MOFA+ analysis - sample level, WM, final metadata
Will Macnair
Neurogenomics, Neuroscience and Rare Diseases, RocheNovember 25, 2021
Last updated: 2021-11-25
Checks: 5 2
Knit directory: MS_lesions/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
The global environment had objects present when the code in the R Markdown file was run. These objects can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment. Use wflow_publish or wflow_build to ensure that the code is always run in an empty environment.
The following objects were defined in the global environment when these results were created:
| Name | Class | Size |
|---|---|---|
| q | function | 1008 bytes |
The command set.seed(20210118) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 74b907e. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rprofile
Ignored: .Rproj.user/
Ignored: ._MS_lesions.sublime-project
Ignored: .log/
Ignored: MS_lesions.sublime-project
Ignored: MS_lesions.sublime-workspace
Ignored: analysis/.__site.yml
Ignored: analysis/fig_muscat_cache/
Ignored: analysis/ms02_doublet_id_cache/
Ignored: analysis/ms03_SampleQC_cache/
Ignored: analysis/ms04_conos_cache/
Ignored: analysis/ms05_splitting_cache/
Ignored: analysis/ms06_sccaf_cache/
Ignored: analysis/ms07_soup_cache/
Ignored: analysis/ms08_modules_cache/
Ignored: analysis/ms08_modules_pseudobulk_cache/
Ignored: analysis/ms09_ancombc_cache/
Ignored: analysis/ms09_ancombc_clean_1e3_cache/
Ignored: analysis/ms09_ancombc_clean_2e3_cache/
Ignored: analysis/ms09_ancombc_mixed_cache/
Ignored: analysis/ms10_muscat_run01_cache/
Ignored: analysis/ms10_muscat_run02_cache/
Ignored: analysis/ms10_muscat_template_broad_cache/
Ignored: analysis/ms10_muscat_template_fine_cache/
Ignored: analysis/ms11_paga_cache/
Ignored: analysis/ms12_markers_cache/
Ignored: analysis/ms13_labelling_cache/
Ignored: analysis/ms14_lesions_cache/
Ignored: analysis/ms15_mofa_sample_gm_cache/
Ignored: analysis/ms15_mofa_sample_gm_final_meta_cache/
Ignored: analysis/ms15_mofa_sample_gm_superclean_cache/
Ignored: analysis/ms15_mofa_sample_gm_w_layers_final_meta_cache/
Ignored: analysis/ms15_mofa_sample_wm_cache/
Ignored: analysis/ms15_mofa_sample_wm_new_meta_cache/
Ignored: analysis/ms15_mofa_sample_wm_superclean_cache/
Ignored: analysis/ms15_patients_cache/
Ignored: analysis/ms15_patients_gm_cache/
Ignored: analysis/ms15_patients_sample_level_cache/
Ignored: analysis/ms15_patients_w_ms_cache/
Ignored: analysis/supp06_sccaf_cache/
Ignored: analysis/supp07_superclean_check_cache/
Ignored: analysis/supp09_ancombc_cache/
Ignored: analysis/supp09_ancombc_mixed_cache/
Ignored: analysis/supp09_ancombc_superclean_cache/
Ignored: analysis/supp10_muscat_cache/
Ignored: analysis/supp10_muscat_ctrl_gm_vs_wm_cache/
Ignored: analysis/supp10_muscat_gm_layers_effects_cache/
Ignored: analysis/supp10_muscat_heatmaps_cache/
Ignored: analysis/supp10_muscat_olg_pc1_cache/
Ignored: analysis/supp10_muscat_olg_pc2_cache/
Ignored: analysis/supp10_muscat_olg_pc_cache/
Ignored: analysis/supp10_muscat_regression_cache/
Ignored: analysis/supp10_muscat_soup_cache/
Ignored: analysis/supp10_muscat_soup_mito_cache/
Ignored: code/._ms10_muscat_fns_recover.R
Ignored: code/.recovery/
Ignored: code/jobs/._muscat_run09_2021-10-11.slurm
Ignored: code/muscat_plan.txt
Ignored: data/
Ignored: figures/
Ignored: output/
Ignored: tmp/
Untracked files:
Untracked: Rplots.pdf
Untracked: _dt = muscat:::.n_cells(pb) ->- as.data.table ->- setnames(c("broad", "sample_id", "n_cells"))
Untracked: analysis/supp09_ancombc_superclean.Rmd
Unstaged changes:
Modified: analysis/ms14_lesions.Rmd
Modified: analysis/ms15_mofa_sample_gm_w_layers_final_meta.Rmd
Modified: analysis/ms15_mofa_sample_wm_final_meta.Rmd
Modified: analysis/ms15_mofa_sample_wm_superclean.Rmd
Modified: analysis/supp07_superclean_check.Rmd
Modified: code/dev_edger_on_mofa_20210804.R
Modified: code/ms10_muscat_runs.R
Modified: code/ms14_lesions.R
Modified: code/ms15_mofa.R
Modified: code/supp10_muscat.R
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/ms15_mofa_sample_wm_final_meta.Rmd) and HTML (docs/ms15_mofa_sample_wm_final_meta.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | c013785 | Macnair | 2021-10-23 | Update MOFA |
| html | c013785 | Macnair | 2021-10-23 | Update MOFA |
| Rmd | 21ca0d3 | Macnair | 2021-10-19 | Tweaks to MOFA barplots |
| html | 21ca0d3 | Macnair | 2021-10-19 | Tweaks to MOFA barplots |
| Rmd | 1fe7571 | Macnair | 2021-10-19 | Fix error in WM MOFA barplots |
| html | 1fe7571 | Macnair | 2021-10-19 | Fix error in WM MOFA barplots |
| Rmd | 8a15881 | Macnair | 2021-10-18 | Update index.Rmd with final MOFA results` |
| Rmd | 47af3ff | Macnair | 2021-10-18 | Finalise MOFA analysis |
| html | 47af3ff | Macnair | 2021-10-18 | Finalise MOFA analysis |
| Rmd | 1552617 | Macnair | 2021-10-05 | Save MOFA genes to xls |
| html | 1552617 | Macnair | 2021-10-05 | Save MOFA genes to xls |
| Rmd | c1a0bff | Macnair | 2021-10-05 | Update GM MOFA with final metadata |
| html | c1a0bff | Macnair | 2021-10-05 | Update GM MOFA with final metadata |
| Rmd | ad82ee3 | Macnair | 2021-10-04 | Update ms09_ancombc with final metadata |
| html | ad82ee3 | Macnair | 2021-10-04 | Update ms09_ancombc with final metadata |
Setup / definitions
Libraries
Helper functions
source('code/ms00_utils.R')
source('code/ms09_ancombc.R')
source('code/ms10_muscat_runs.R')
source('code/ms15_mofa.R')
knitr::knit_hooks$set(webgl = hook_webgl)Inputs
# specify what goes into muscat run
meta_f = "data/metadata/metadata_checked_assumptions_2021-10-08.xlsx"
olg_grps_f = 'data/metadata/oligo_groupings.txt'
comp_grps_f = 'output/ms09_ancombc/clr_clustering_WM_2021-10-19.txt'
labels_f = 'data/byhand_markers/validation_markers_2021-05-31.csv'
labelled_f = 'output/ms13_labelling/conos_labelled_2021-05-31.txt.gz'
pb_f = file.path(soup_dir, 'pb_sum_broad_2021-10-11.rds')
pb_fine_f = file.path(soup_dir, 'pb_sum_fine_2021-10-11.rds')
soup_f = 'data/ambient/ambient.100UMI.txt'
# define run to load
run_tag = 'run09'
time_stamp = '2021-10-13'
# define files
model_dir = file.path('output/ms10_muscat', run_tag)
muscat_f = '%s/muscat_res_dt_%s_%s.txt.gz' %>%
sprintf(model_dir, run_tag, time_stamp)
anova_f = '%s/muscat_goodness_dt_%s_%s.txt.gz' %>%
sprintf(model_dir, run_tag, time_stamp)
params_f = '%s/muscat_params_%s_%s.rds' %>%
sprintf(model_dir, run_tag, time_stamp)
ranef_dt_f = sprintf('%s/muscat_ranef_dt_%s_%s.txt.gz',
model_dir, run_tag, time_stamp)
mds_sep_f = sprintf('%s/mds_sep_dt_%s_%s.txt.gz',
model_dir, run_tag, time_stamp)Outputs
# where to save
save_dir = 'output/ms15_mofa'
date_tag = '2021-10-14'
if (!dir.exists(save_dir))
dir.create(save_dir)
# parameters for gene selection
min_sd = log(2)
min_fc = log(2)
max_p = 0.01
n_factors = 5
sel_cl = c("OPCs / COPs", "Oligodendrocytes", "Astrocytes", "Microglia",
"Endothelial cells", "Pericytes", "Immune")
fgsea_cut = 0.1
log_p_mad = 2
n_paths = 50
n_cores = 8
# parameters for plotting
min_var = 5
# output files
mofa_f = sprintf('%s/mofa_%s_%s.hdf5', save_dir, run_tag, date_tag)
fgsea_pat = sprintf('%s/mofa_fgsea_%s_%s_%s.txt',
save_dir, run_tag, '%s', date_tag)
interesting_f = sprintf('%s/mofa_interesting_genes_%s_%s.xlsx',
save_dir, run_tag, date_tag)
