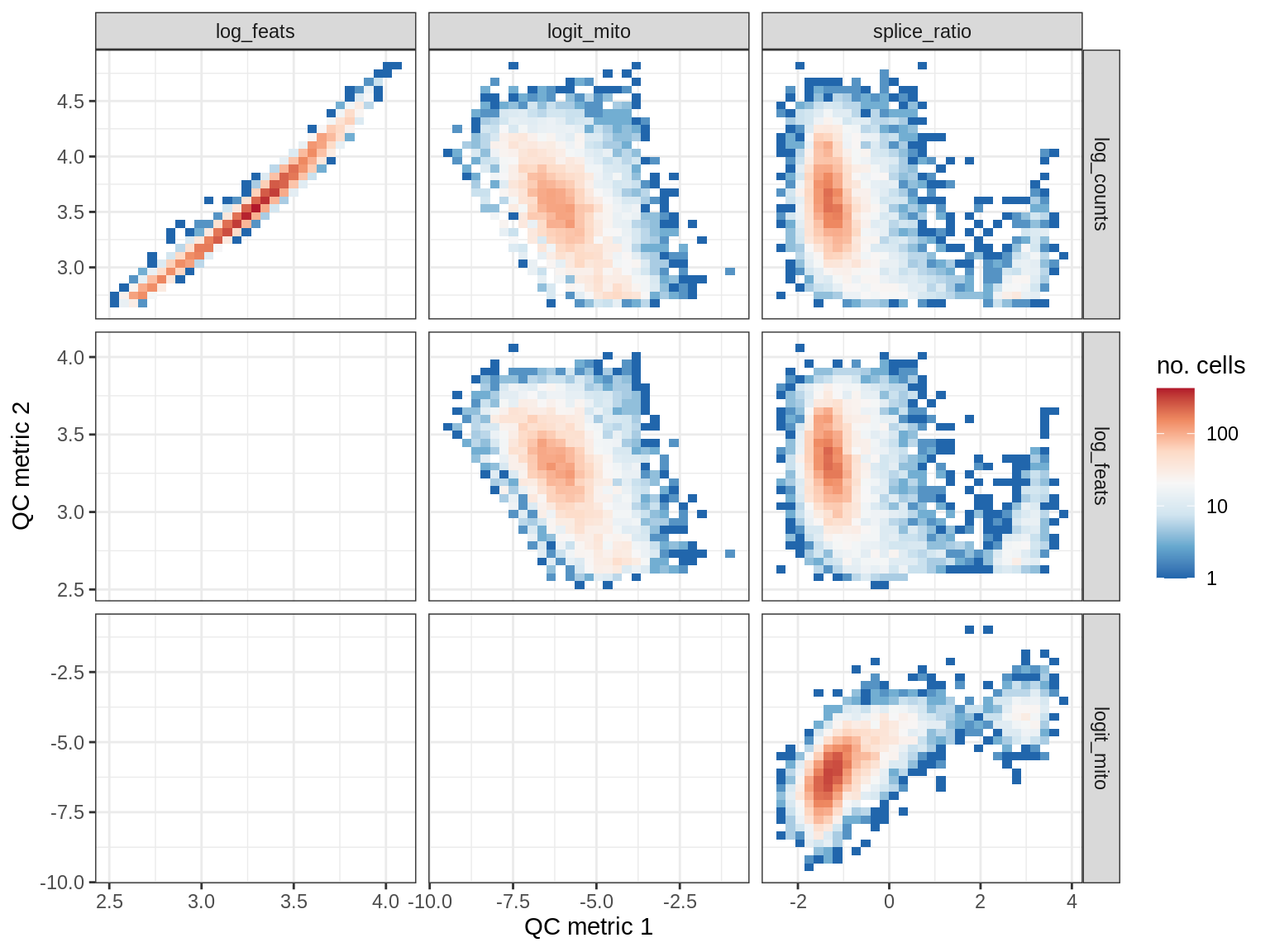
WM159
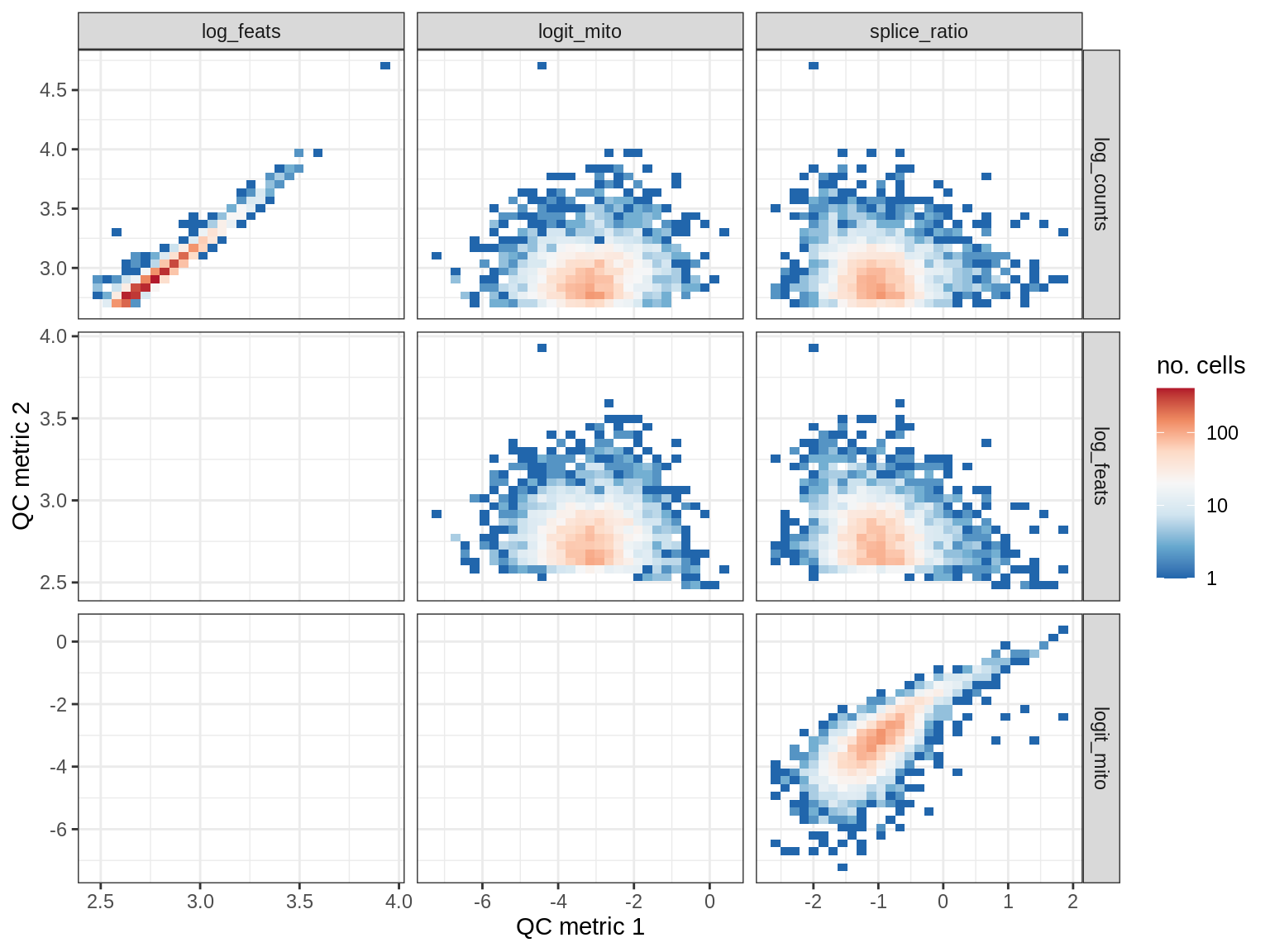
WM160
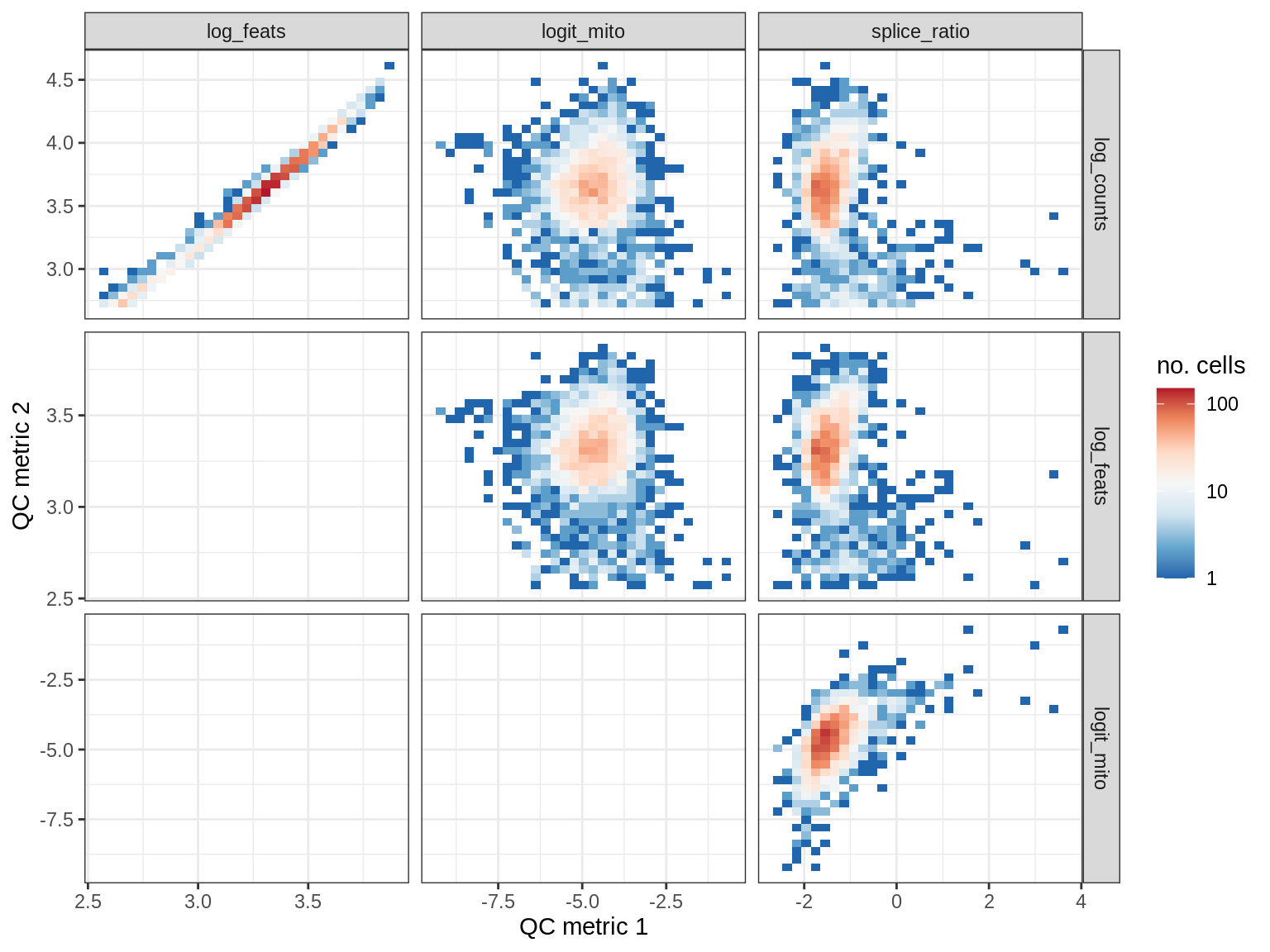
WM161
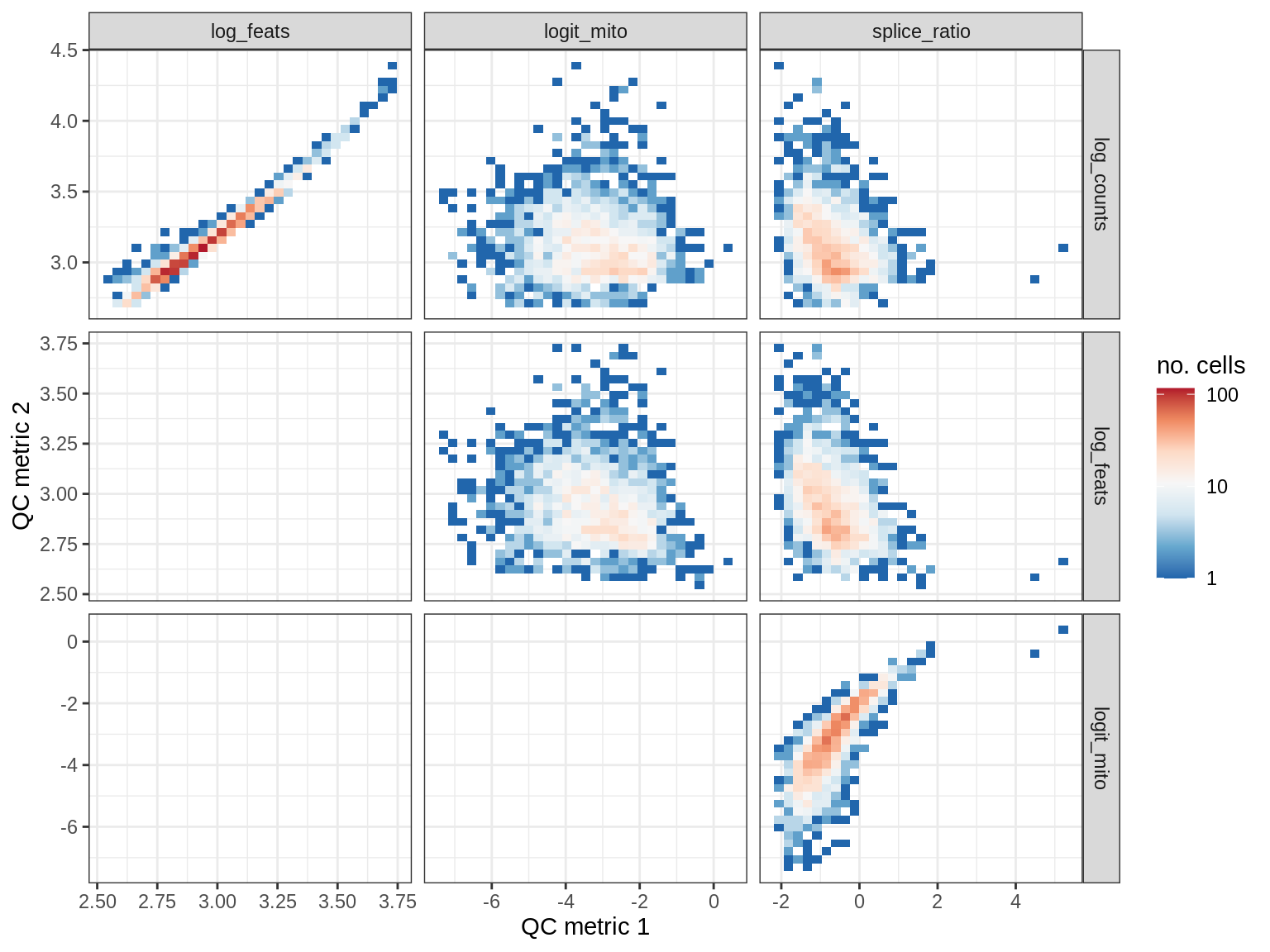
WM162
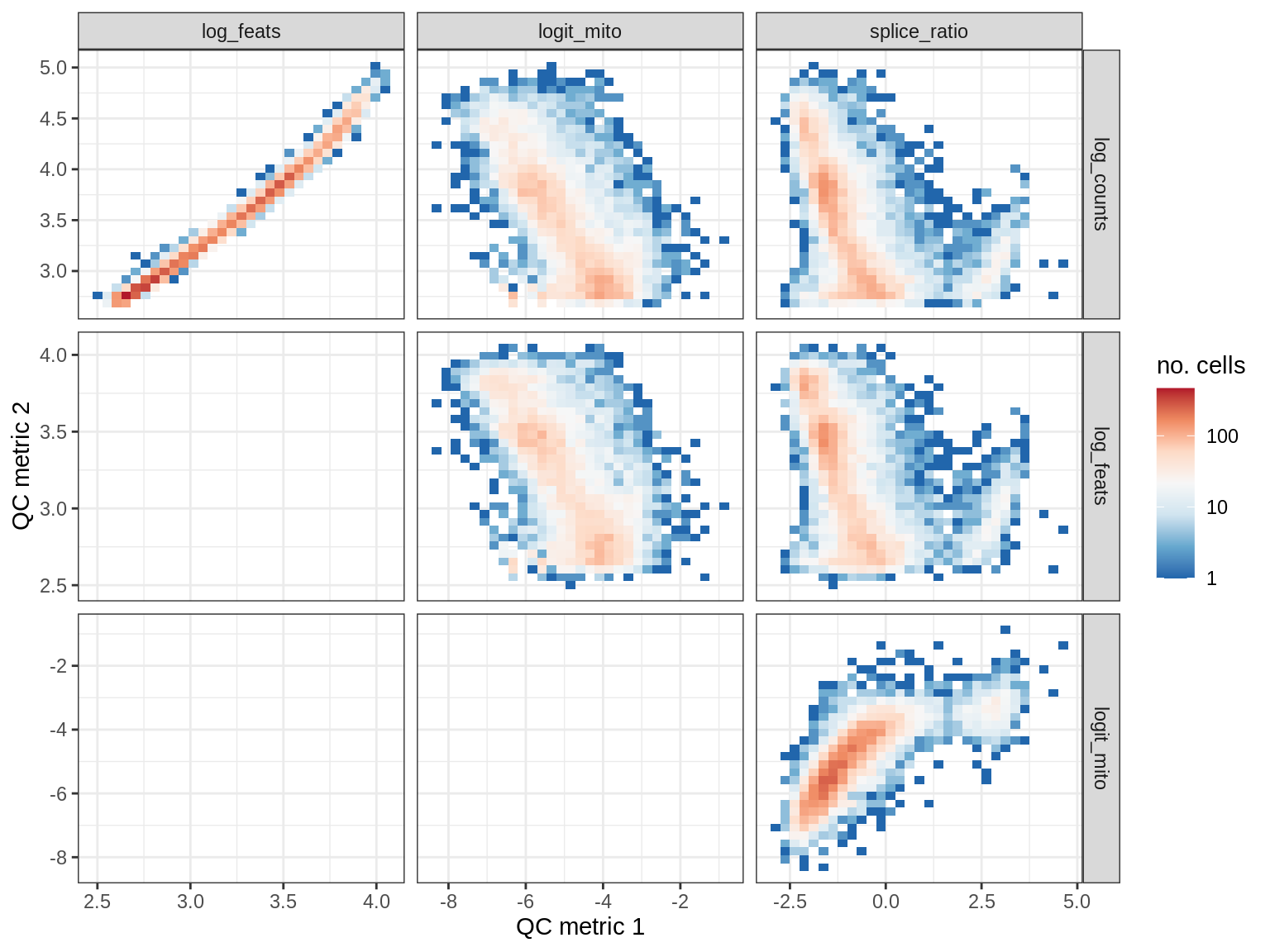
WM163
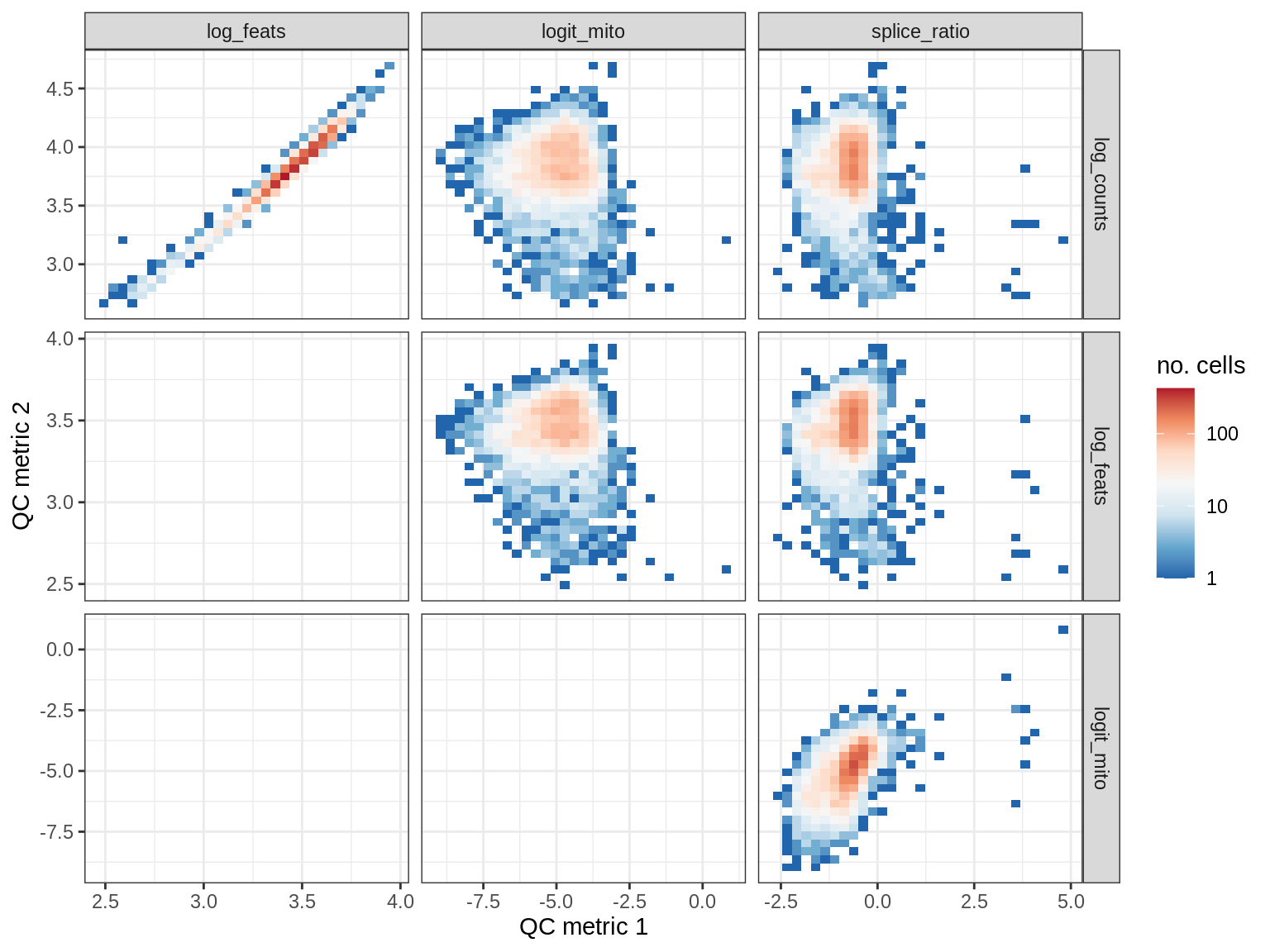
WM164
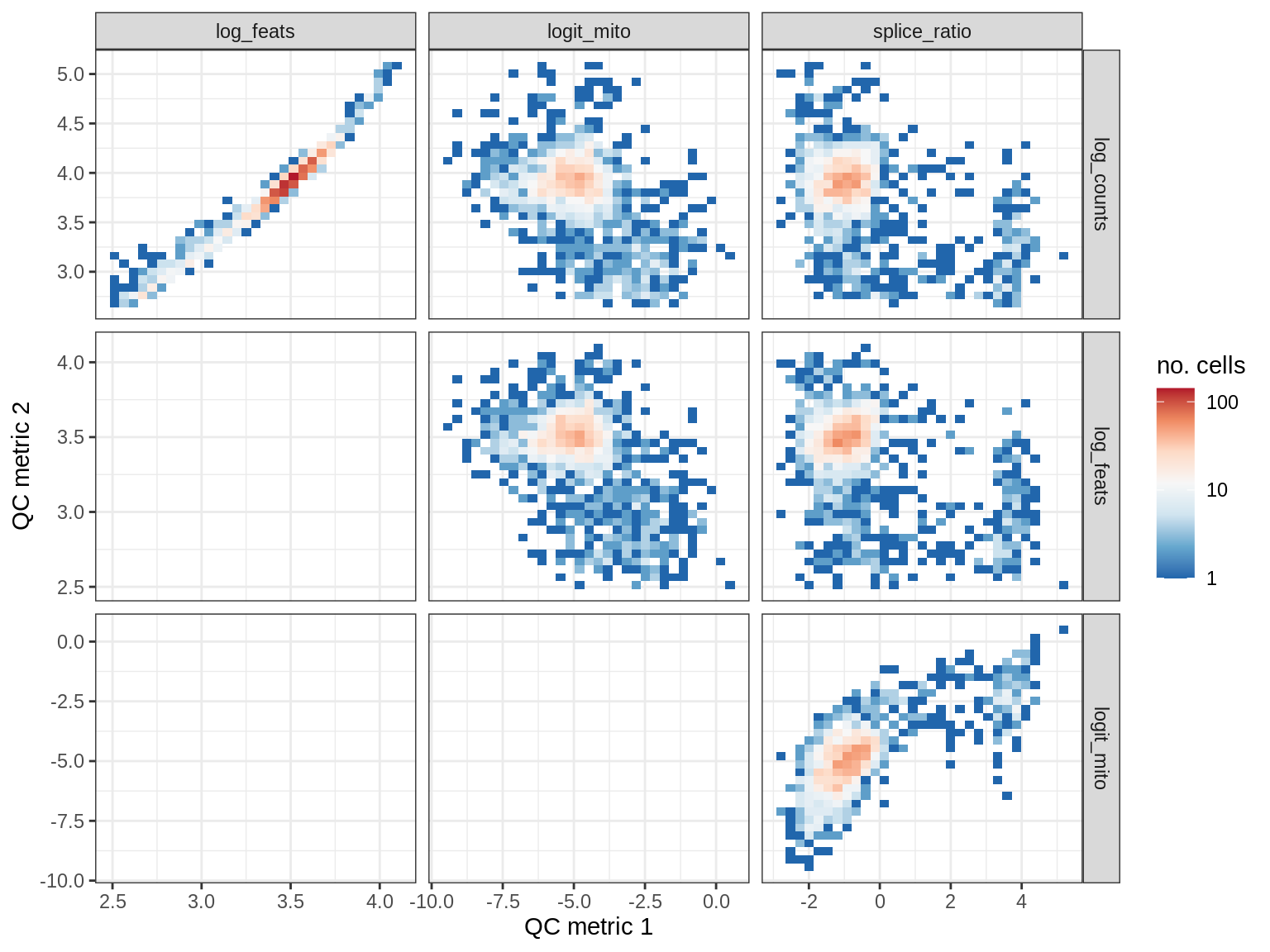
WM166
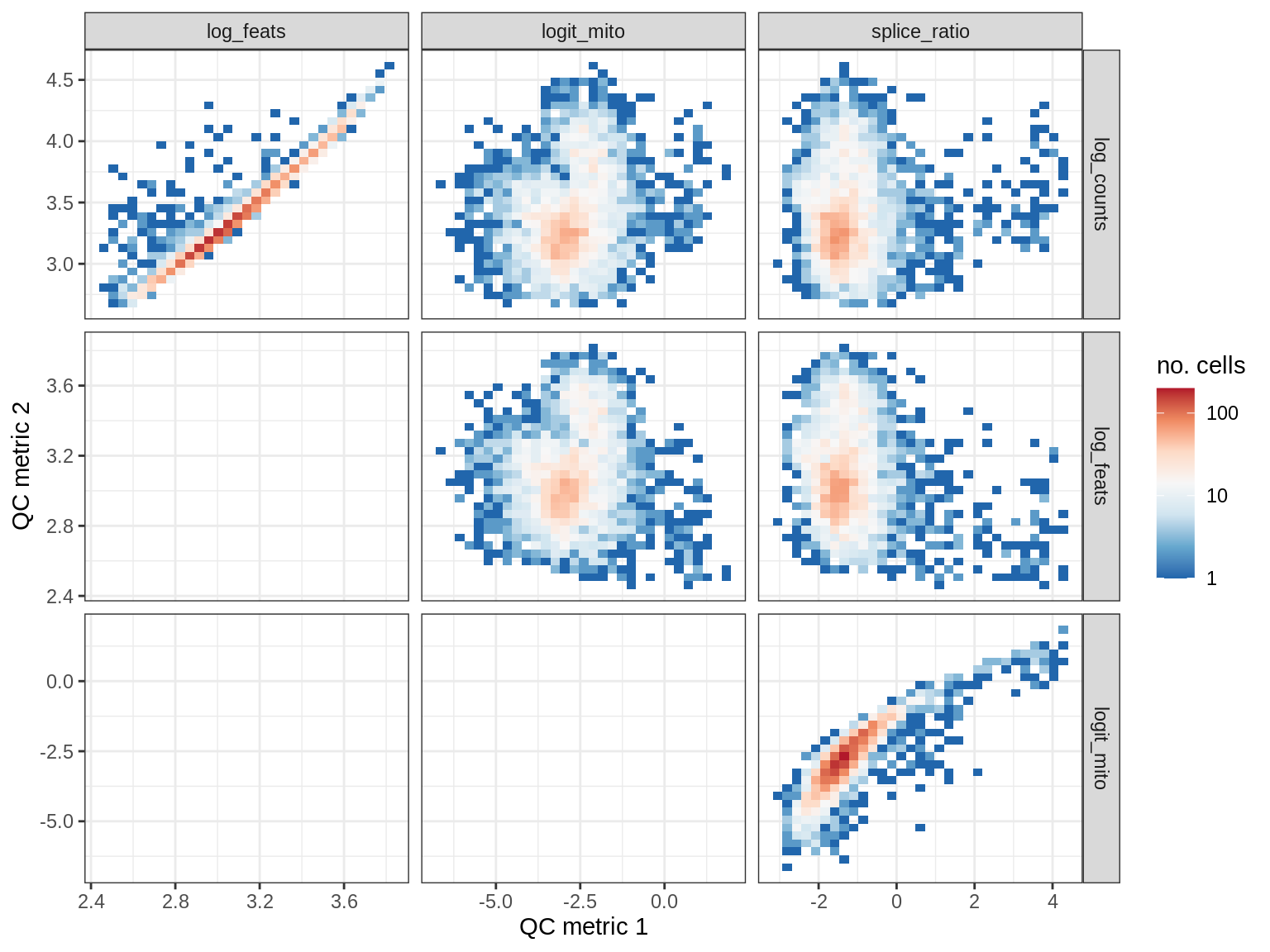
WM167
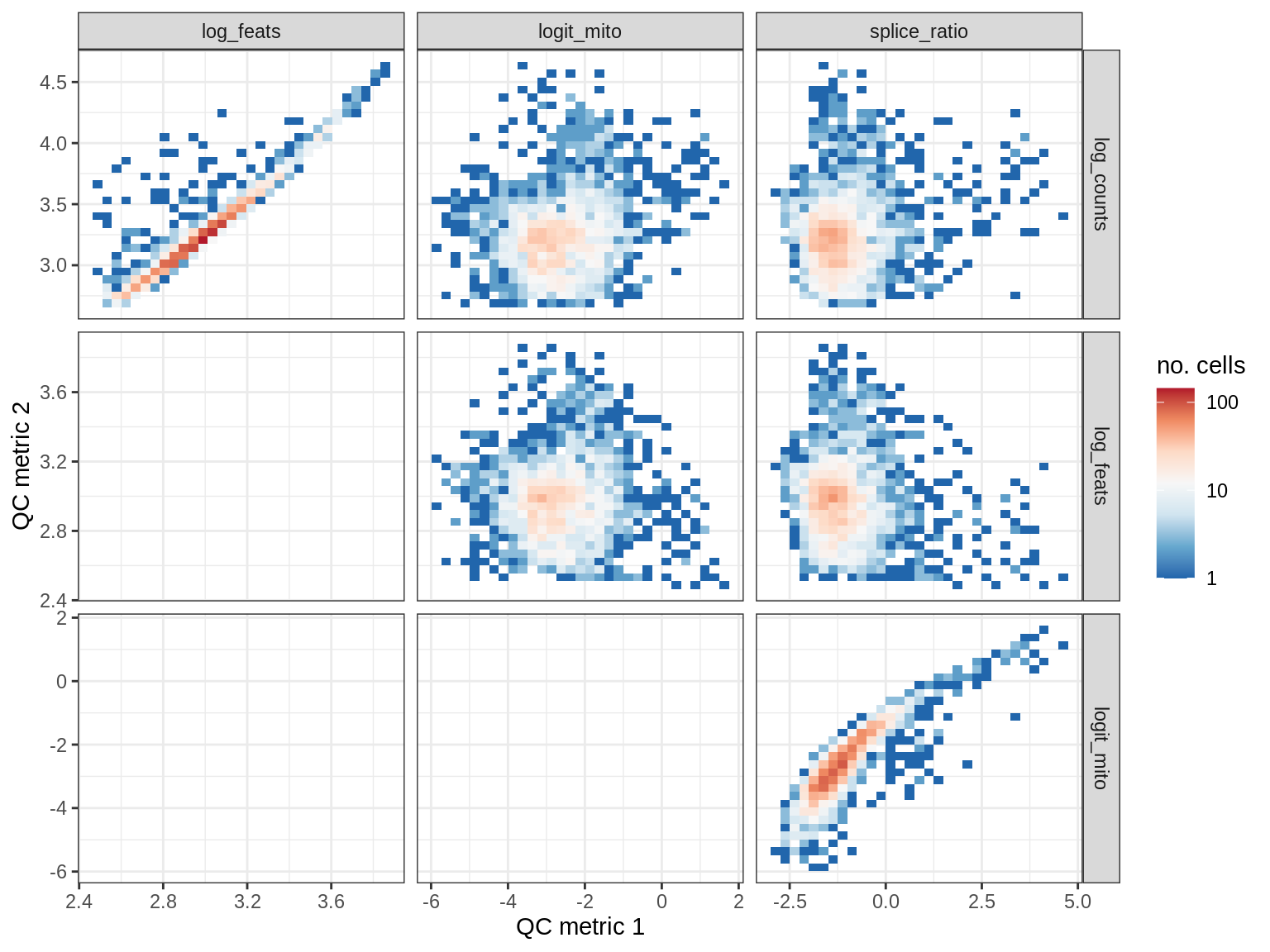
WM168