# what to use to illustrate random effects concept?
example_cl = 'Oligodendrocytes'
example_gs = c("NHLH1_ENSG00000171786", "CASP7_ENSG00000165806",
"RELN_ENSG00000189056", "KLB_ENSG00000134962", "NRTN_ENSG00000171119",
"EVI5L_ENSG00000142459", "PWP2_ENSG00000241945", "GRID2_ENSG00000152208",
"MET_ENSG00000105976")Load inputs
# load parameters
params = params_f %>% readRDS
# load pseudobulk object
pb = readRDS(params$pb_f) %>% .subset_pb(params$subset_spec) %>%
subset_pb_celltypes(sel_cl) subsetting pb object restricting to samples that meet subset criteria updating factors to remove levels no longer observed# check for any massive outliers
outliers_dt = calc_log_prop_outliers(pb, mad_cut = log_p_mad)no samples have half or more of celltypes with very extreme (2 > MADs)
log proportionsok_samples = outliers_dt[ props_ok == TRUE ]$sample_id
pb = pb[ , ok_samples ]
# load other useful things
labels_dt = .load_labels_dt(labels_f, params$cluster_var)
magma_dt = .load_magma_dt(magma_f, pb)
tfs_dt = .load_tfs_dt(tfs_f, pb)
lof_dt = .load_lof_dt(lof_f, pb)
# load annotations
annots_dt = .get_cols_dt(pb) %>%