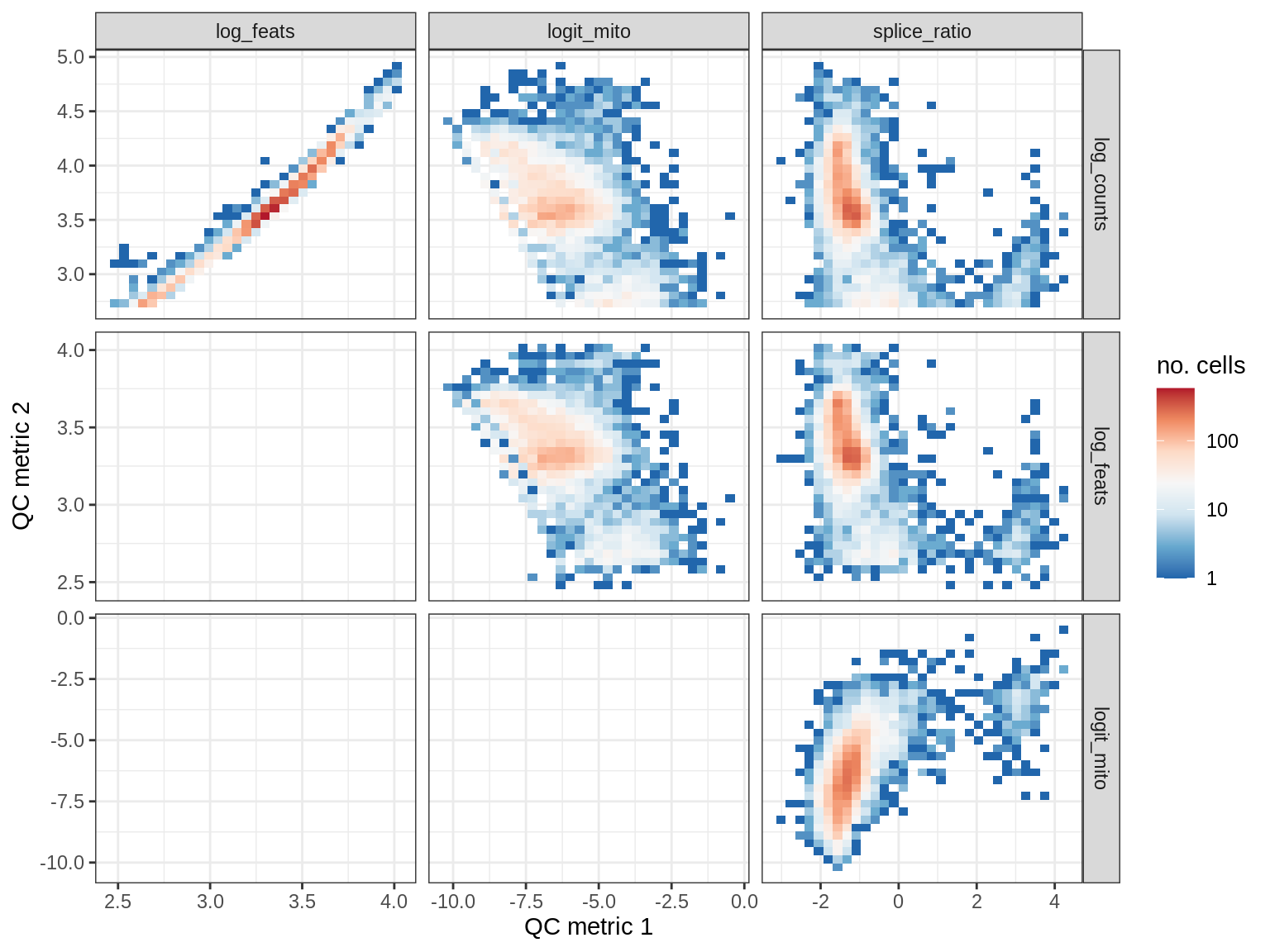
WM169
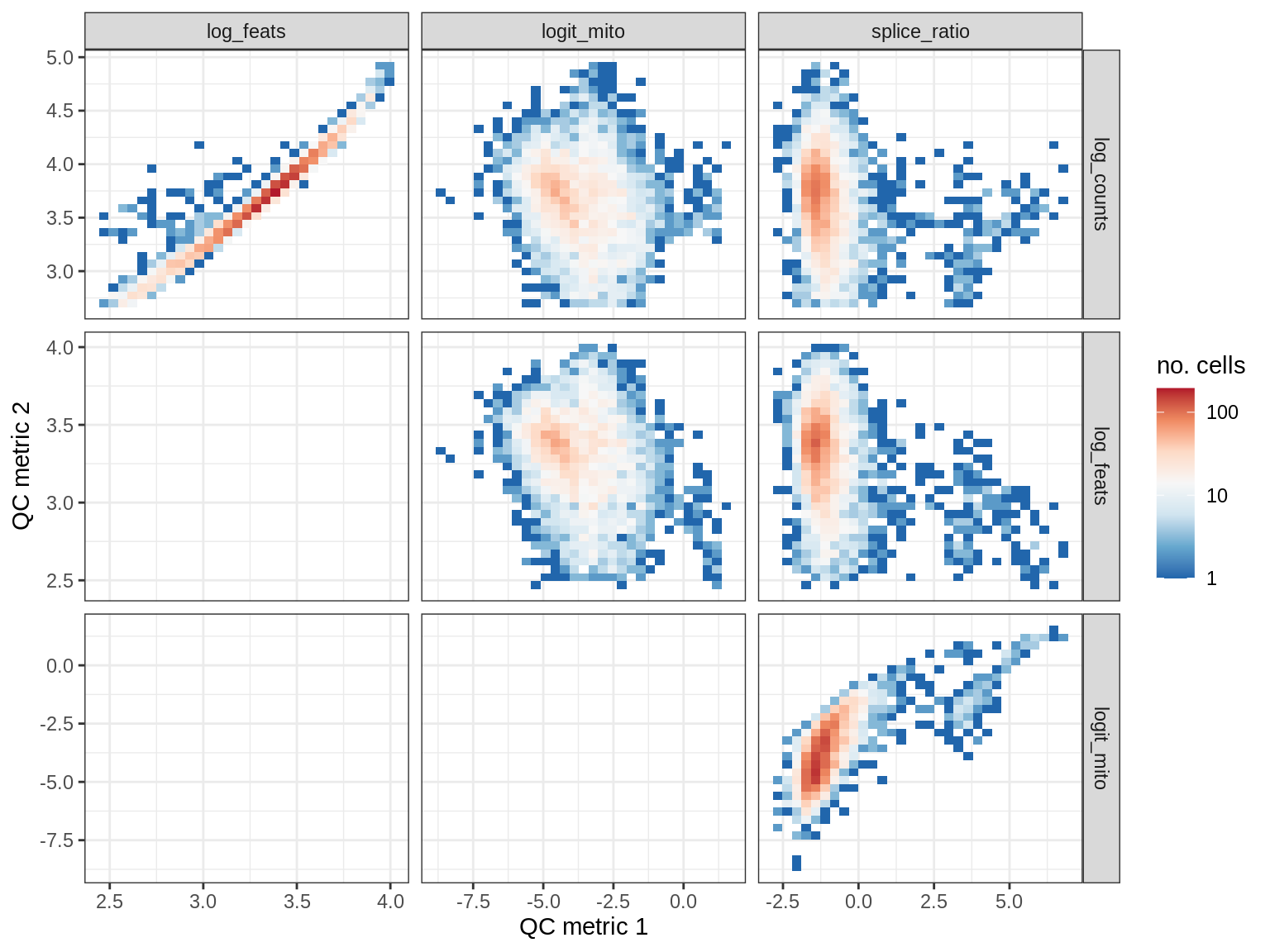
WM170
WM171
WM172
WM173
WM174
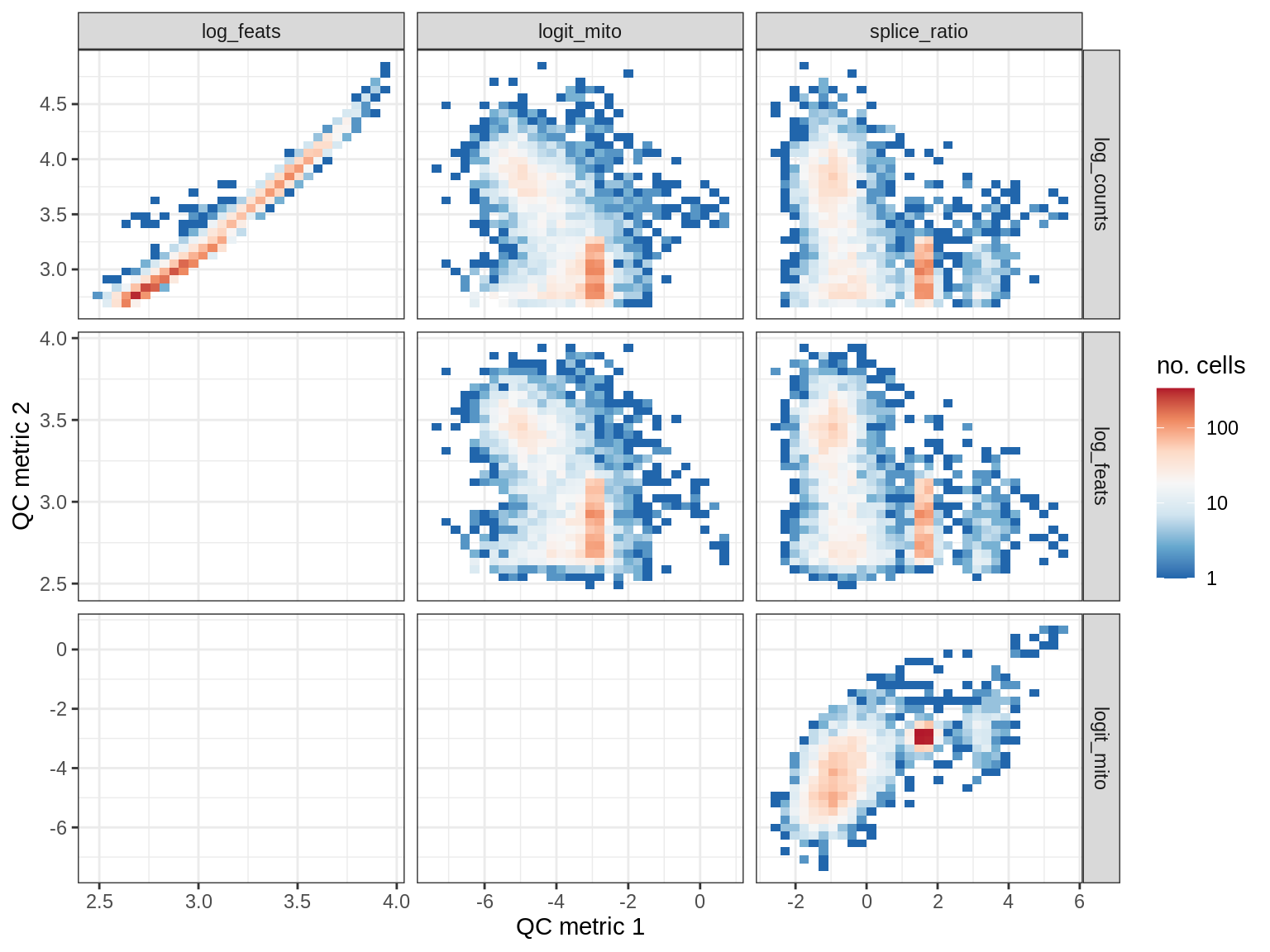
WM175
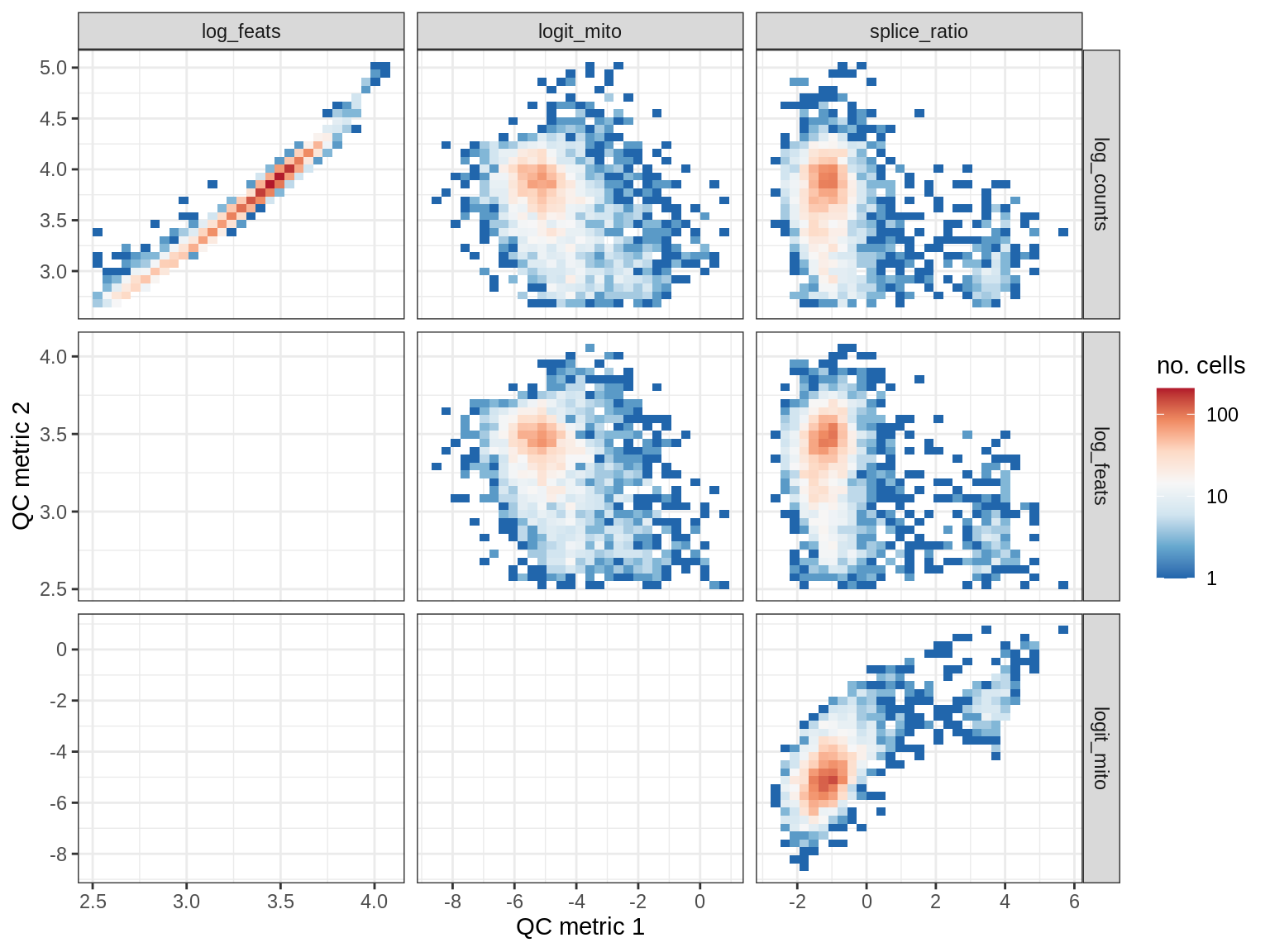
WM176
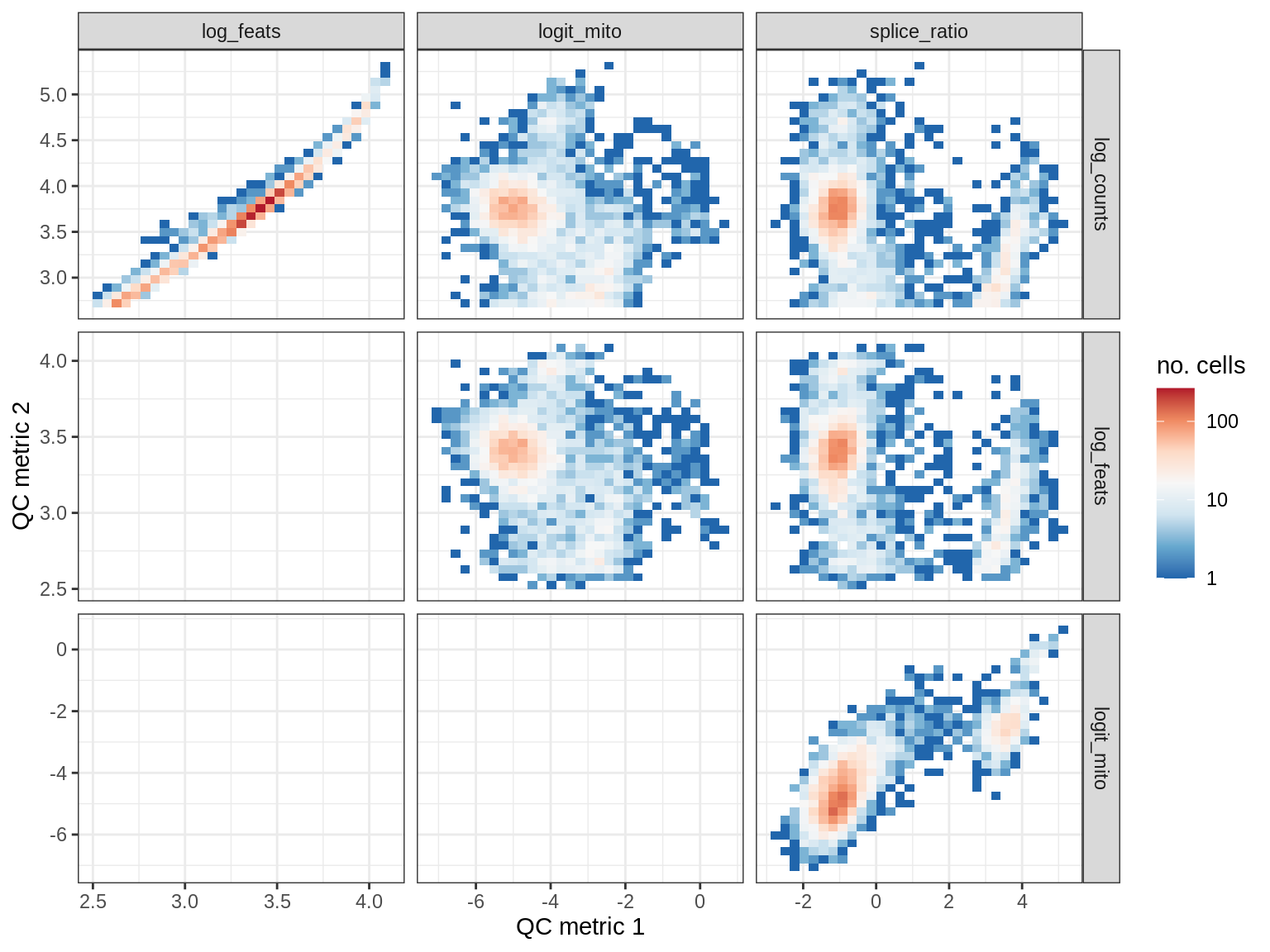
WM177
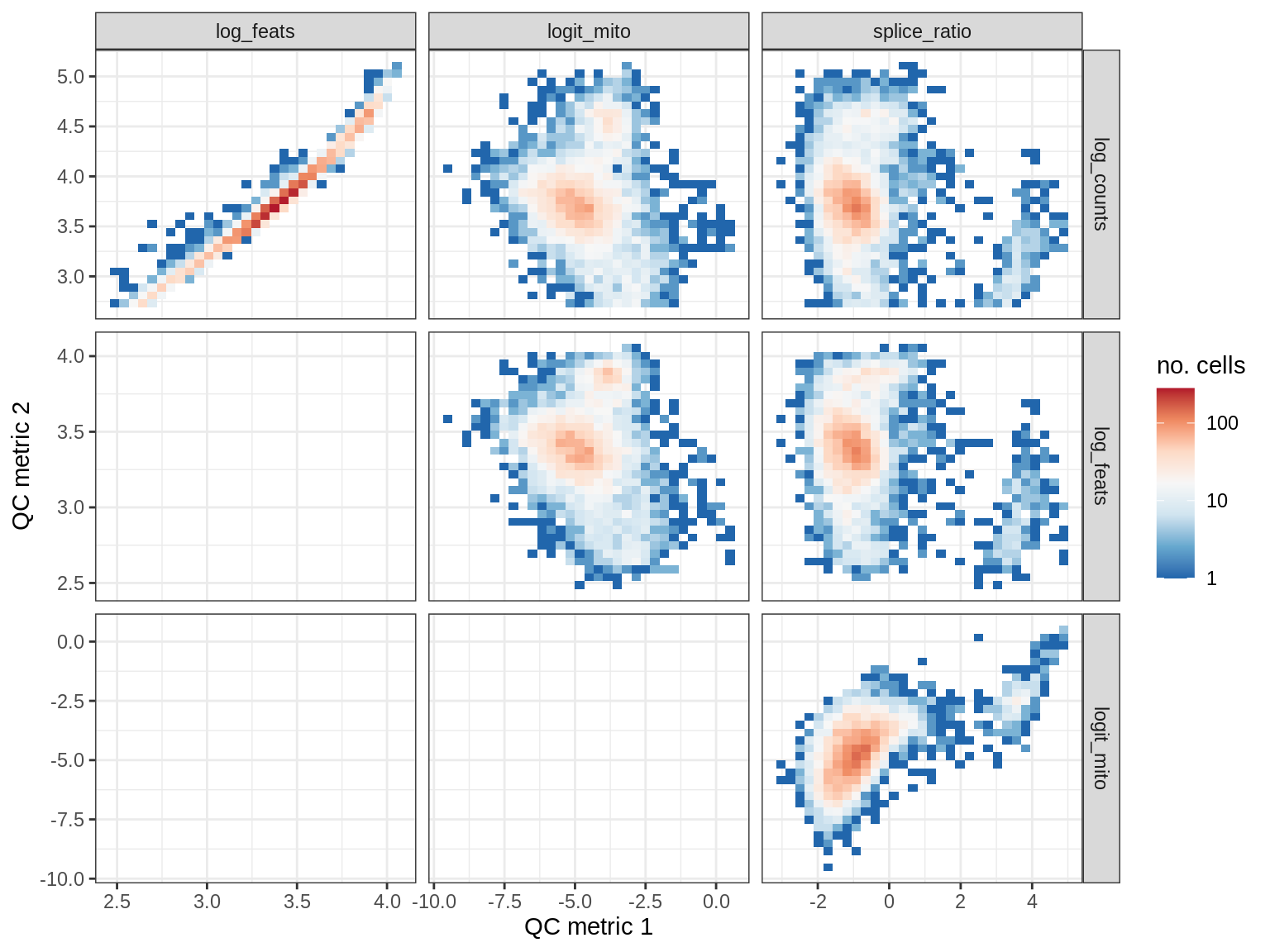
WM178
WM179
WM180
WM181
WM182
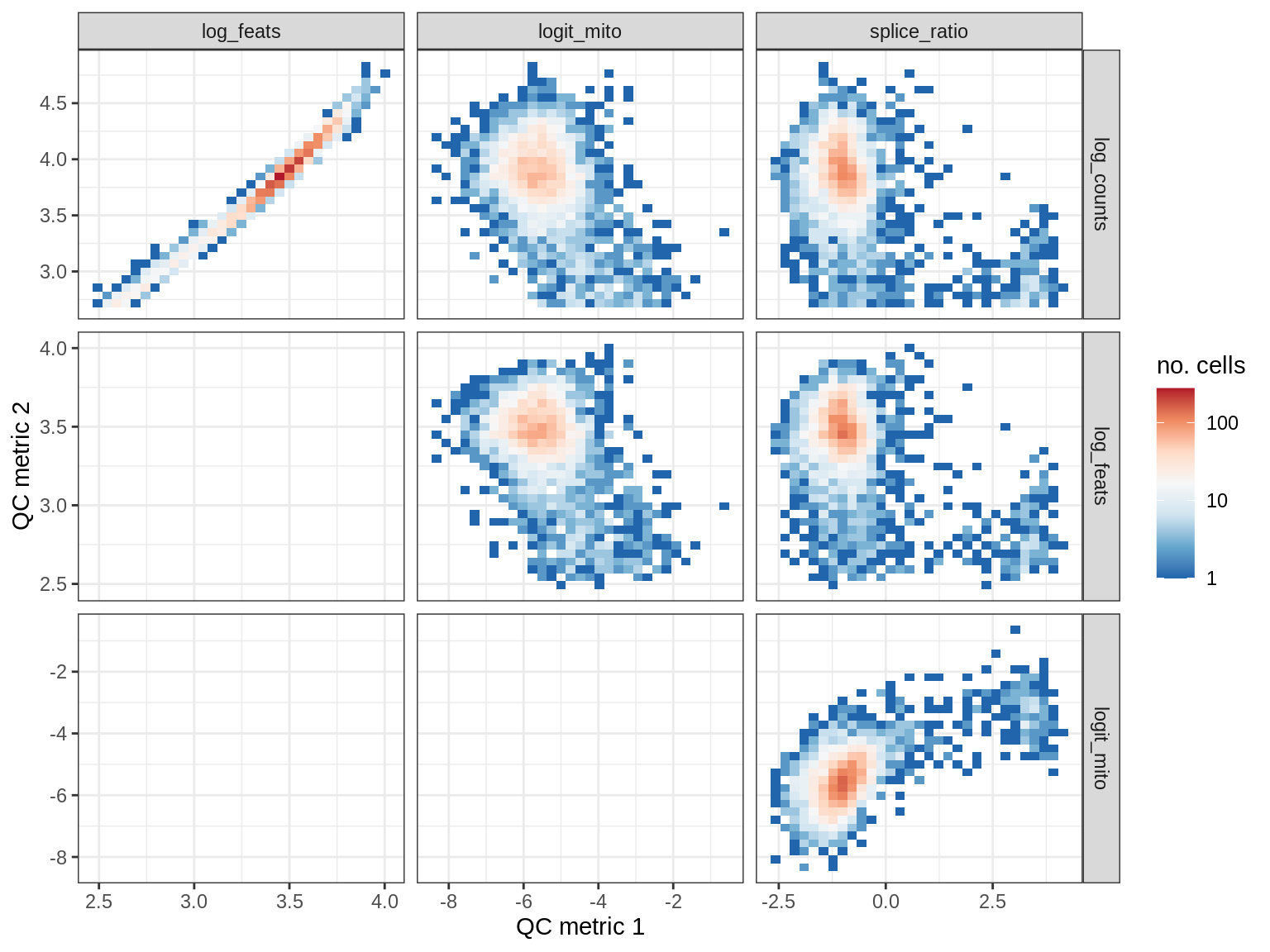
WM183
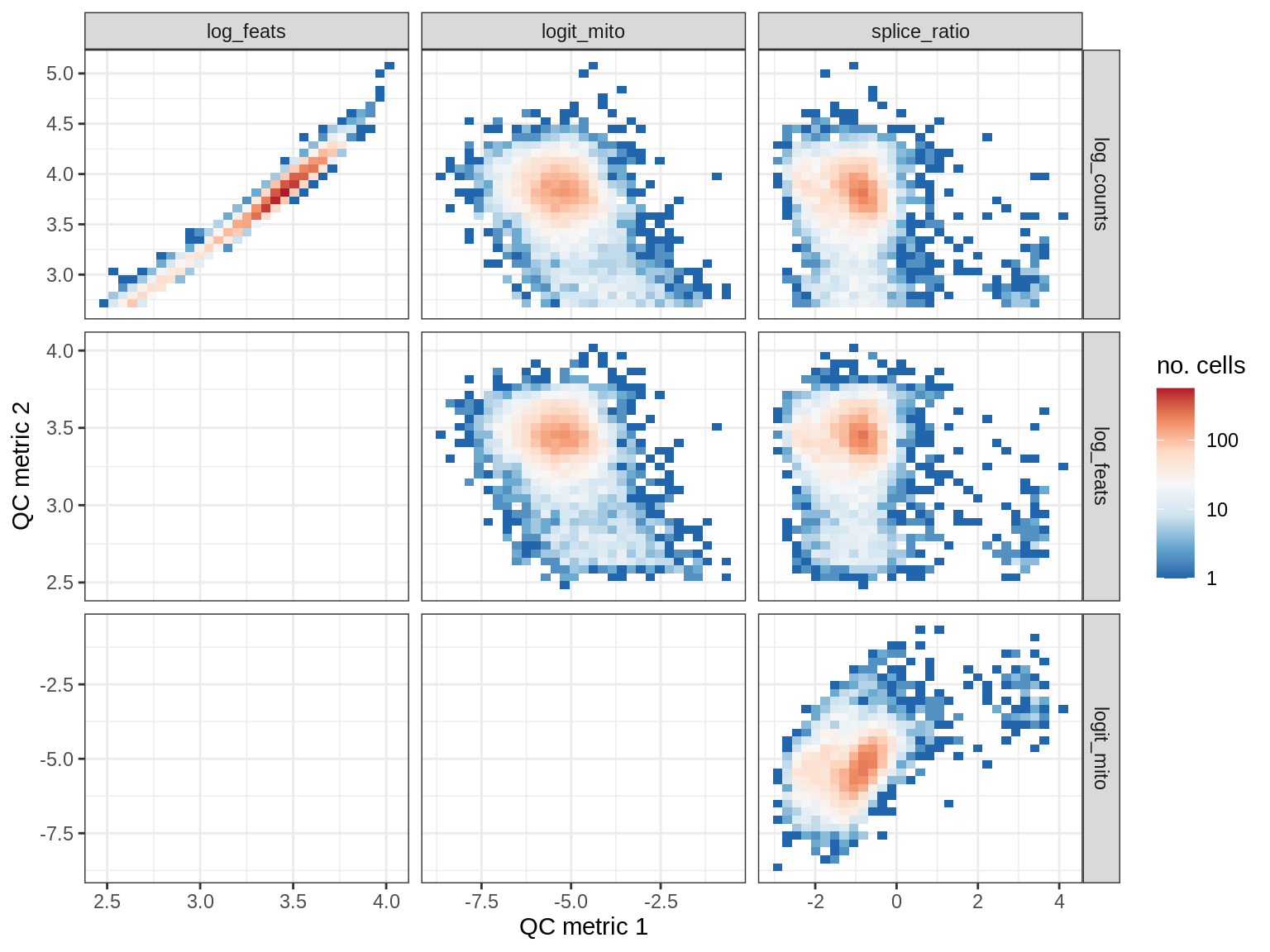
WM184
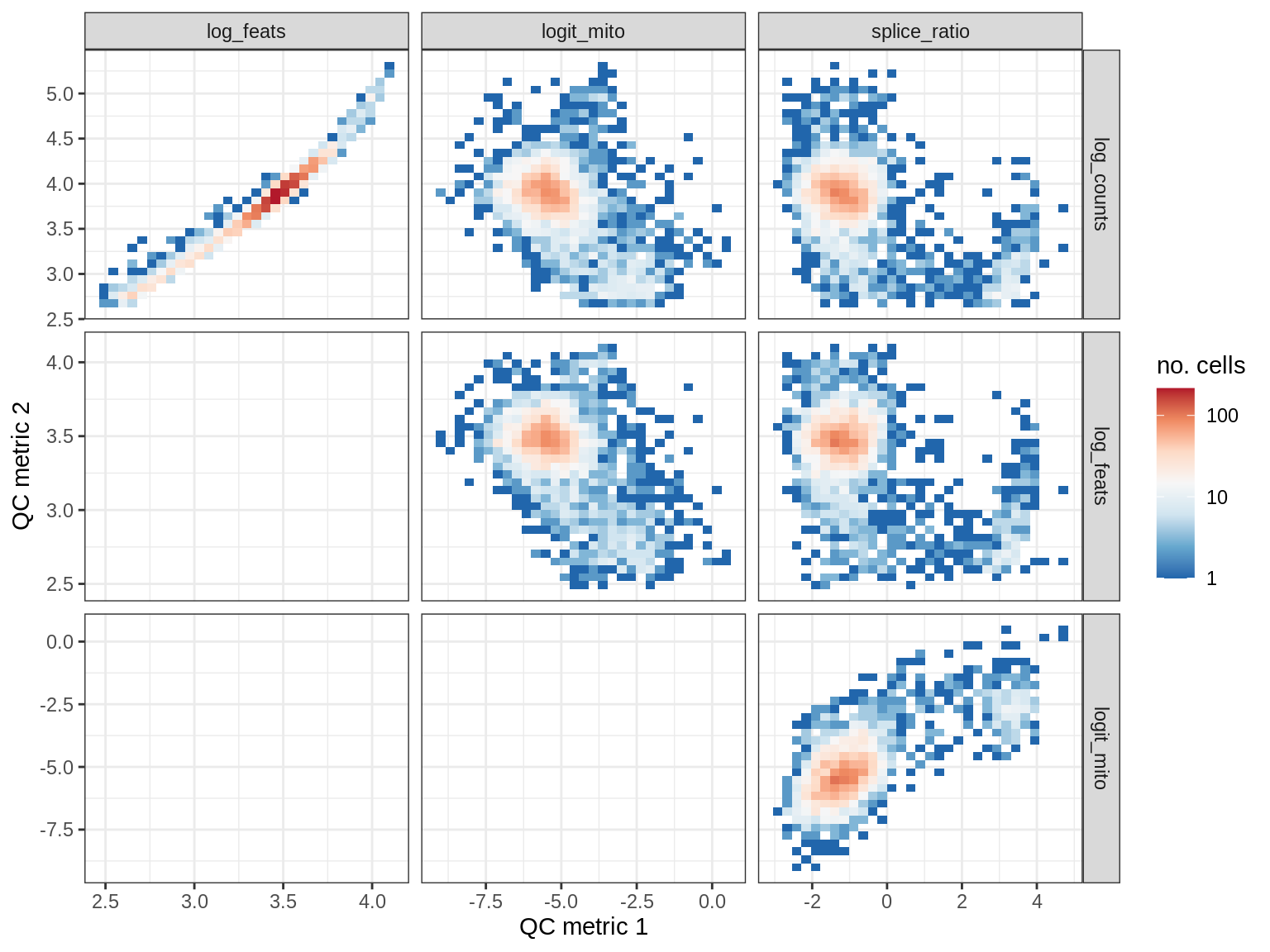