.[, sample := sample_id ] %>% .[, group := 'single_group'] %>%
.[, .(sample, sample_id, subject_id, subject_orig, sample_source,
age_at_death, years_w_ms, diagnosis, lesion_type, sex, pmi_cat, smoker )]
annots_dt = add_oligo_groups(annots_dt, olg_grps_f)
annots_dt = add_compositional_groups(annots_dt, comp_grps_f)
# get random effects
ranef_dt = .load_ranef_dt(ranef_dt_f, labels_dt, pb)
# get results
res_dt = muscat_f %>% fread %>%
.load_muscat_results(labels_dt, params) %>%
.[, .(cluster_id, gene_id, symbol, var_type, coef, test_var,
logCPM, mean_soup, padj = p_adj.soup, logFC)] %>%
.[ !is.na(padj) ]
# get anova results
anova_dt = .load_anova_dt(anova_f, res_dt) %>%
.[ is.na(full), full := 1 ]
# get MDS outputs
mds_sep_dt = mds_sep_f %>% fread
if (params$cluster_var == 'type_broad')
mds_sep_dt[, cluster_id := factor(cluster_id, levels = broad_ord)]# get random effects
sd_dt = ranef_dt %>% calc_ranef_melt %>% calc_sd_dt
filter_dt = calc_filter_dt(res_dt, sd_dt, pb, anova_dt,
max_p = max_p, min_sd = min_sd, min_fc = min_fc)
filtered_dt = filter_dt[ ( (ms_signif == 'signif') & (ms_effect == 'big') ) |
( (pt_signif == 'signif') & (pt_variab == 'variable')) ] %>%