WM185
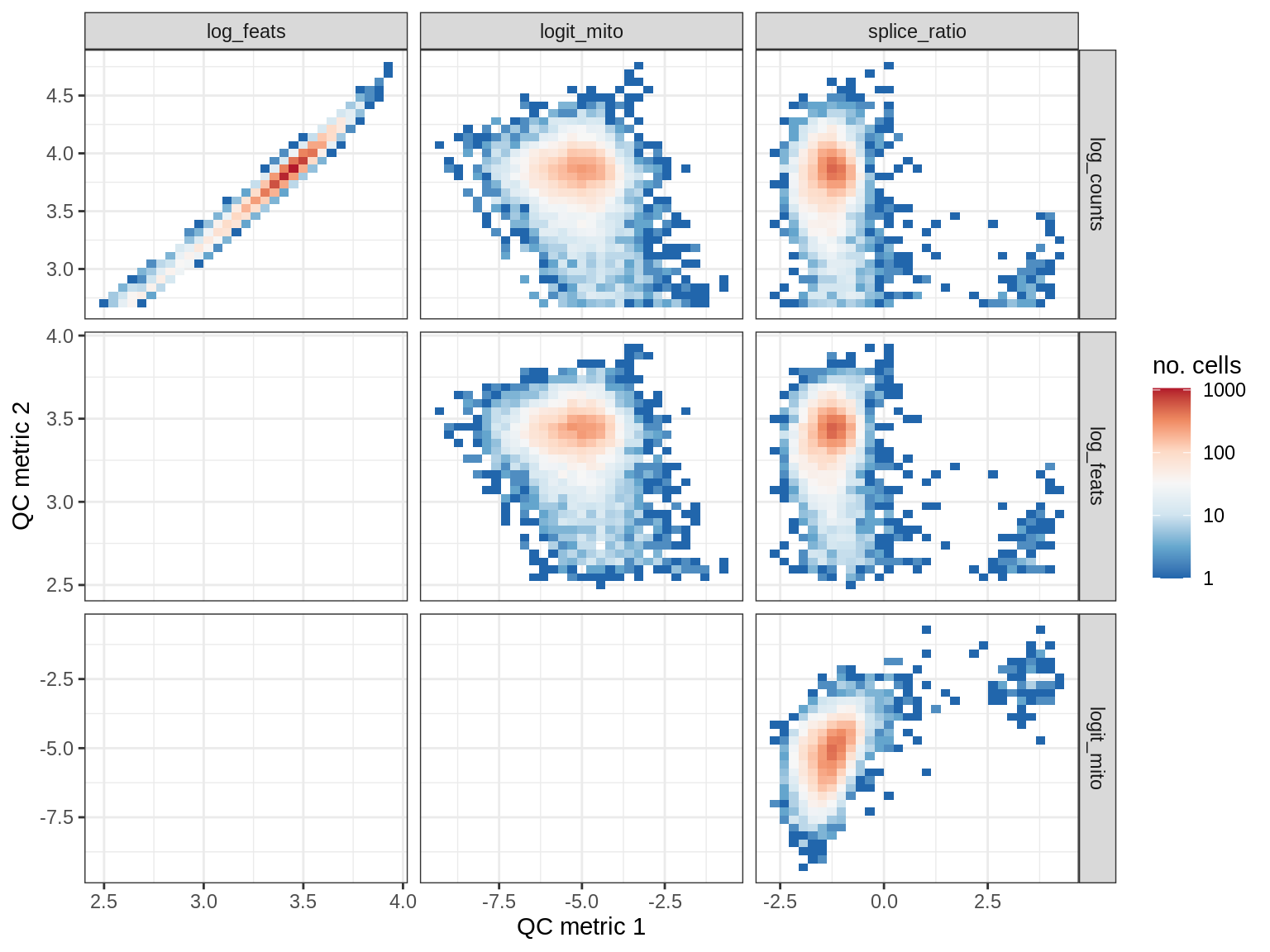
WM186
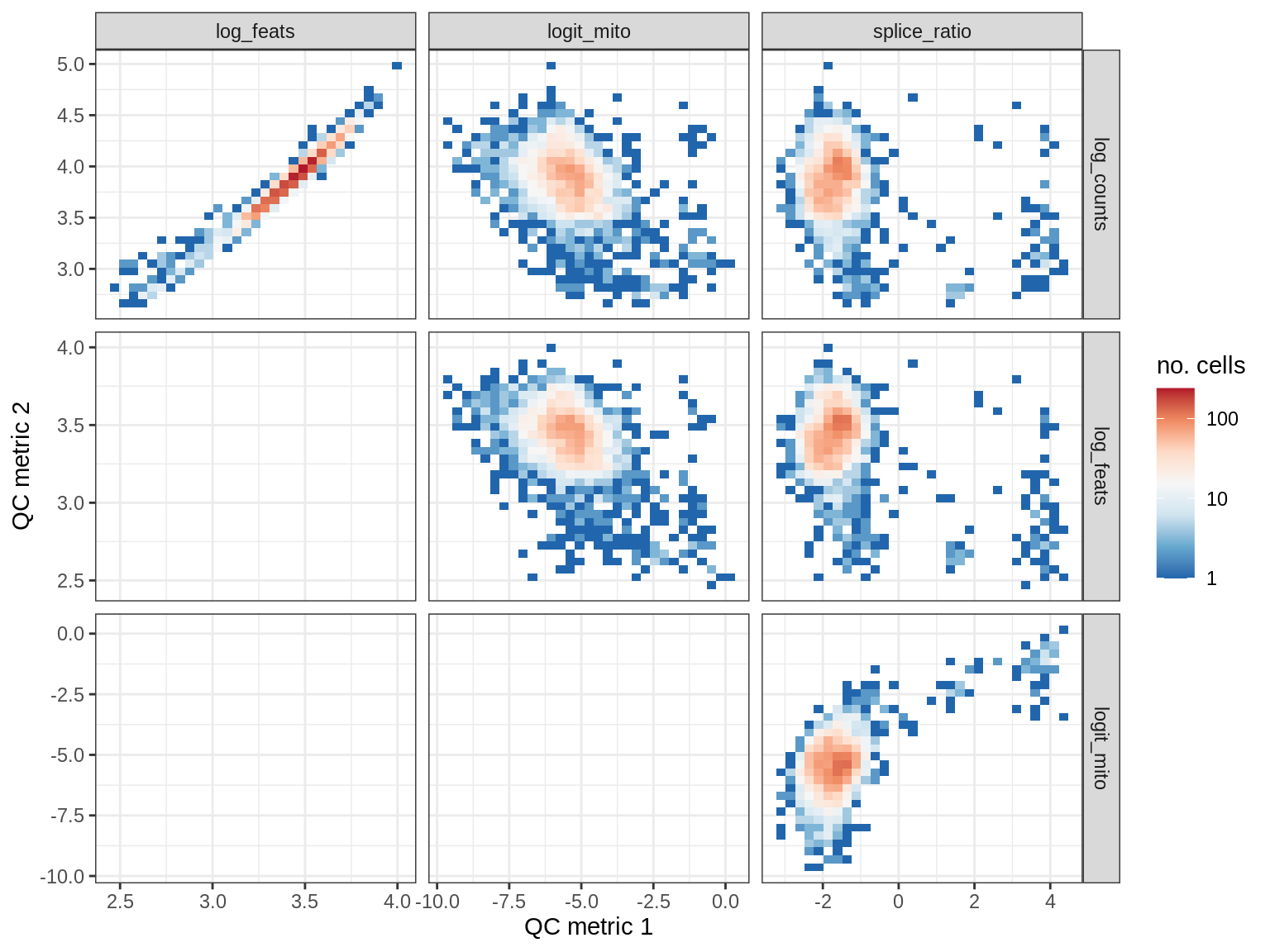
WM187
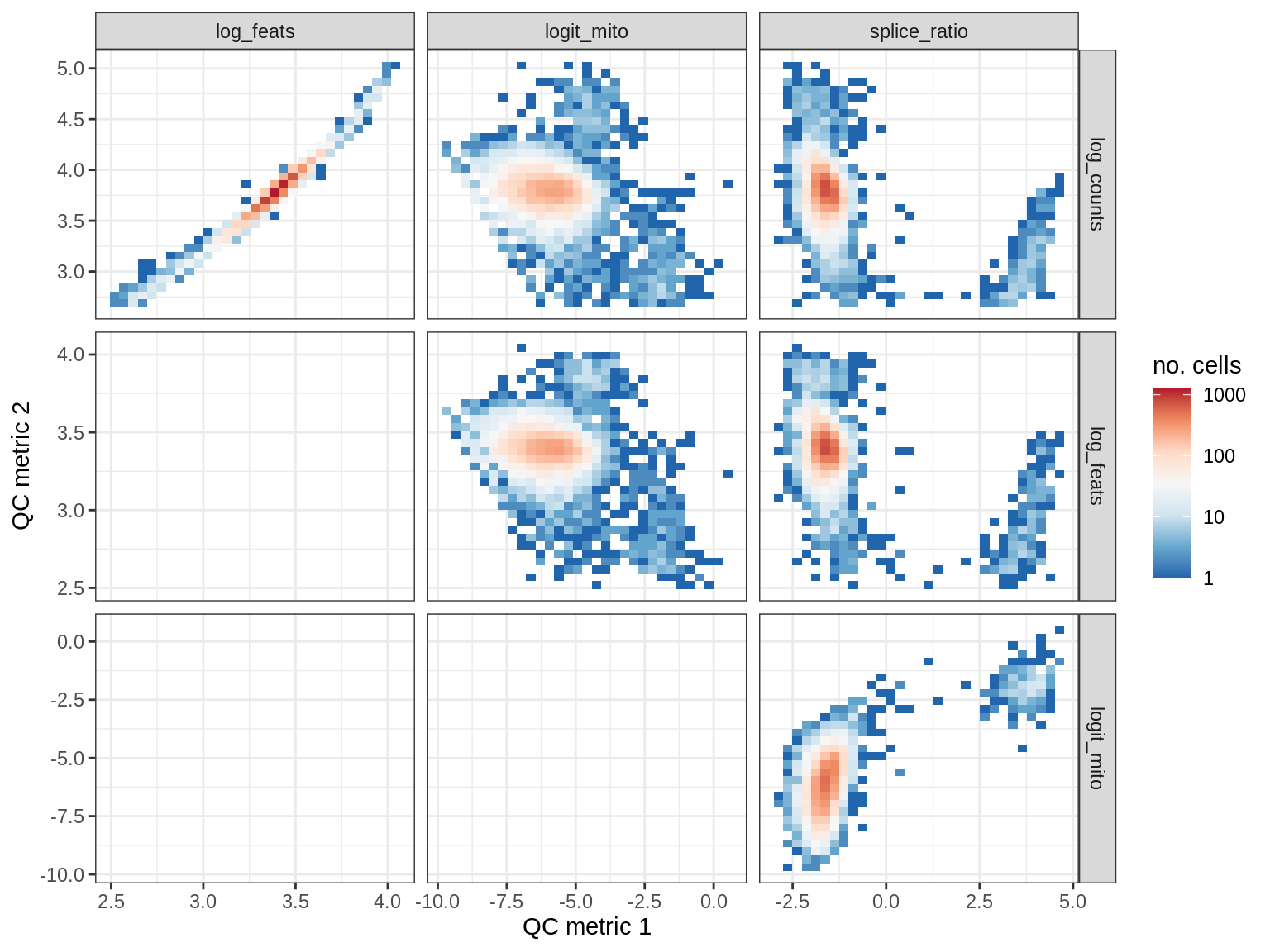
WM188
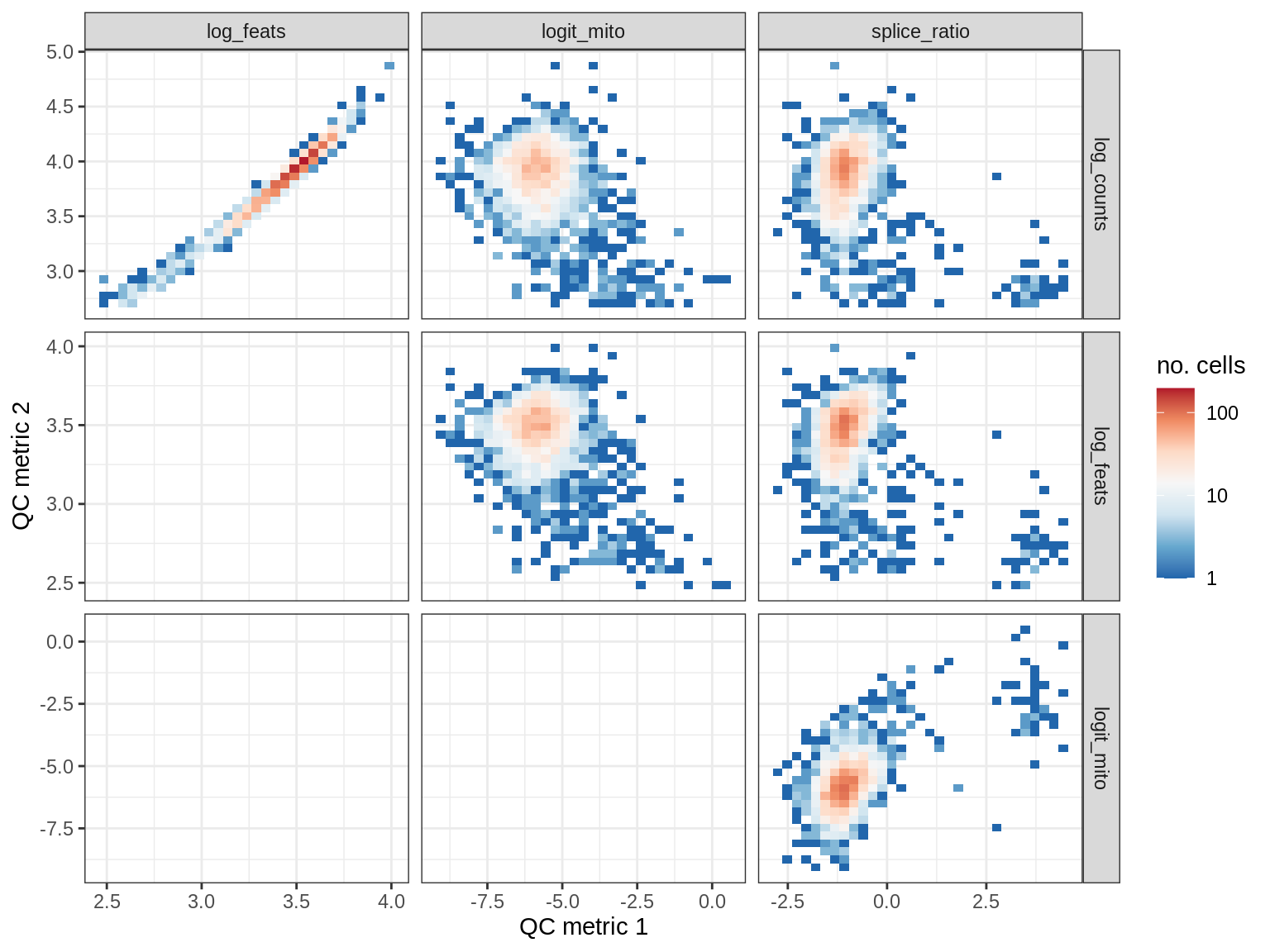
WM189
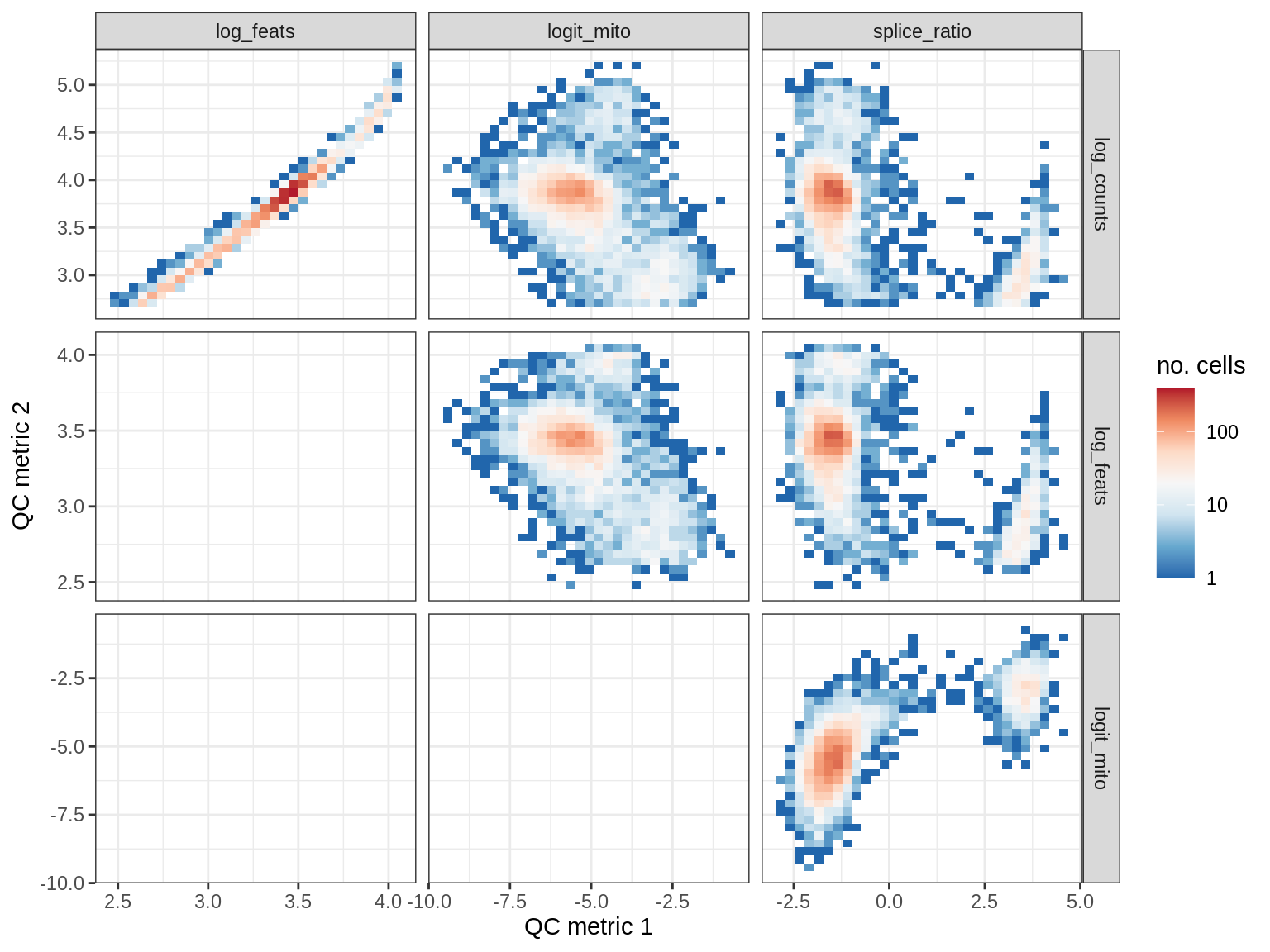