.[ cluster_id %in% sel_cl ] %>%
.[, is_ms := ifelse(ms_effect == "big" & ms_signif == "signif", "ms", "not") ] %>%
.[, is_pt := ifelse(pt_signif == "signif" & pt_variab == "variable", "pt", "not") ]
# check what we've got
filtered_dt[, .N, by = .(cluster_id, is_ms, is_pt)] %>%
.[, total := sum( N ), by = cluster_id ] %>%
dcast.data.table(cluster_id + total ~ is_ms + is_pt, fill = 0, value.var = "N") cluster_id total ms_not ms_pt not_pt
1: OPCs / COPs 97 20 2 75
2: Oligodendrocytes 507 259 16 232
3: Astrocytes 794 559 29 206
4: Microglia 667 255 48 364
5: Endothelial cells 86 2 0 84
6: Pericytes 27 1 0 26
7: Immune 27 4 0 23n_cells_dt = calc_n_cells_dt(pb_fine_f, annots_dt, sel_cl)soup_dt = get_soup_logcpms(soup_f, pb)Processing / calculations
mofa_obj = make_mofa_obj_samples(pb, filtered_dt, sel_cl)Creating MOFA object from a data.frame...# set up
data_opts = get_default_data_options(mofa_obj)
model_opts = get_default_model_options(mofa_obj)
train_opts = get_default_training_options(mofa_obj)
# specify how many factors
model_opts$num_factors = n_factors
# train mofa
mofa_obj = prepare_mofa(
object = mofa_obj,
data_options = data_opts,
model_options = model_opts,
training_options = train_opts
)Checking data options...Checking training options...Checking model options...model = run_mofa(mofa_obj, mofa_f)Warning: Output file output/ms15_mofa/mofa_run09_2021-10-14.hdf5 already exists, it will be replacedConnecting to the mofapy2 python package using reticulate (use_basilisk = FALSE)...
Please make sure to manually specify the right python binary when loading R with reticulate::use_python(..., force=TRUE) or the right conda environment with reticulate::use_condaenv(..., force=TRUE)
If you prefer to let us automatically install a conda environment with 'mofapy2' installed using the 'basilisk' package, please use the argument 'use_basilisk = TRUE'Warning in .quality_control(object, verbose = verbose): Factor(s) 1 are strongly correlated with the total number of expressed features for at least one of your omics. Such factors appear when there are differences in the total 'levels' between your samples, *sometimes* because of poor normalisation in the preprocessing steps.# update metadata
model = add_metadata(model, annots_dt)
# put weights and scores in MS order
model = put_model_in_ms_order(model)var_exp_dt = get_variance_explained(model, as.data.frame = TRUE) %>%
as.data.table %>%
.[, .(
view = r2_per_factor.view %>% factor(levels = broad_short),
factor = r2_per_factor.factor,
var_exp = r2_per_factor.value
)]
to_plot_dt = var_exp_dt[ var_exp > min_var ] %>% .[order(factor, -var_exp)]w_dt = extract_weights(model, sd_dt)
fgsea_fs = sapply(names(paths_list)[1:2], function(p) sprintf(fgsea_pat, p))
if (all(file.exists(fgsea_fs))) {
# gsea_list = lapply(fgsea_fs, fread)
gsea_list = lapply(fgsea_fs, fread)
} else {
# do fgsea for these
bpparam = MulticoreParam(workers = n_cores,
progressbar = TRUE, tasks = 50)
bpstart()
gsea_list = calc_mofa_fgsea(paths_list[1:2], w_dt, fgsea_pat, fgsea_cut, bpparam)
bpstop()
}
# restrict to interesting ones
gsea_main = gsea_list %>% map( ~.x[ main_path == TRUE ]) %>% rbindlistr2_dt = calc_r2_for_factors(model, annots_dt)Analysis
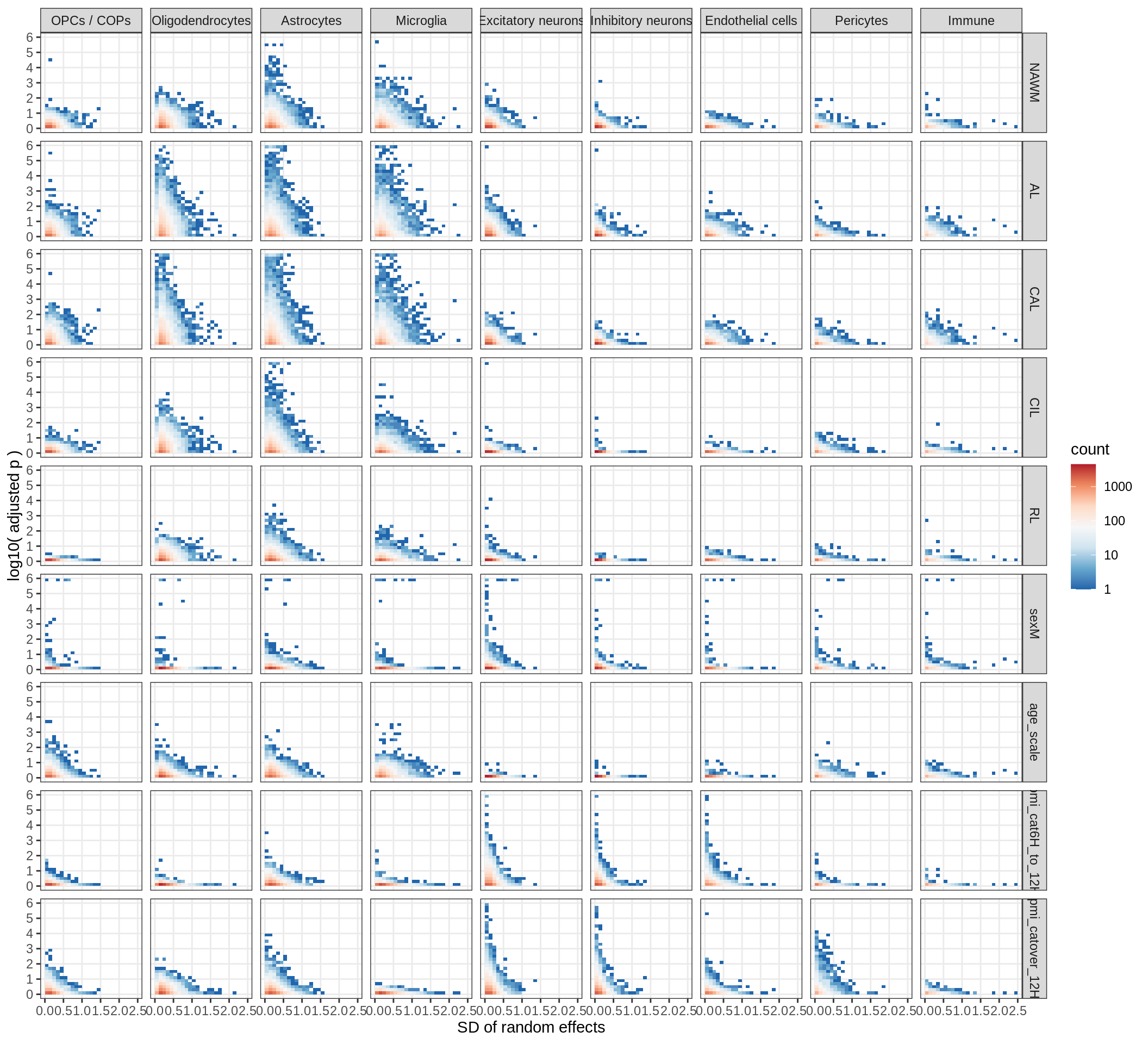
muscat results vs SD
for (what in c('log10_padj', 'log2FC')) {
cat('### ', what, '\n', sep = '')
print(plot_muscat_vs_sd(res_dt, sd_dt, NULL, what = what))