WM190
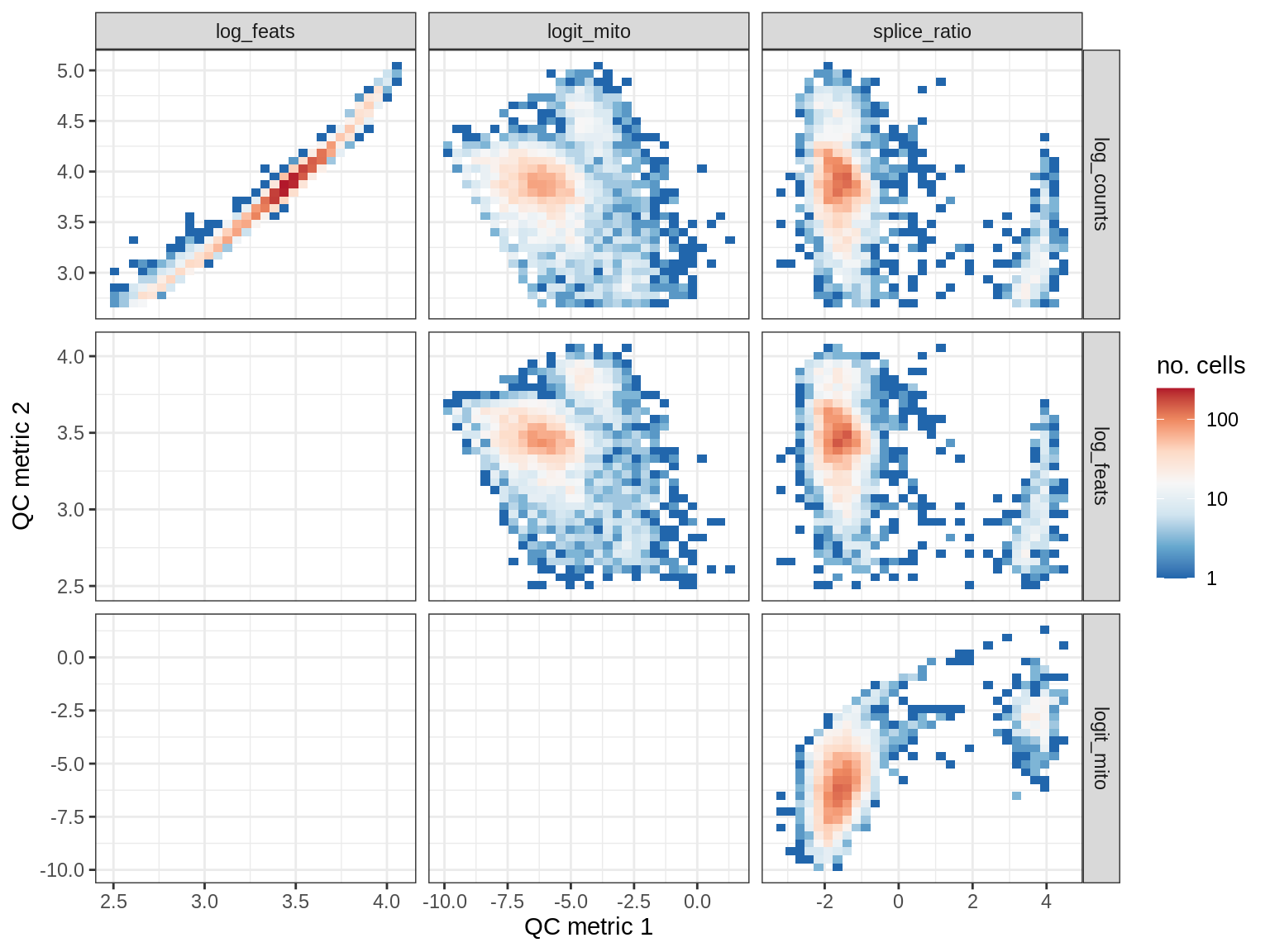
WM191
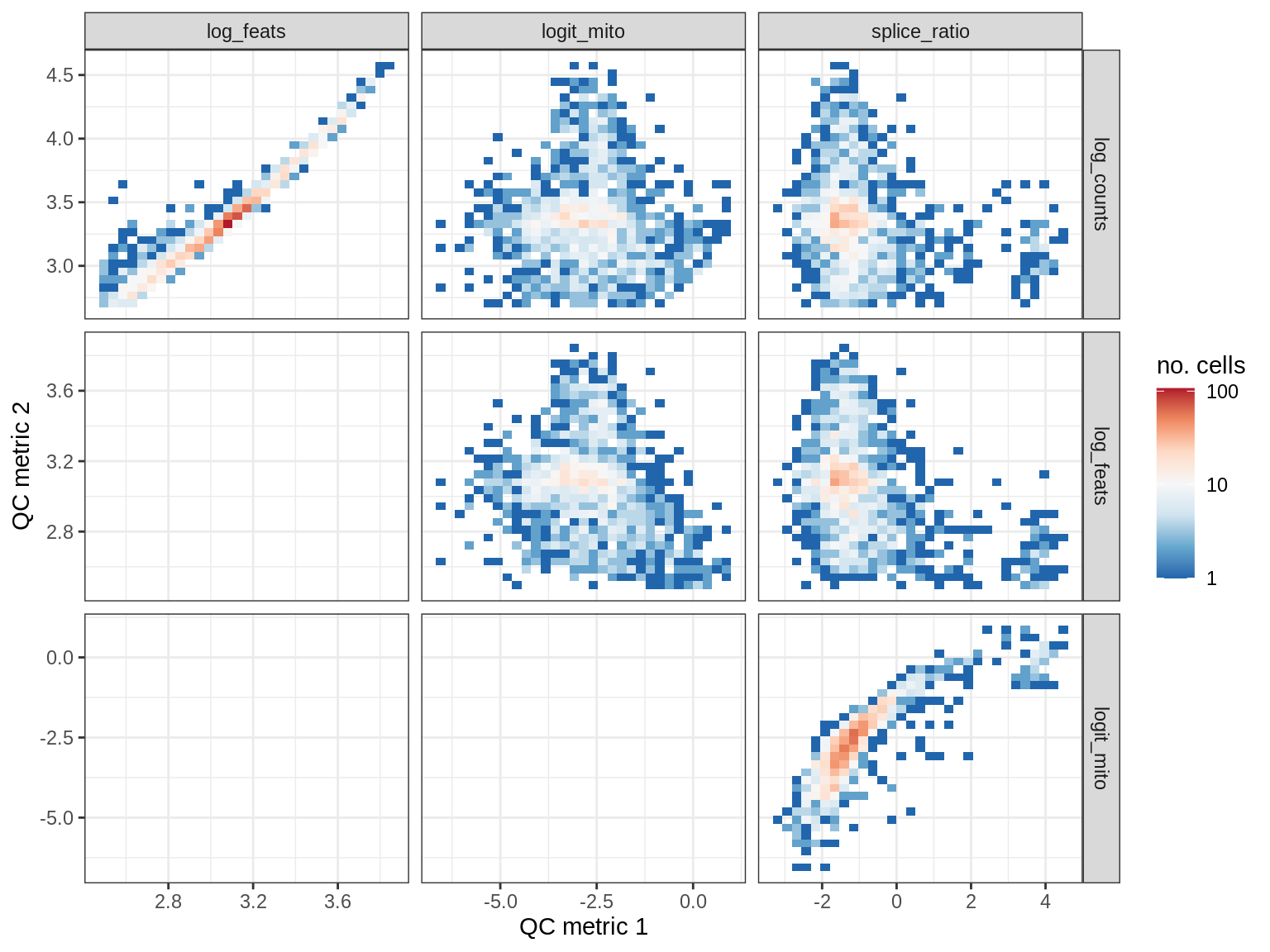
WM192
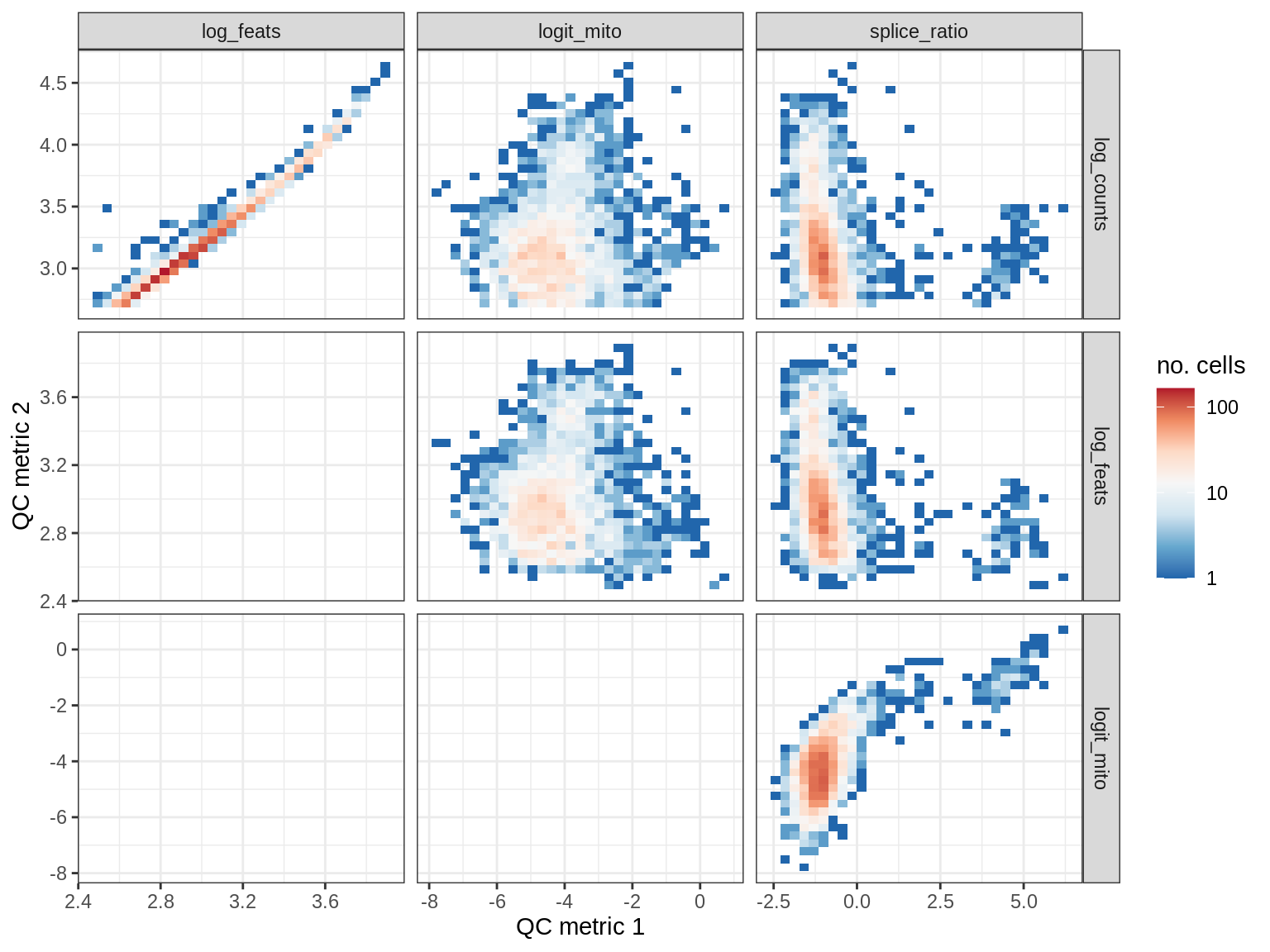
WM193
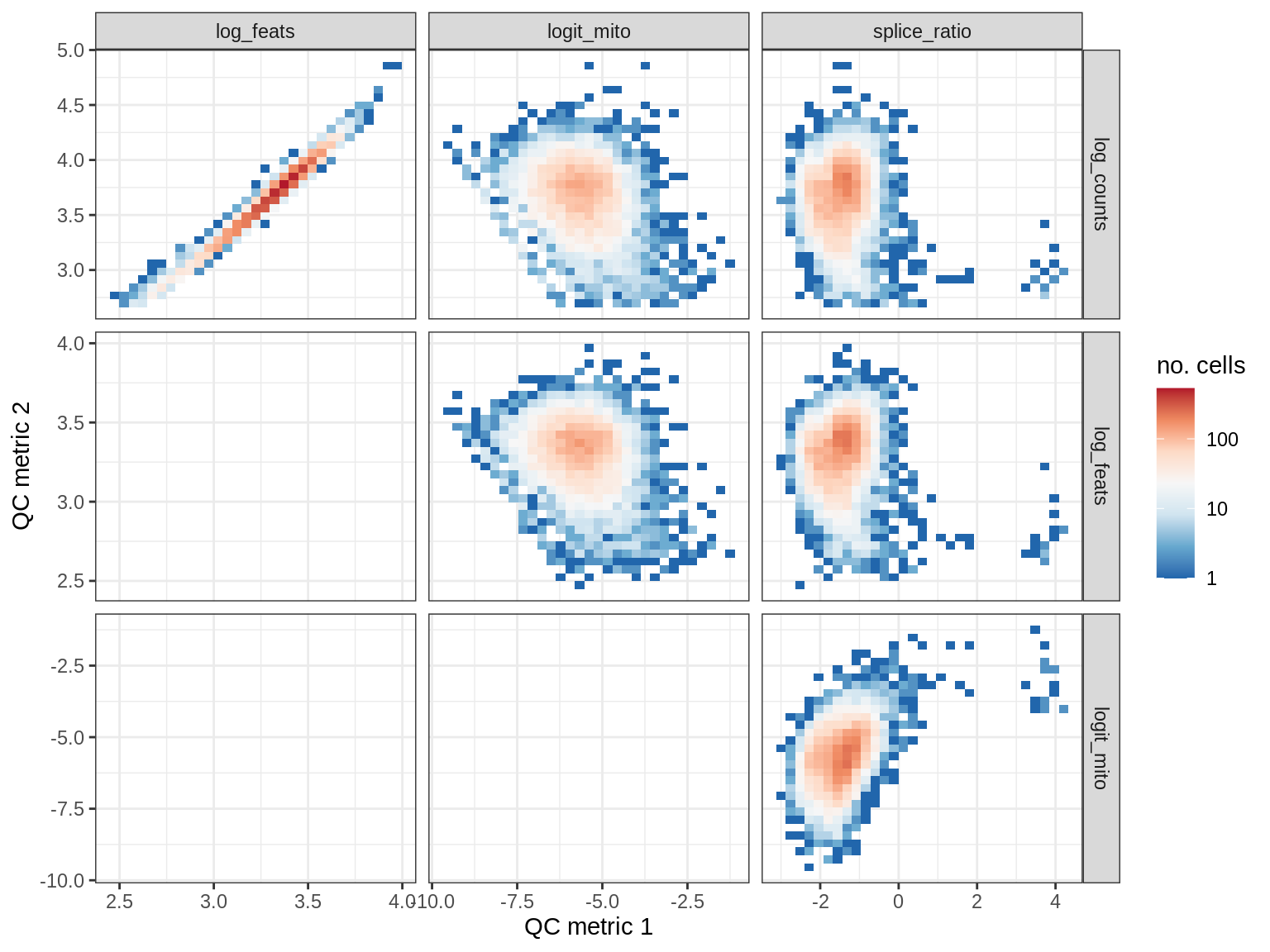
WM194