cat('\n\n')
}

Cytokine effects
cyto_gs = unique(res_dt$gene_id) %>% str_subset('(^IL[0-9]+|^CCL|^CXCL|^IFN|^TGF|^TNF|^CSF)')
(plot_muscat_vs_sd_min(res_dt[ gene_id %in% cyto_gs ], sd_dt[ gene_id %in% cyto_gs ],
sel_cl, min_sd, max_p, do_labels = TRUE))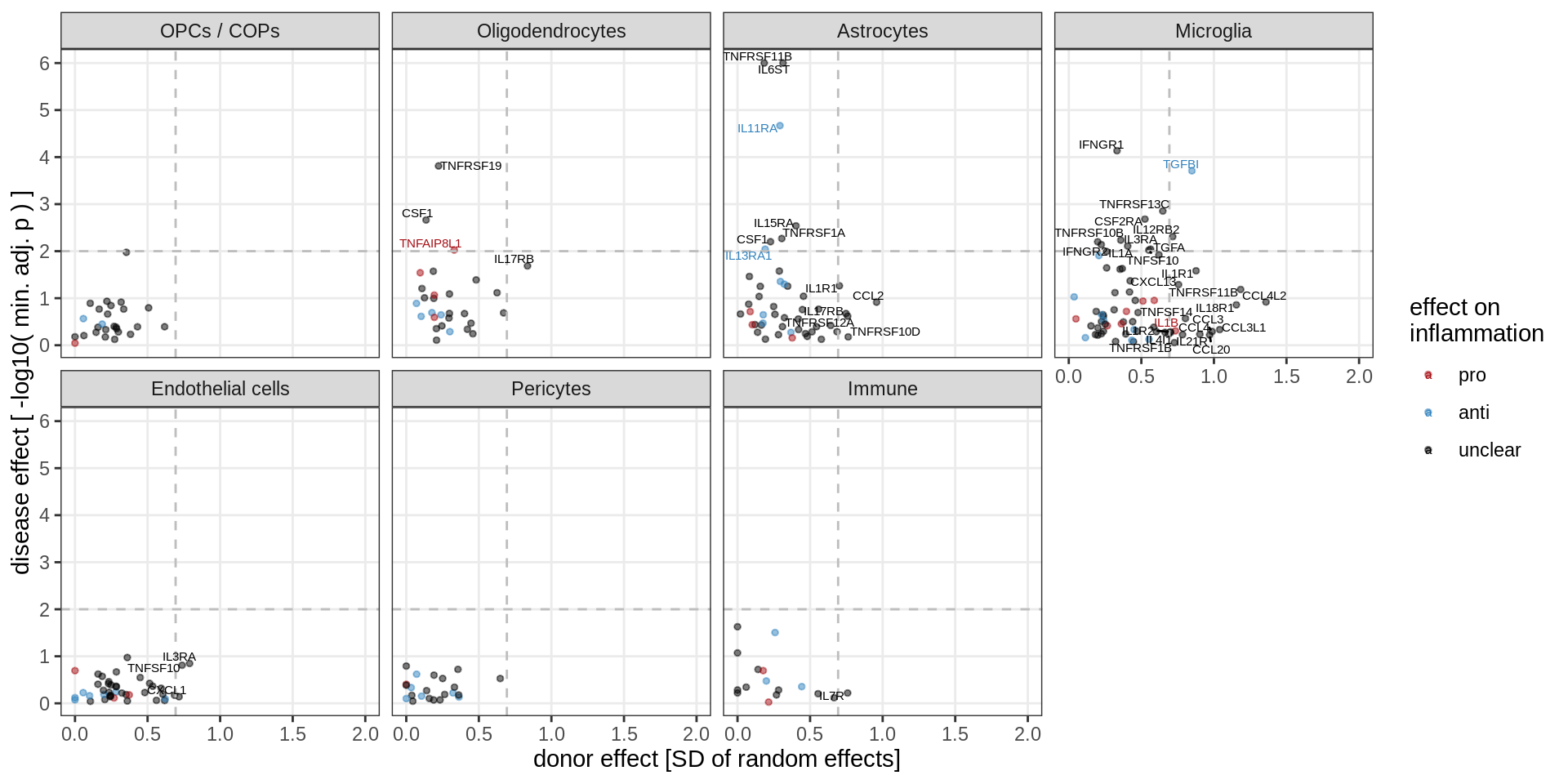
Ages vs duration of MS
(plot_age_duration(annots_dt))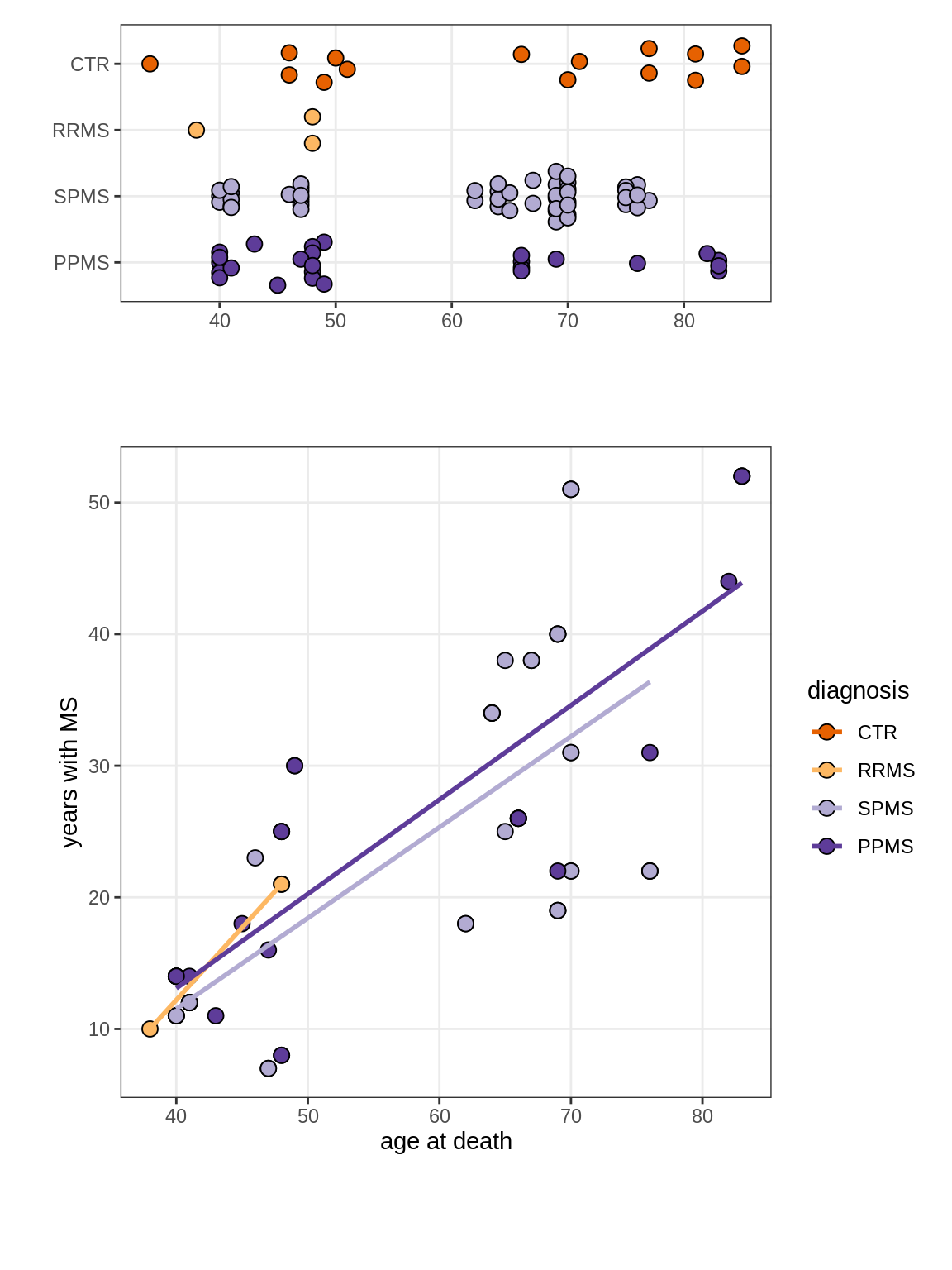
| Version | Author | Date |
|---|---|---|
| ad82ee3 | Macnair | 2021-10-04 |
Data overview
(plot_data_overview(mofa_obj))Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> =
"none")` instead.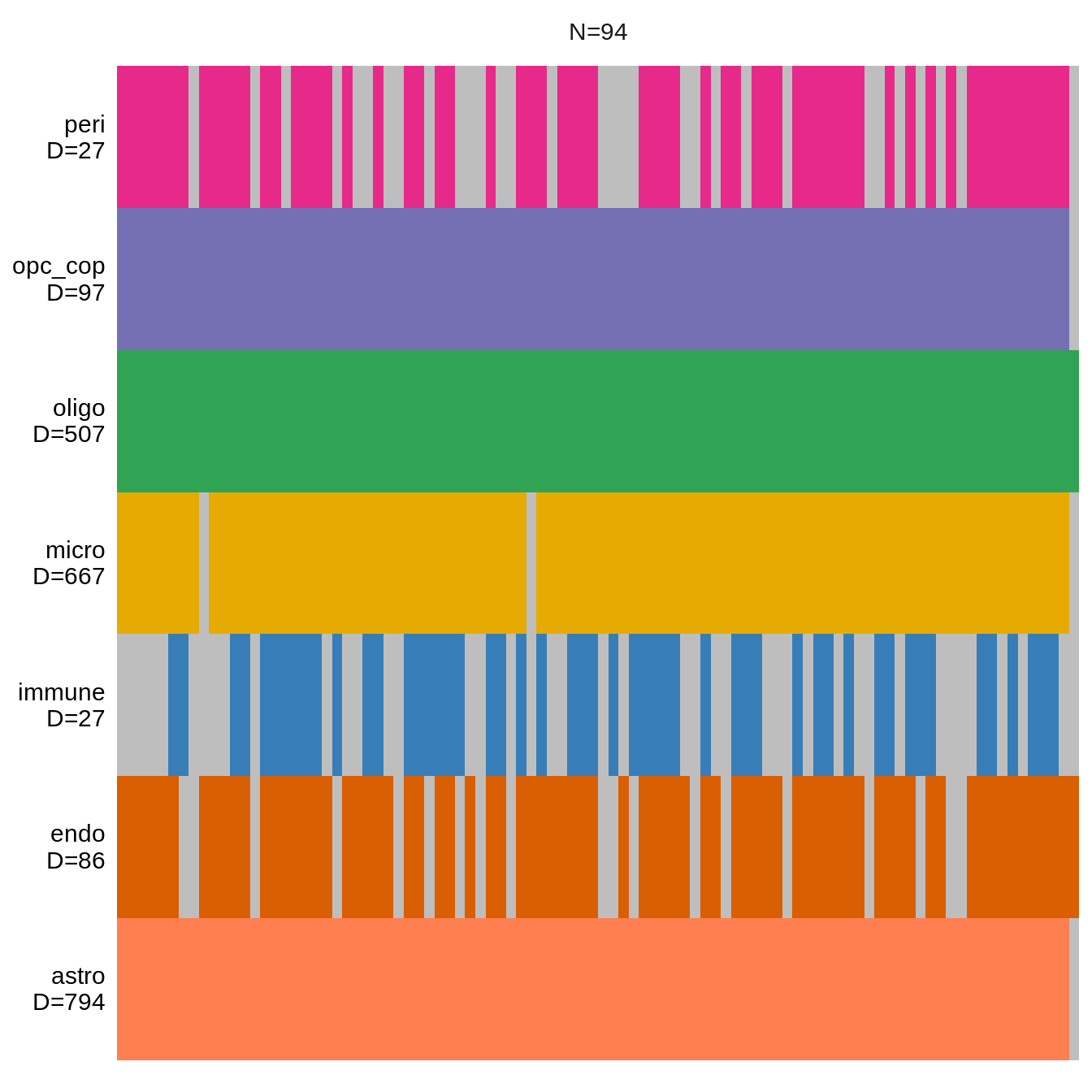
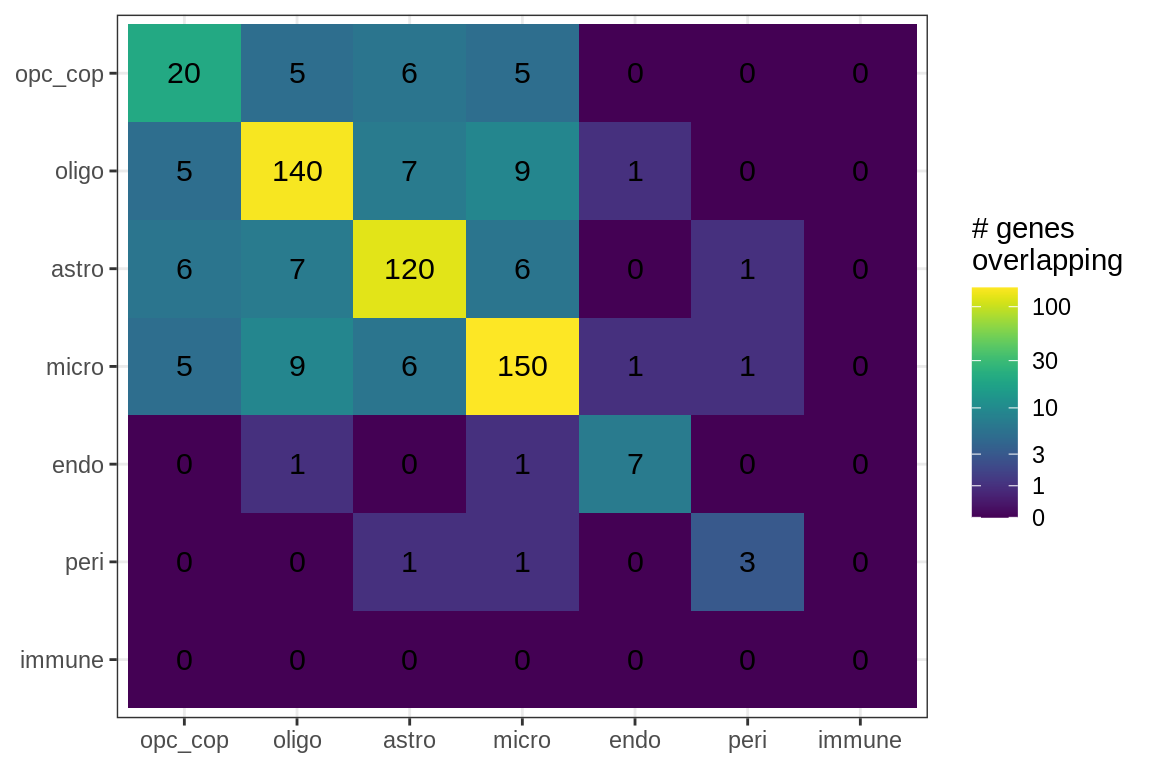
Overlapping genes
cat('### All genes\n')All genes
suppressWarnings(print(plot_gene_overlap(model)))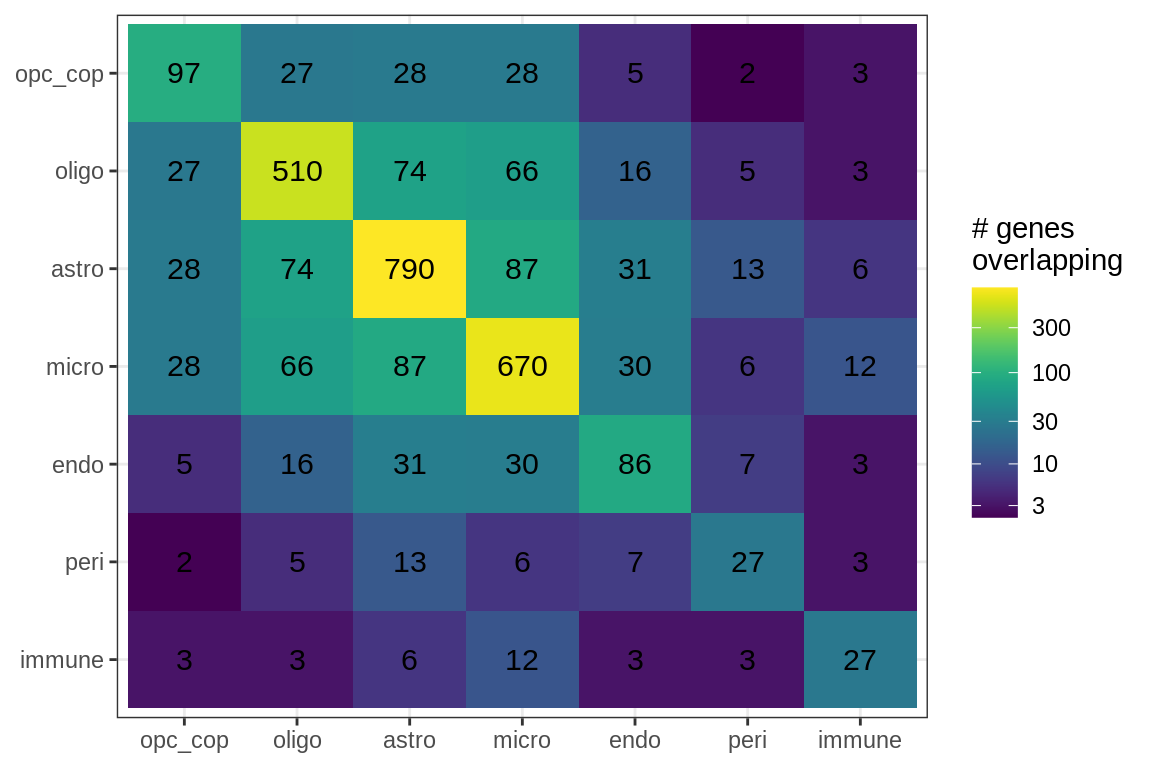
cat('\n\n')for (sel_f in factors_names(model)) {
cat('### Genes in ', sel_f, '\n', sep = '')
suppressWarnings(print(plot_gene_overlap(model, sel_f = sel_f, w_cut = 0.2)))
cat('\n\n')
}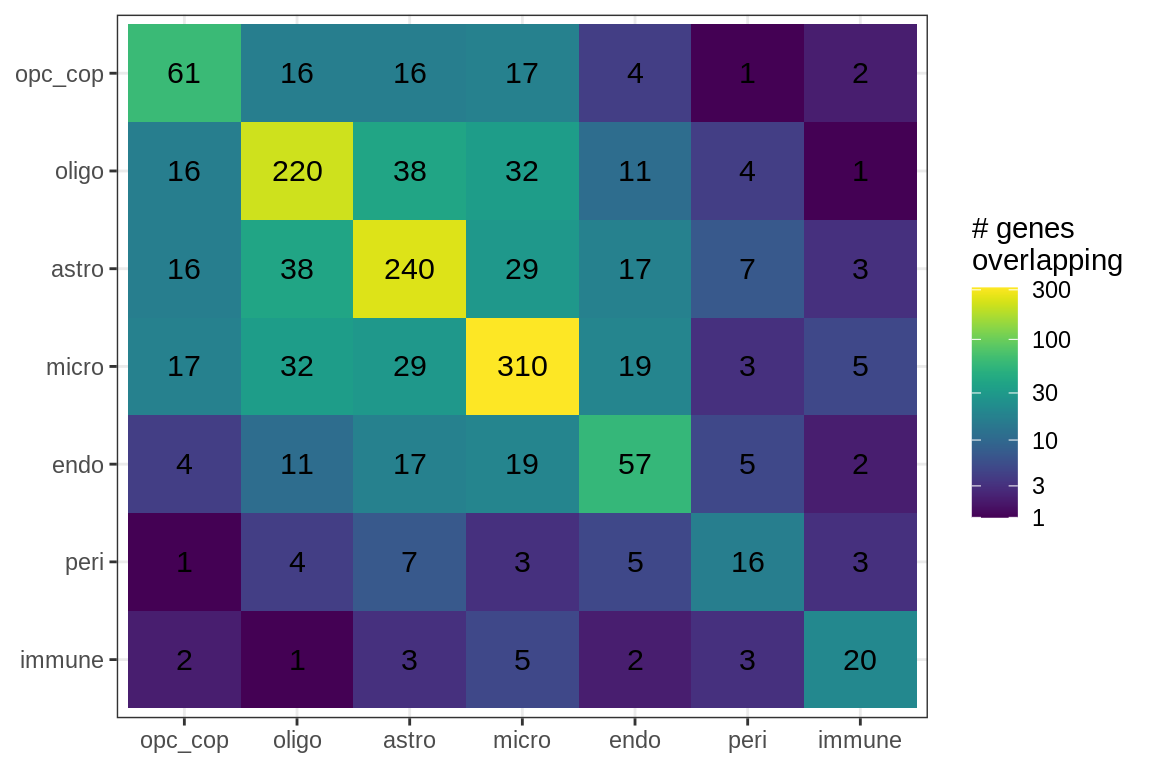
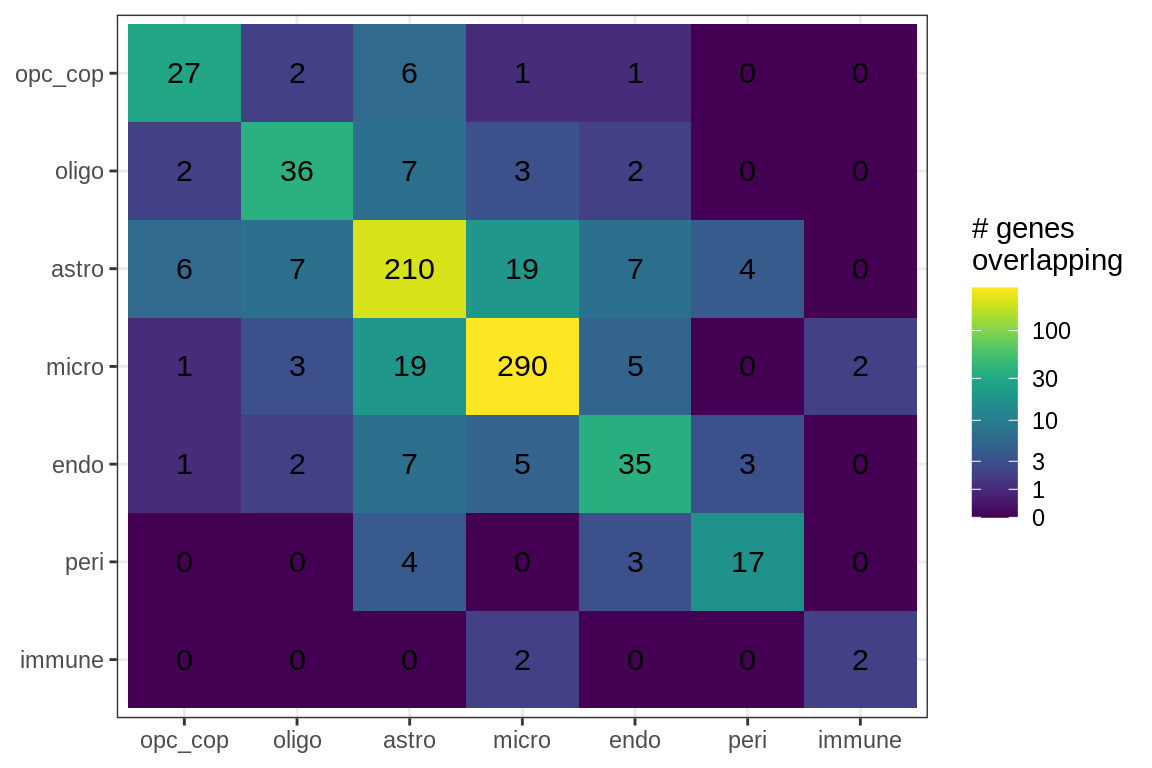
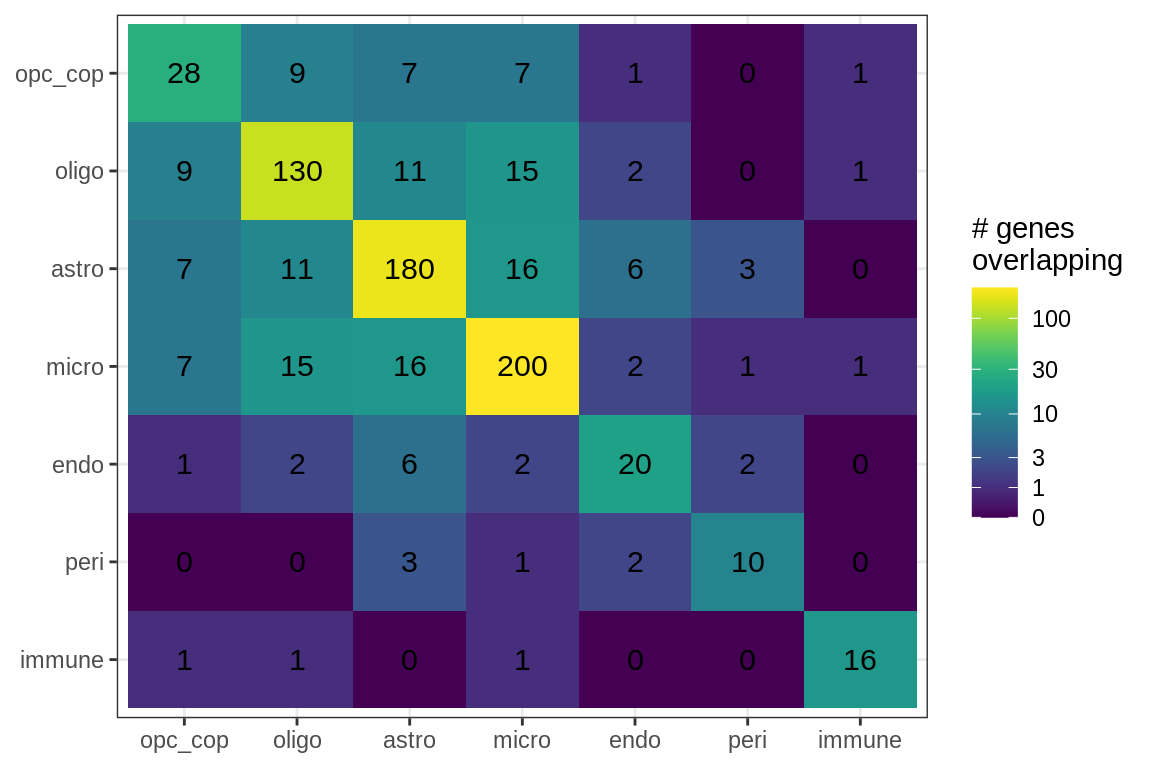