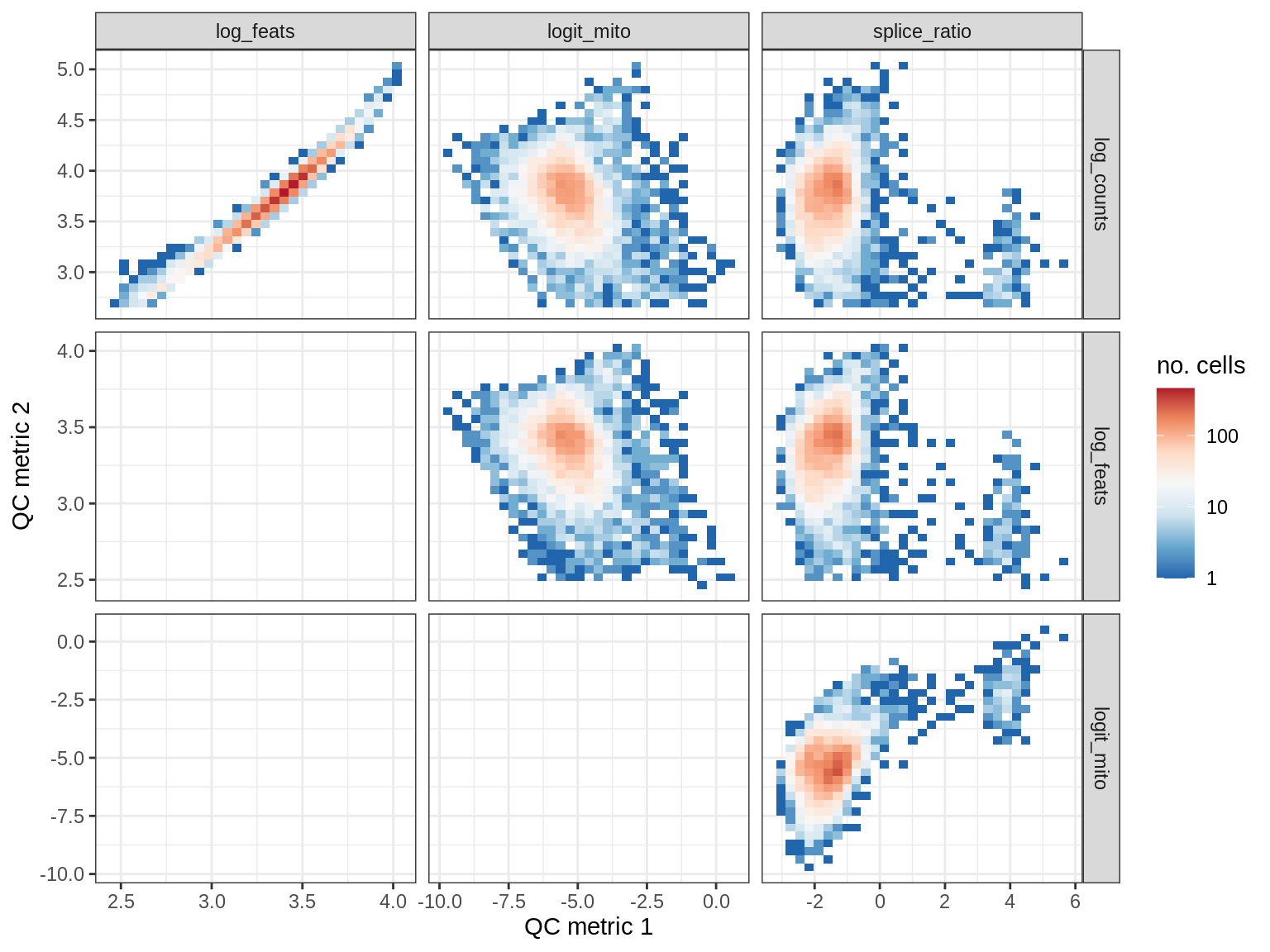
WM195

WM196
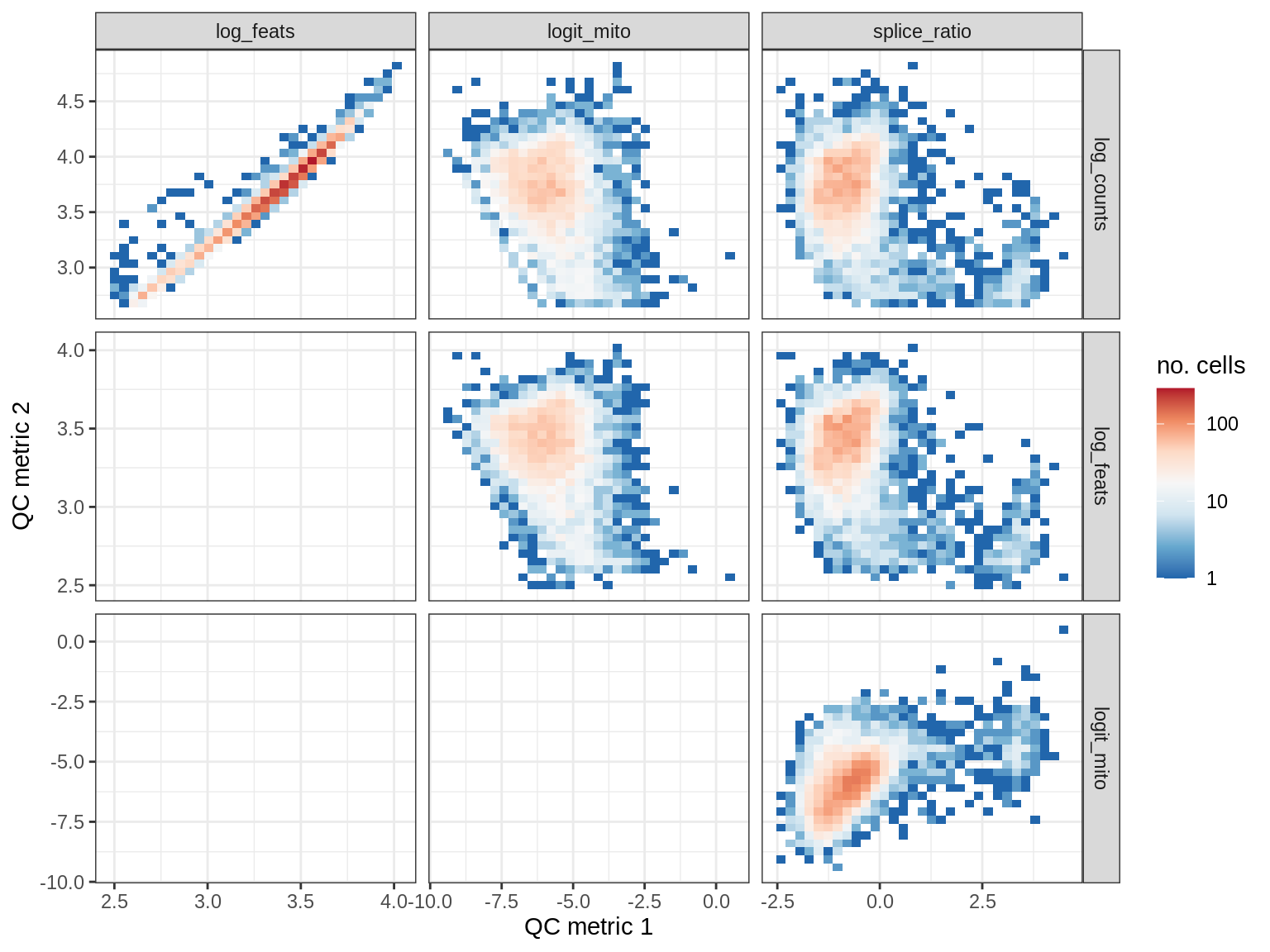
WM197
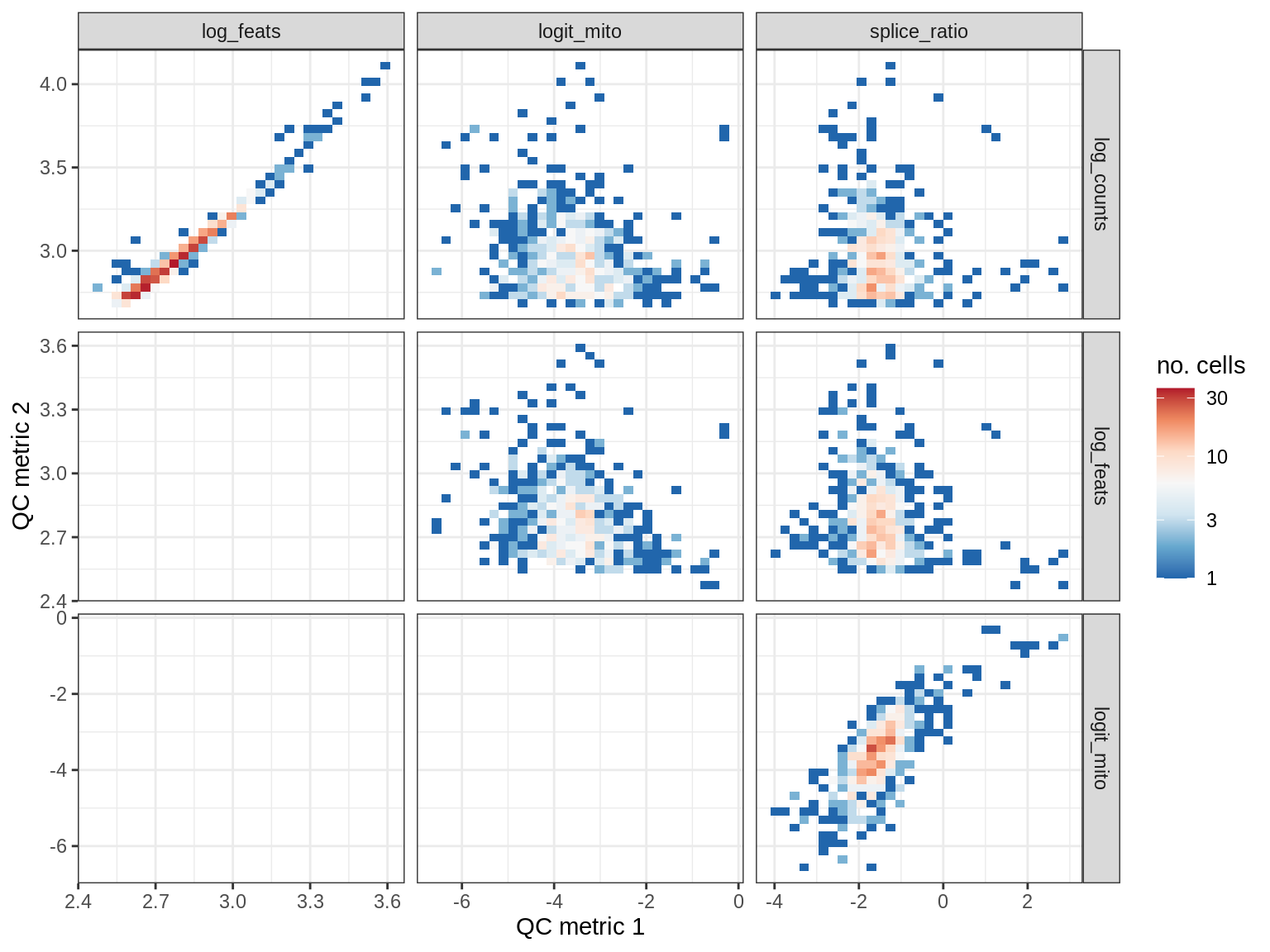
WM198
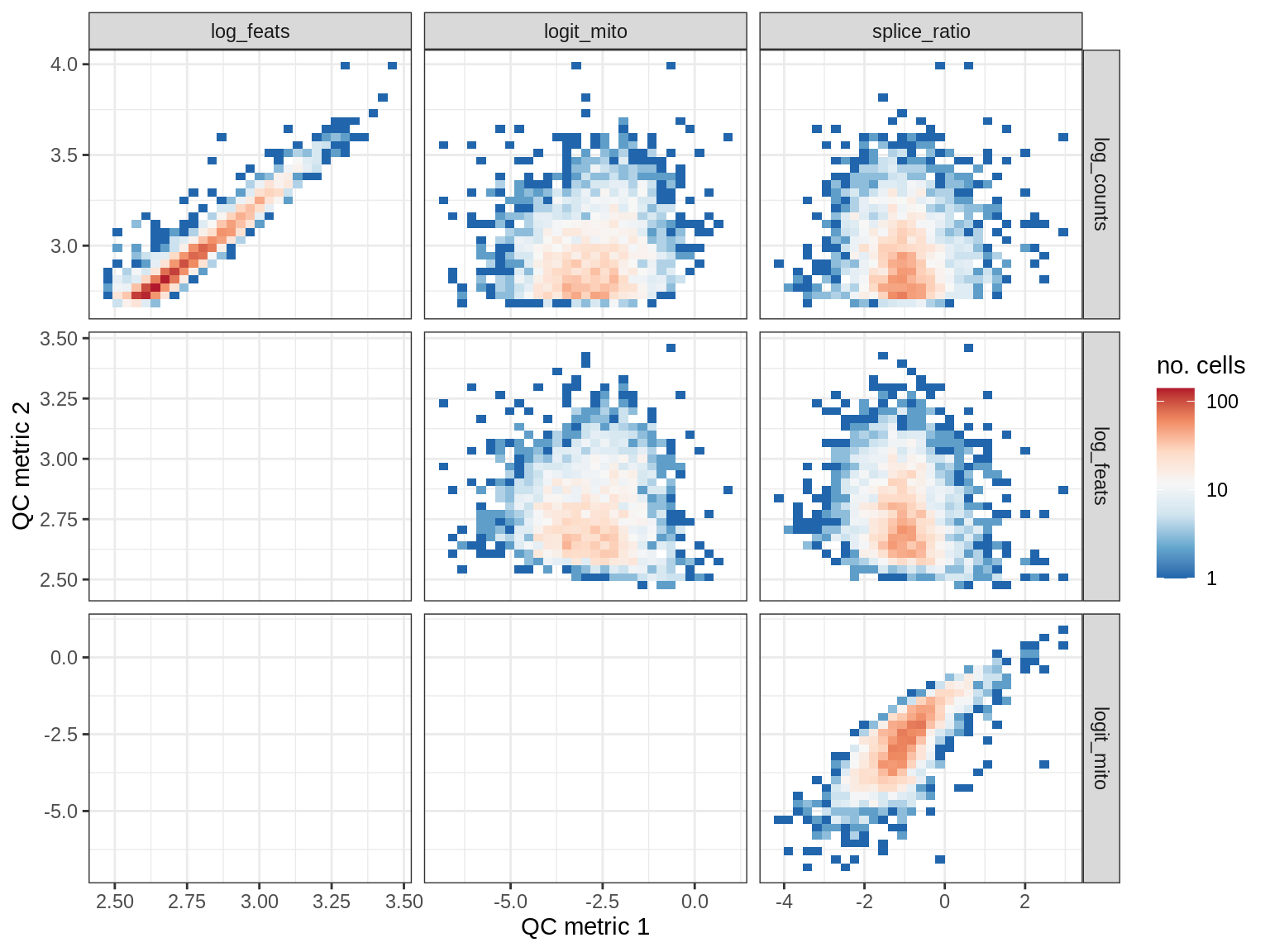
WM199
How many cells and samples retained?
(plot_totals_split_by_meta(pre_dt = dbl_dt, post_dt = keep_dt, meta_f))Warning in melt.data.table(., measure = c("n_cells", "n_samples",
"n_patients"), : 'measure.vars' [n_cells, n_samples, n_patients, ...] are not
all of the same type. By order of hierarchy, the molten data value column will
be of type 'double'. All measure variables not of type 'double' will be coerced
too. Check DETAILS in ?melt.data.table for more on coercion.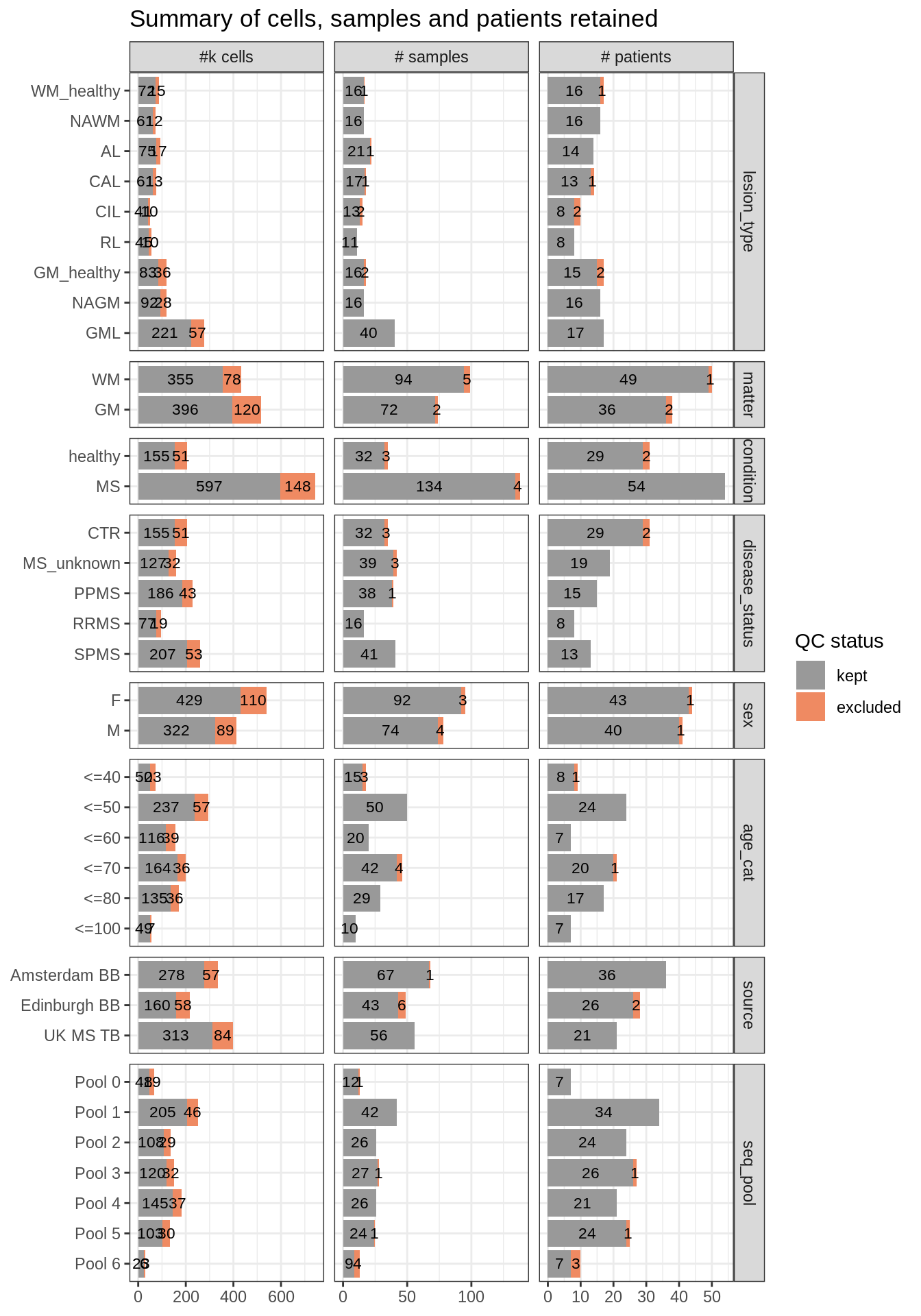
message('pre QC:')pre QC:print(calc_summary_dt(dbl_dt, meta_f)) matter condition n_cells n_samples n_patients
1: WM healthy 86997 17 17
2: WM MS 346512 82 33
3: GM healthy 119310 18 17
4: GM MS 397625 56 21message('post QC:')post QC:print(calc_summary_dt(keep_dt, meta_f)) matter condition n_cells n_samples n_patients
1: WM healthy 71624 16 16
2: WM MS 283389 78 33
3: GM healthy 83309 16 15
4: GM MS 313127 56 21Outputs
if (!file.exists(report_f)) {
# render the report
make_SampleQC_report(qc_obj, save_dir, proj_name)
}fwrite(outliers_dt, file=outliers_f)
fwrite(keep_dt, file=keep_f)devtools::session_info()- Session info ---------------------------------------------------------------
setting value
version R version 4.0.3 (2020-10-10)
os CentOS Linux 7 (Core)
system x86_64, linux-gnu
ui X11
language (EN)
collate en_US.UTF-8
ctype C
tz Europe/Zurich
date 2021-04-28
- Packages -------------------------------------------------------------------
package * version date lib
assertthat * 0.2.1 2019-03-21 [2]
Biobase * 2.50.0 2020-10-27 [1]
BiocGenerics * 0.36.1 2021-04-16 [1]
BiocManager 1.30.12 2021-03-28 [1]
BiocParallel 1.24.1 2020-11-06 [1]
BiocStyle * 2.18.1 2020-11-24 [1]
bit 4.0.4 2020-08-04 [2]
bit64 4.0.5 2020-08-30 [2]
bitops 1.0-6 2013-08-17 [2]
bslib 0.2.4 2021-01-25 [2]
cachem 1.0.4 2021-02-13 [2]
callr 3.6.0 2021-03-28 [2]
circlize * 0.4.12 2021-01-08 [1]
cli 2.4.0 2021-04-05 [2]
codetools 0.2-18 2020-11-04 [2]
colorout * 1.2-2 2021-04-15 [1]
colorspace 2.0-0 2020-11-11 [2]
crayon 1.4.1 2021-02-08 [2]
data.table * 1.14.0 2021-02-21 [2]
DBI 1.1.1 2021-01-15 [2]
DelayedArray 0.16.3 2021-03-24 [1]
desc 1.3.0 2021-03-05 [2]
devtools 2.4.0 2021-04-07 [1]
digest 0.6.27 2020-10-24 [2]
dplyr 1.0.5 2021-03-05 [2]
ellipsis 0.3.1 2020-05-15 [2]
evaluate 0.14 2019-05-28 [2]
fansi 0.4.2 2021-01-15 [2]
farver 2.1.0 2021-02-28 [2]
fastmap 1.1.0 2021-01-25 [2]
forcats * 0.5.1 2021-01-27 [2]
fs 1.5.0 2020-07-31 [2]
generics 0.1.0 2020-10-31 [2]
GenomeInfoDb * 1.26.7 2021-04-08 [1]
GenomeInfoDbData 1.2.4 2021-04-15 [1]
GenomicRanges * 1.42.0 2020-10-27 [1]
ggplot2 * 3.3.3 2020-12-30 [2]
git2r 0.28.0 2021-01-10 [1]
GlobalOptions 0.1.2 2020-06-10 [1]
glue 1.4.2 2020-08-27 [2]
gridExtra 2.3 2017-09-09 [2]
gtable 0.3.0 2019-03-25 [2]
gtools 3.8.2 2020-03-31 [2]
hdf5r * 1.3.3 2020-08-18 [2]
highr 0.9 2021-04-16 [2]
htmltools 0.5.1.1 2021-01-22 [2]
httpuv 1.5.5 2021-01-13 [2]
igraph 1.2.6 2020-10-06 [2]
IRanges * 2.24.1 2020-12-12 [1]
iterators * 1.0.13 2020-10-15 [2]
itertools * 0.1-3 2014-03-12 [1]
jquerylib 0.1.3 2020-12-17 [2]
jsonlite 1.7.2 2020-12-09 [2]
kernlab 0.9-29 2019-11-12 [1]
knitr 1.32 2021-04-14 [1]
labeling 0.4.2 2020-10-20 [2]
later 1.1.0.1 2020-06-05 [2]
lattice 0.20-41 2020-04-02 [2]
lifecycle 1.0.0 2021-02-15 [2]
loomR * 0.2.0 2021-04-15 [1]
magrittr * 2.0.1 2020-11-17 [1]
MASS 7.3-53.1 2021-02-12 [2]
Matrix * 1.3-2 2021-01-06 [2]
MatrixGenerics * 1.2.1 2021-01-30 [1]
matrixStats * 0.58.0 2021-01-29 [2]
mclust 5.4.7 2020-11-20 [1]
memoise 2.0.0 2021-01-26 [1]
mixtools 1.2.0 2020-02-07 [1]
munsell 0.5.0 2018-06-12 [2]
mvnfast 0.2.5.1 2020-10-14 [1]
mvtnorm 1.1-1 2020-06-09 [1]
patchwork * 1.1.1 2020-12-17 [2]
pillar 1.6.0 2021-04-13 [2]
pkgbuild 1.2.0 2020-12-15 [1]
pkgconfig 2.0.3 2019-09-22 [2]
pkgload 1.2.1 2021-04-06 [2]
prettyunits 1.1.1 2020-01-24 [2]
processx 3.5.1 2021-04-04 [2]
promises 1.2.0.1 2021-02-11 [2]
ps 1.6.0 2021-02-28 [2]
purrr 0.3.4 2020-04-17 [2]
R6 * 2.5.0 2020-10-28 [2]
RColorBrewer * 1.1-2 2014-12-07 [2]
Rcpp 1.0.6 2021-01-15 [2]
RCurl 1.98-1.3 2021-03-16 [1]
remotes 2.3.0 2021-04-01 [1]
rlang 0.4.10 2020-12-30 [2]
rmarkdown 2.7 2021-02-19 [2]
rprojroot 2.0.2 2020-11-15 [2]
S4Vectors * 0.28.1 2020-12-09 [1]
SampleQC * 0.4.5 2021-04-15 [1]
sass 0.3.1 2021-01-24 [2]
scales * 1.1.1 2020-05-11 [2]
segmented 1.3-3 2021-03-08 [1]
sessioninfo 1.1.1 2018-11-05 [1]
shape 1.4.5 2020-09-13 [2]
SingleCellExperiment * 1.12.0 2020-10-27 [1]
stringi 1.5.3 2020-09-09 [2]
stringr * 1.4.0 2019-02-10 [2]
SummarizedExperiment * 1.20.0 2020-10-27 [1]
survival 3.2-10 2021-03-16 [2]
testthat 3.0.2 2021-02-14 [2]
tibble 3.1.1 2021-04-18 [2]
tidyselect 1.1.0 2020-05-11 [2]
usethis 2.0.1 2021-02-10 [1]
utf8 1.2.1 2021-03-12 [2]
uwot 0.1.10 2020-12-15 [2]
vctrs 0.3.7 2021-03-29 [2]
viridis * 0.6.0 2021-04-15 [1]
viridisLite * 0.4.0 2021-04-13 [2]
whisker 0.4 2019-08-28 [1]
withr 2.4.2 2021-04-18 [2]
workflowr * 1.6.2 2020-04-30 [1]
xfun 0.22 2021-03-11 [1]
XVector 0.30.0 2020-10-27 [1]
yaml 2.2.1 2020-02-01 [2]
zlibbioc 1.36.0 2020-10-27 [1]
source
CRAN (R 4.0.0)
Bioconductor
Bioconductor
CRAN (R 4.0.3)
Bioconductor
Bioconductor
CRAN (R 4.0.2)
CRAN (R 4.0.2)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Github (jalvesaq/colorout@79931fd)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.2)
CRAN (R 4.0.3)
Bioconductor
Bioconductor
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.2)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Github (mojaveazure/loomR@df0144b)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Bioconductor
Github (wmacnair/SampleQC@5e3f021)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.2)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.3)
Bioconductor
[1] /pstore/home/macnairw/lib/conda_r3.12
[2] /pstore/home/macnairw/.conda/envs/r_4.0.3/lib/R/library
sessionInfo()R version 4.0.3 (2020-10-10)
Platform: x86_64-conda-linux-gnu (64-bit)
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS/LAPACK: /pstore/home/macnairw/.conda/envs/r_4.0.3/lib/libopenblasp-r0.3.12.so
locale:
[1] LC_CTYPE=C LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] SampleQC_0.4.5 SingleCellExperiment_1.12.0
[3] SummarizedExperiment_1.20.0 Biobase_2.50.0
[5] GenomicRanges_1.42.0 GenomeInfoDb_1.26.7
[7] IRanges_2.24.1 S4Vectors_0.28.1
[9] BiocGenerics_0.36.1 MatrixGenerics_1.2.1
[11] matrixStats_0.58.0 Matrix_1.3-2
[13] loomR_0.2.0 itertools_0.1-3
[15] iterators_1.0.13 hdf5r_1.3.3
[17] R6_2.5.0 patchwork_1.1.1
[19] forcats_0.5.1 ggplot2_3.3.3
[21] scales_1.1.1 viridis_0.6.0
[23] viridisLite_0.4.0 assertthat_0.2.1
[25] stringr_1.4.0 data.table_1.14.0
[27] magrittr_2.0.1 circlize_0.4.12
[29] RColorBrewer_1.1-2 BiocStyle_2.18.1
[31] colorout_1.2-2 workflowr_1.6.2
loaded via a namespace (and not attached):
[1] colorspace_2.0-0 ellipsis_0.3.1 mclust_5.4.7
[4] rprojroot_2.0.2 XVector_0.30.0 GlobalOptions_0.1.2
[7] fs_1.5.0 rstudioapi_0.13 farver_2.1.0
[10] remotes_2.3.0 bit64_4.0.5 fansi_0.4.2
[13] mvtnorm_1.1-1 codetools_0.2-18 splines_4.0.3
[16] cachem_1.0.4 knitr_1.32 pkgload_1.2.1
[19] jsonlite_1.7.2 kernlab_0.9-29 uwot_0.1.10
[22] BiocManager_1.30.12 compiler_4.0.3 fastmap_1.1.0
[25] cli_2.4.0 later_1.1.0.1 htmltools_0.5.1.1
[28] prettyunits_1.1.1 tools_4.0.3 igraph_1.2.6
[31] gtable_0.3.0 glue_1.4.2 GenomeInfoDbData_1.2.4
[34] dplyr_1.0.5 Rcpp_1.0.6 jquerylib_0.1.3
[37] vctrs_0.3.7 xfun_0.22 ps_1.6.0
[40] testthat_3.0.2 lifecycle_1.0.0 gtools_3.8.2
[43] devtools_2.4.0 zlibbioc_1.36.0 MASS_7.3-53.1
[46] promises_1.2.0.1 yaml_2.2.1 mvnfast_0.2.5.1
[49] memoise_2.0.0 gridExtra_2.3 sass_0.3.1
[52] segmented_1.3-3 stringi_1.5.3 highr_0.9
[55] desc_1.3.0 pkgbuild_1.2.0 BiocParallel_1.24.1
[58] shape_1.4.5 rlang_0.4.10 pkgconfig_2.0.3
[61] bitops_1.0-6 evaluate_0.14 lattice_0.20-41
[64] purrr_0.3.4 labeling_0.4.2 bit_4.0.4
[67] tidyselect_1.1.0 processx_3.5.1 generics_0.1.0
[70] DelayedArray_0.16.3 DBI_1.1.1 pillar_1.6.0
[73] whisker_0.4 withr_2.4.2 mixtools_1.2.0
[76] survival_3.2-10 RCurl_1.98-1.3 tibble_3.1.1
[79] crayon_1.4.1 utf8_1.2.1 rmarkdown_2.7
[82] usethis_2.0.1 grid_4.0.3 callr_3.6.0
[85] git2r_0.28.0 digest_0.6.27 httpuv_1.5.5
[88] munsell_0.5.0 bslib_0.2.4 sessioninfo_1.1.1