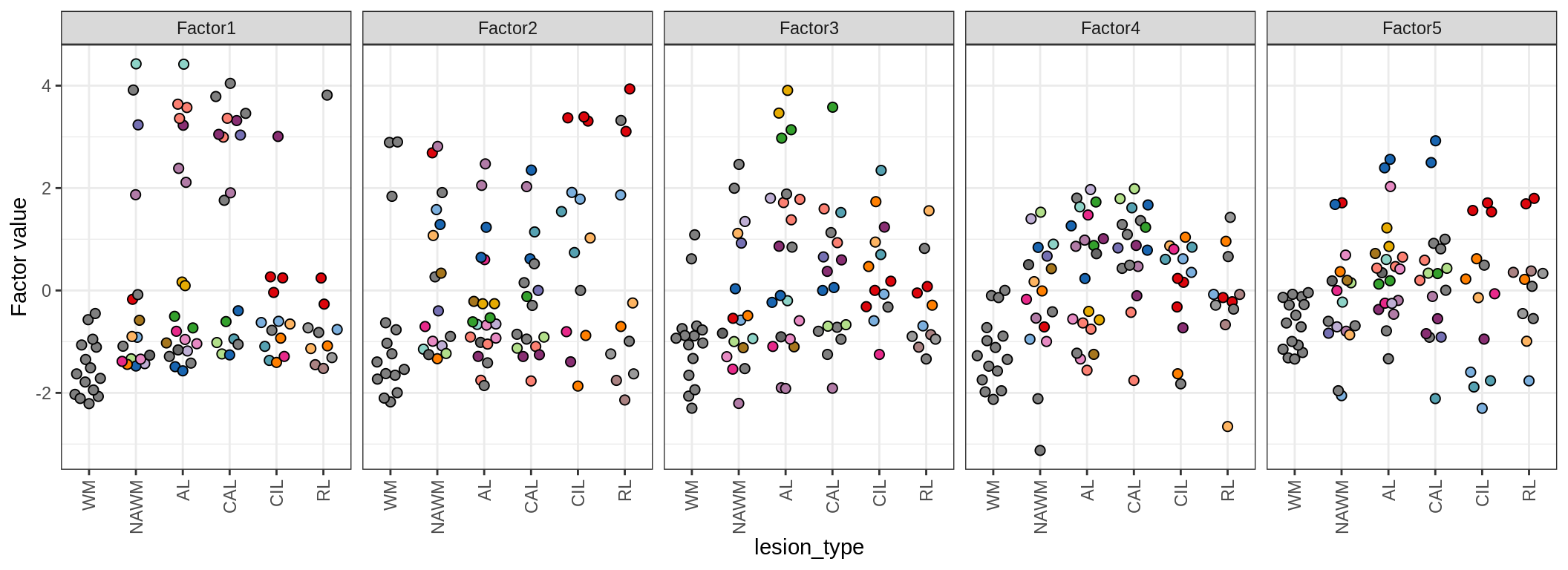
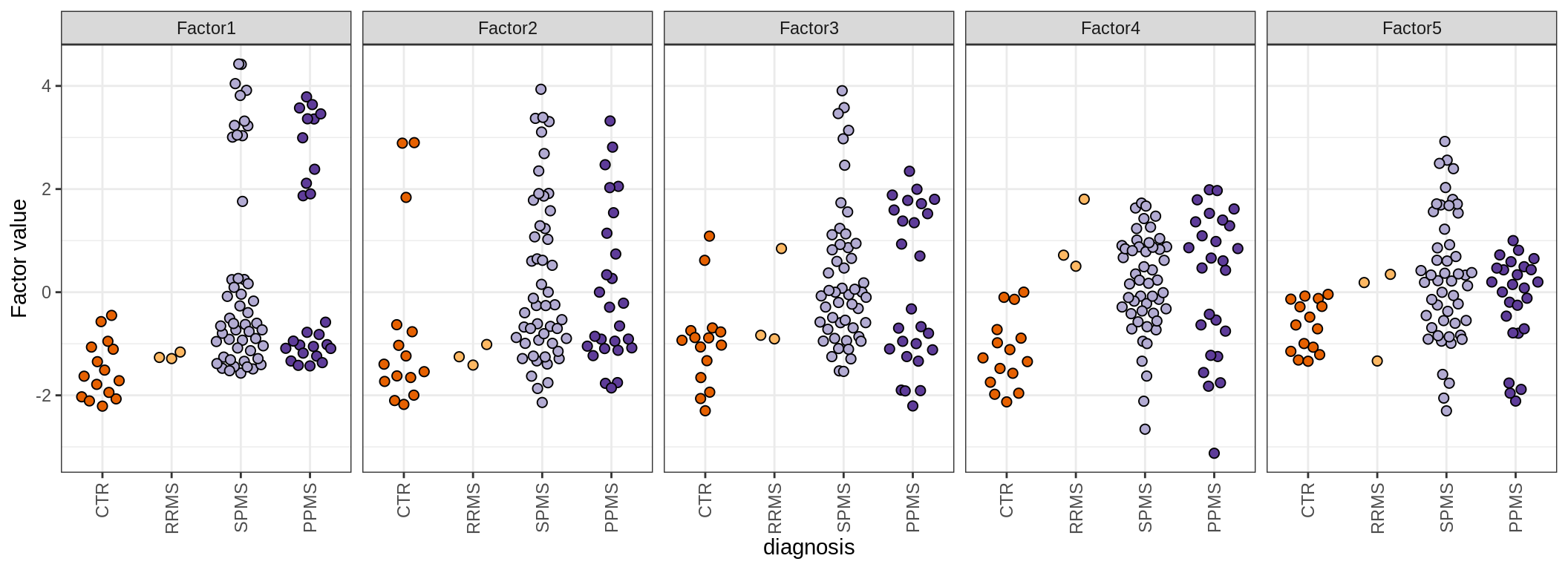
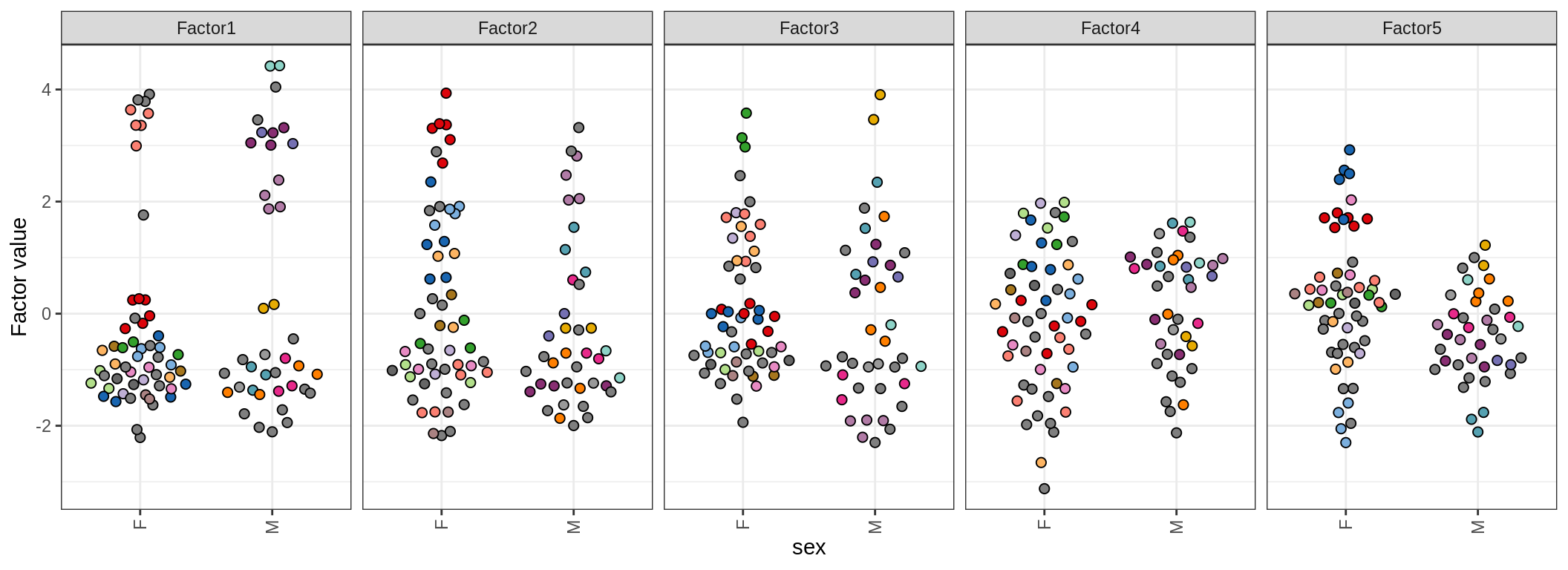
Factor distributions
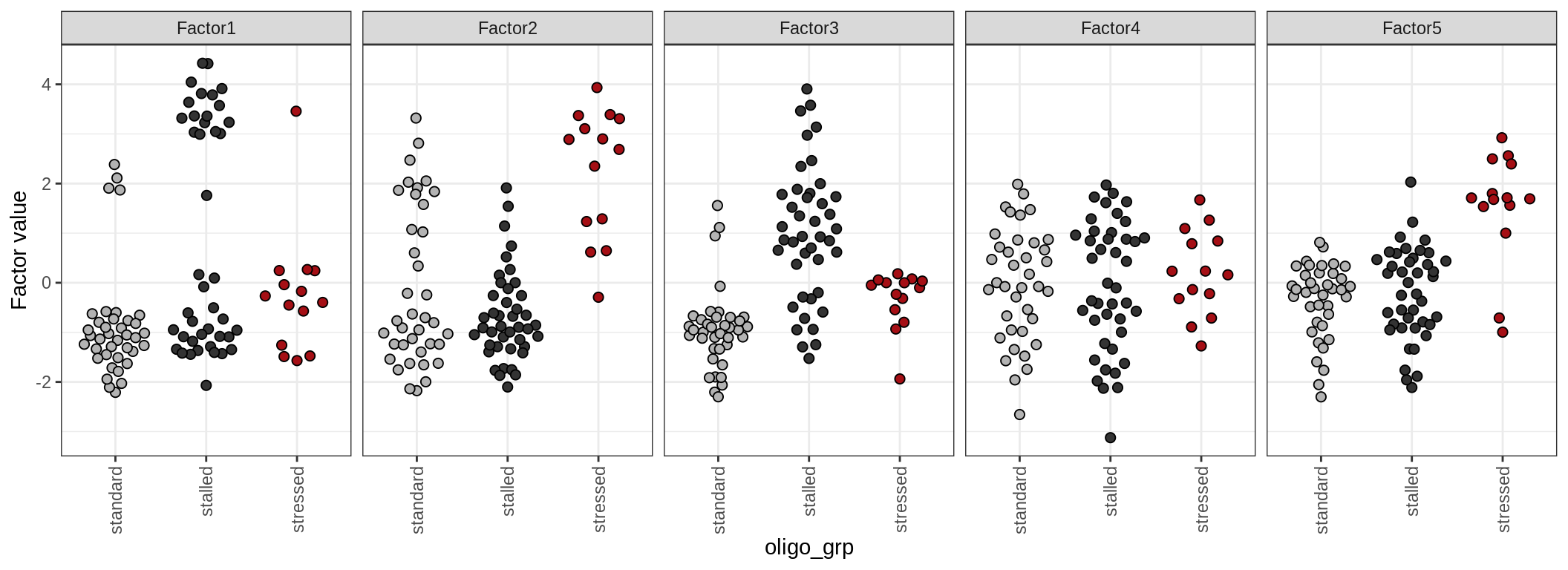
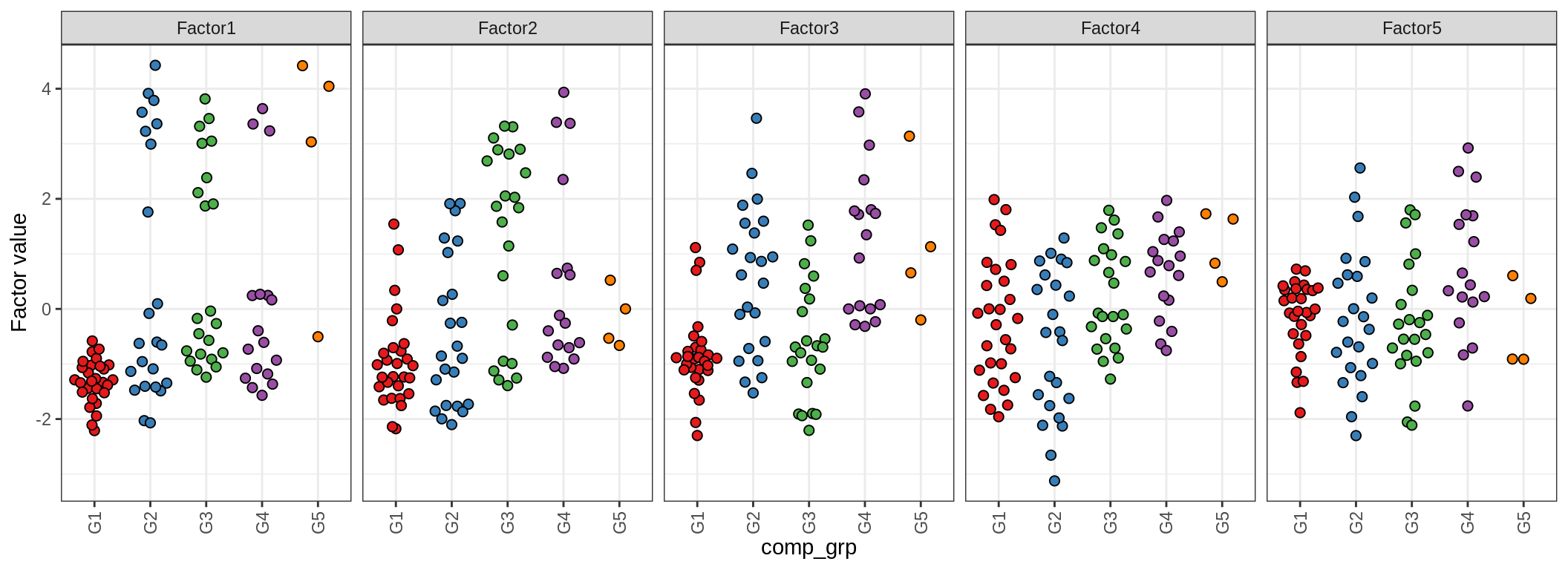
for (annot in c('lesion_type', 'diagnosis', 'sex', 'sample_source', 'smoker', 'oligo_grp', 'comp_grp')) {
cat('### by ', annot, '\n', sep = '')
print(plot_factors_univariate(model, annots_dt, pb, by = annot))
cat('\n\n')
}by lesion_type
by diagnosis
by sex
by sample_source

by smoker
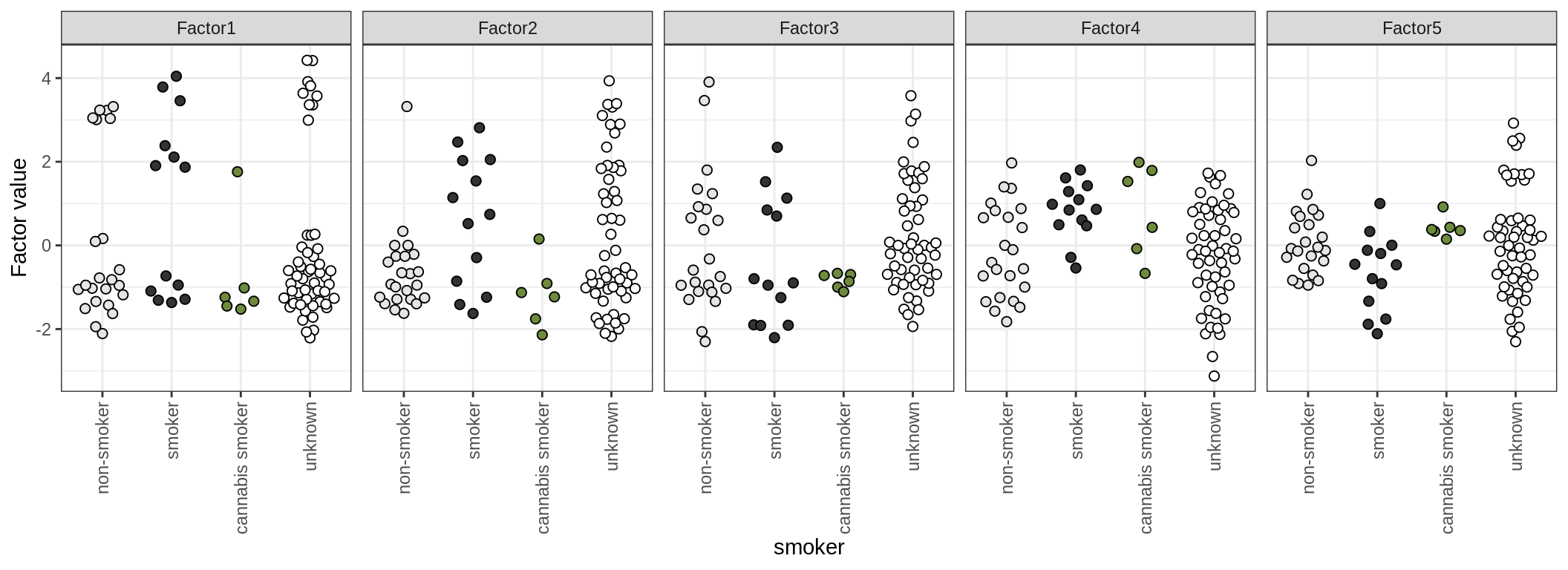
by oligo_grp
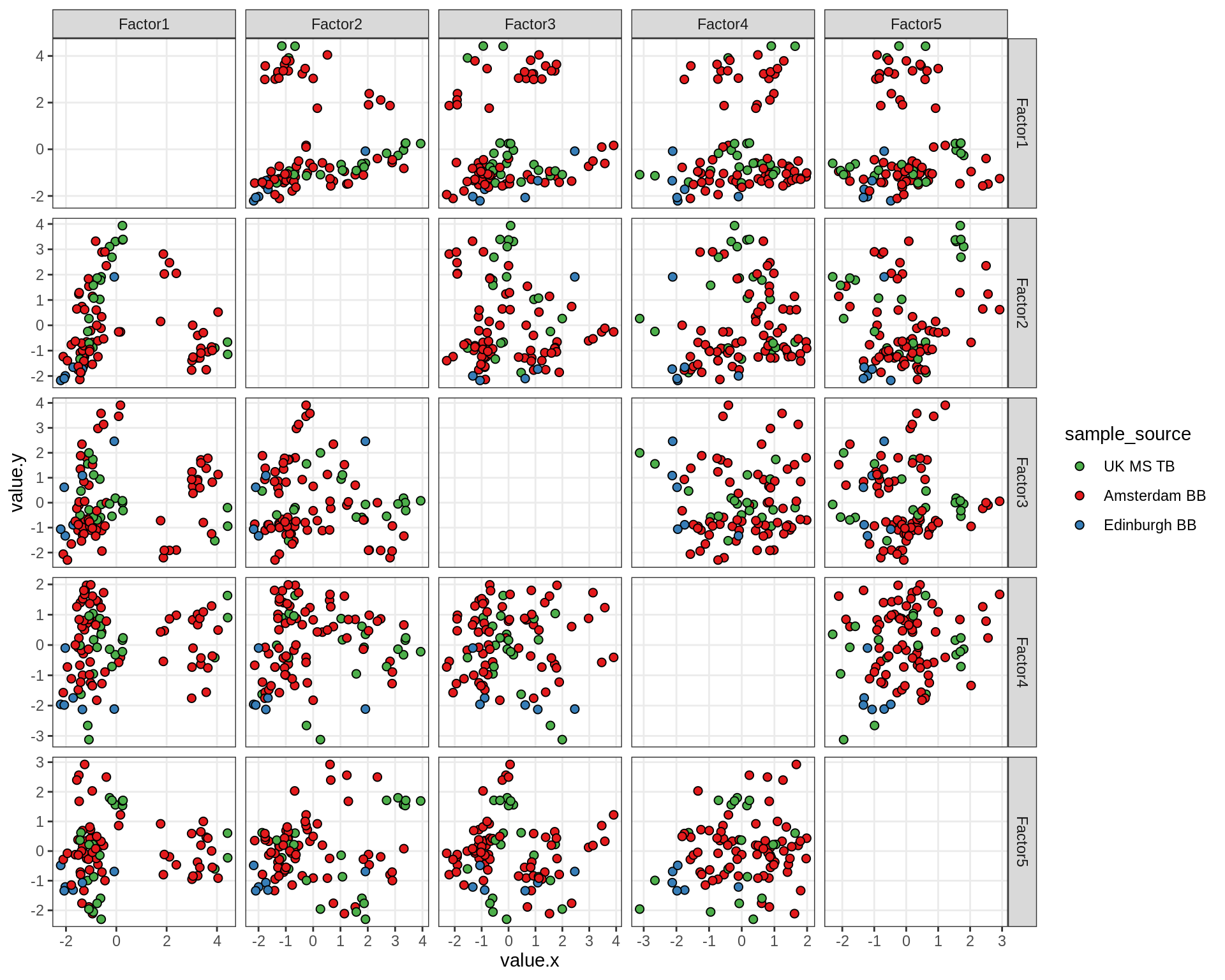
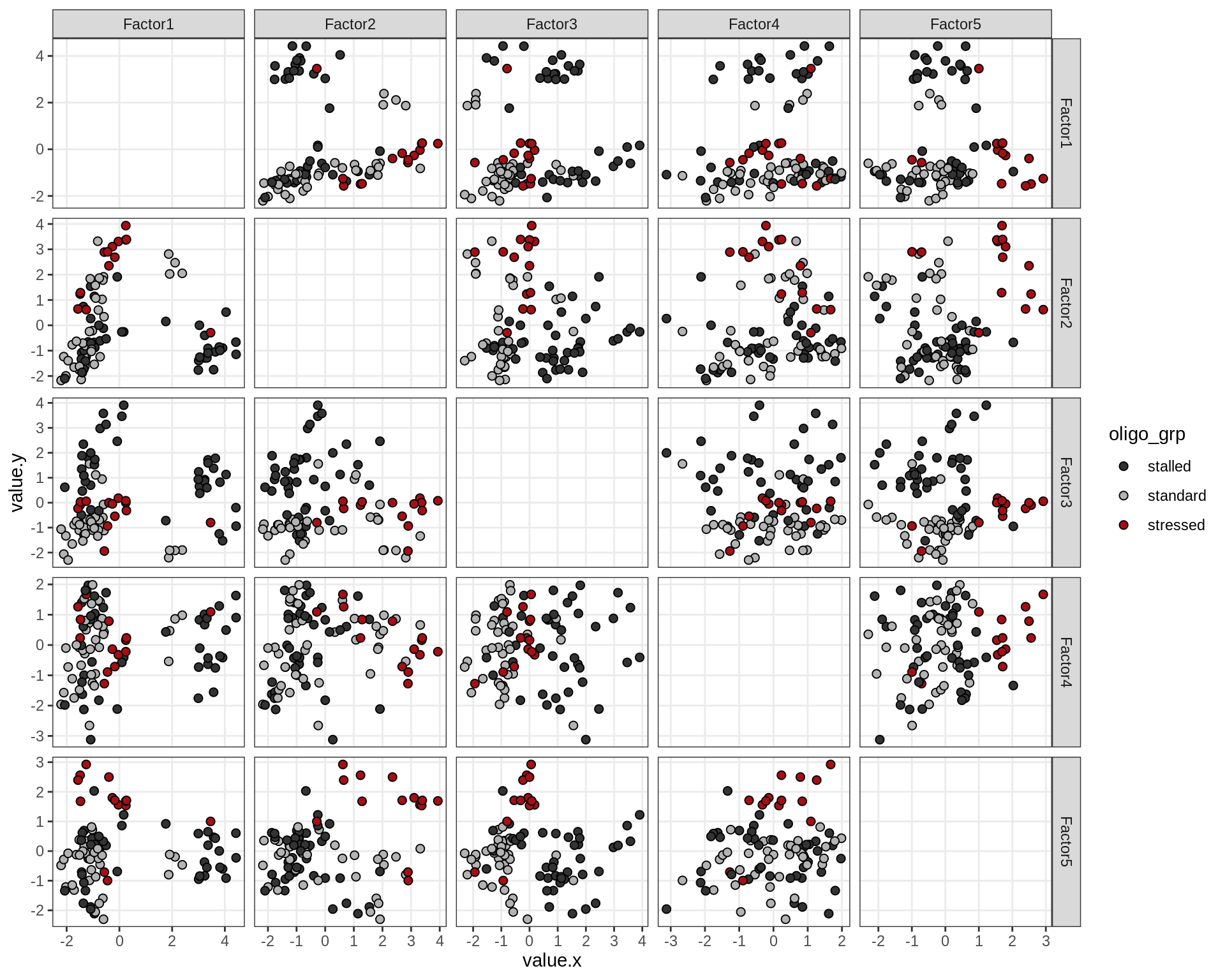
Factor distributions - pairwise
for (annot in c('subject_id', 'lesion_type', 'diagnosis', 'sex', 'sample_source', 'smoker', 'oligo_grp', 'comp_grp')) {
cat('### by ', annot, '\n', sep = '')
print(plot_factors_pairwise(model, annots_dt, pb, by = annot))
cat('\n\n')
}by subject_id
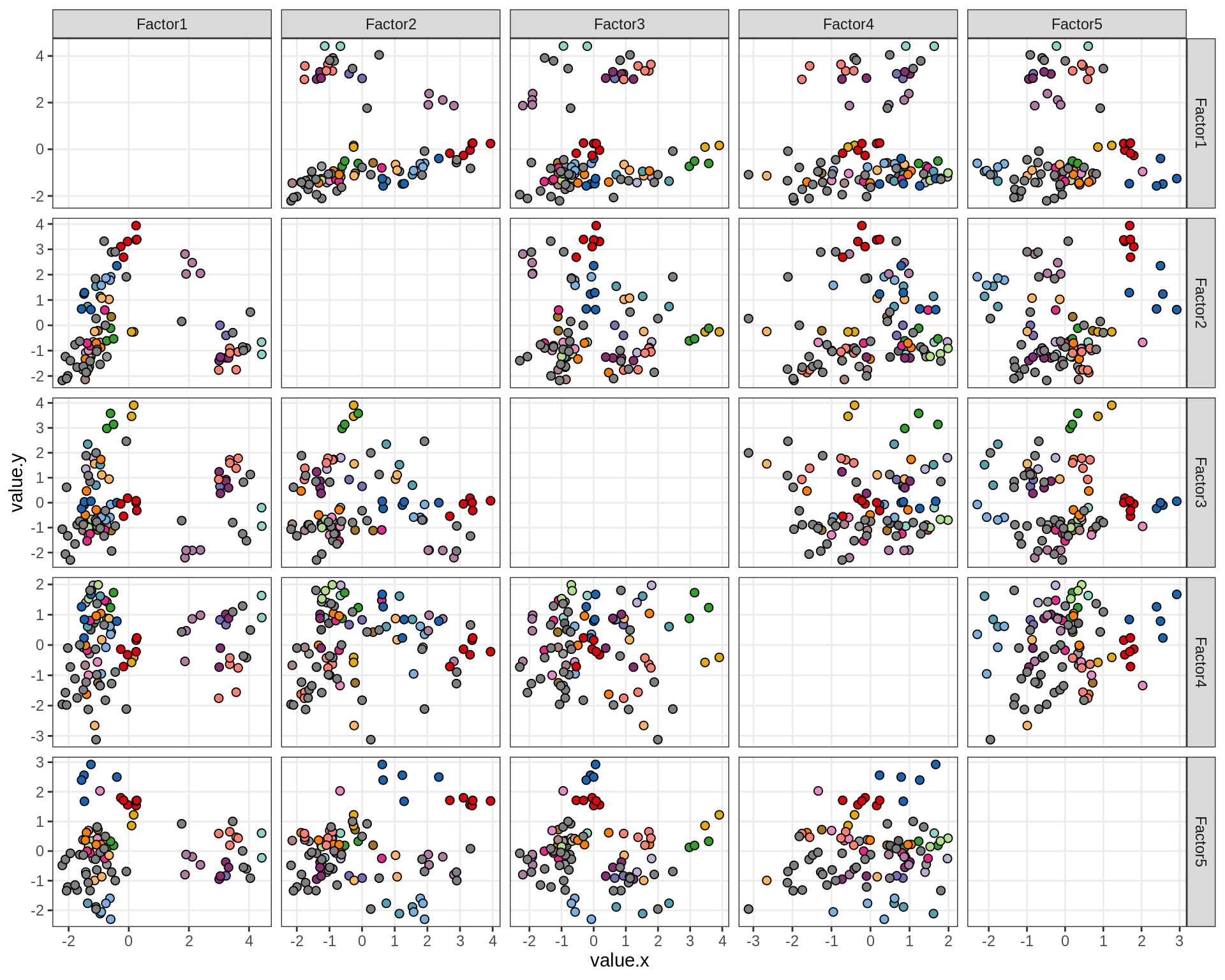
by lesion_type

by diagnosis
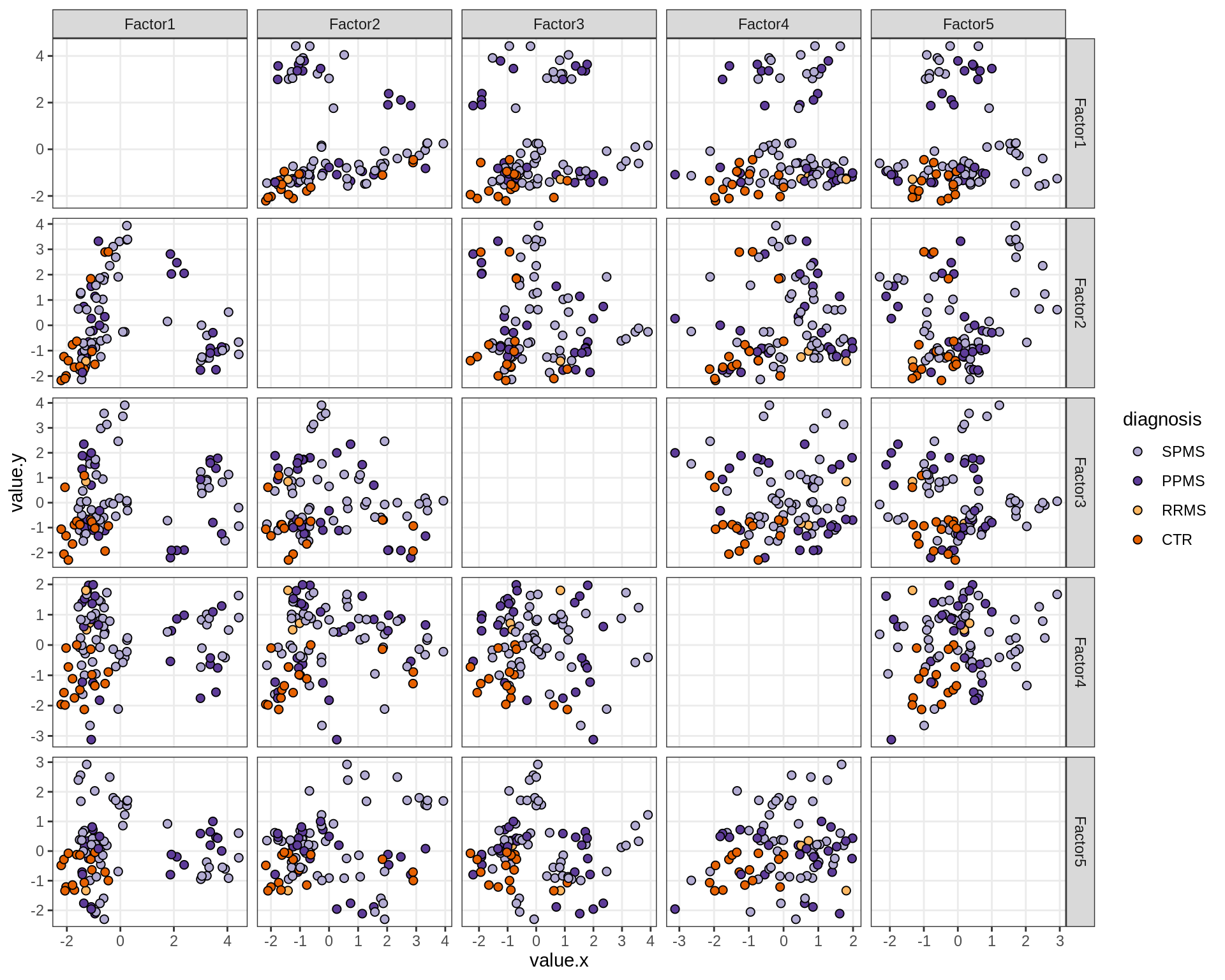
by sex
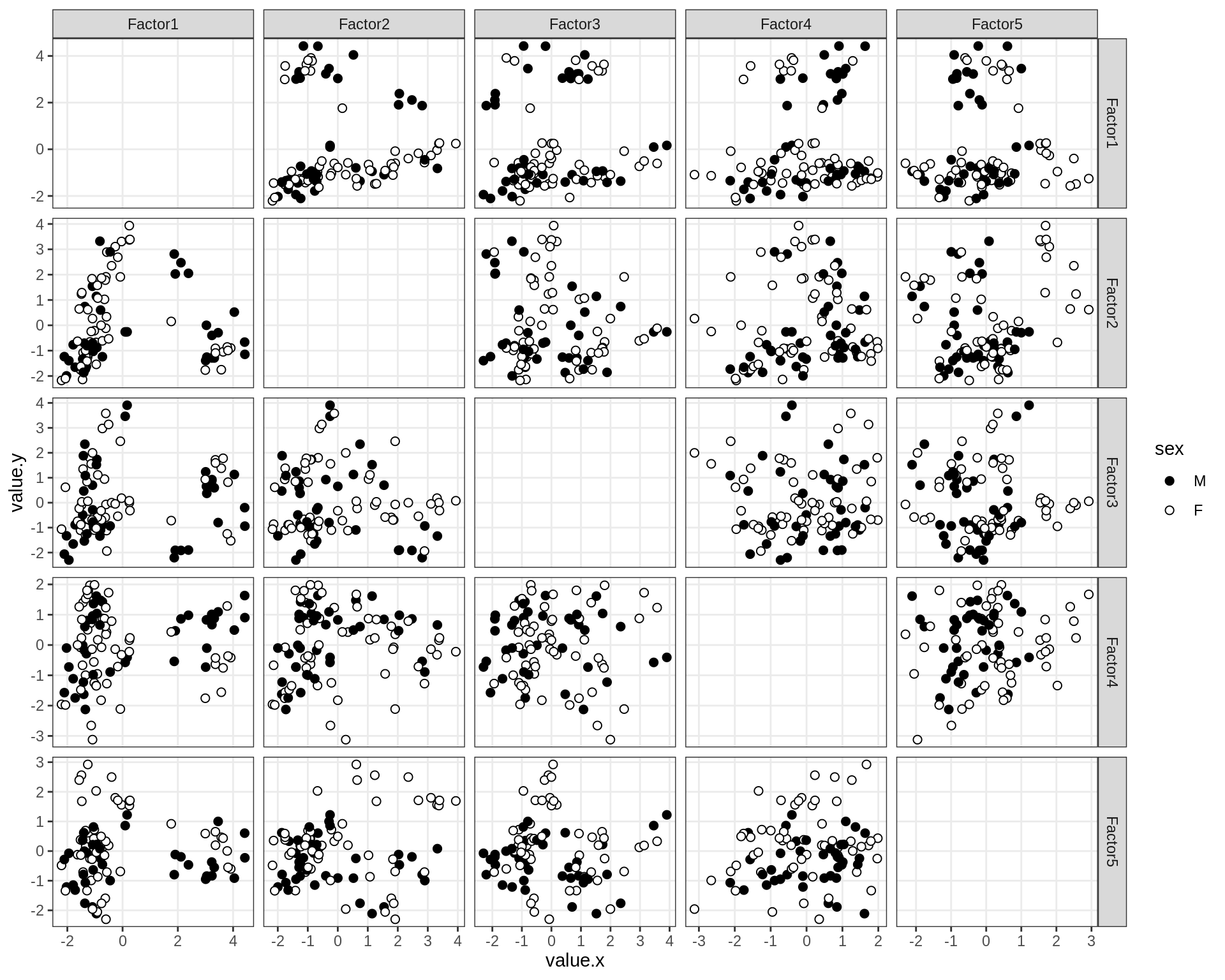
by sample_source
by smoker

by oligo_grp
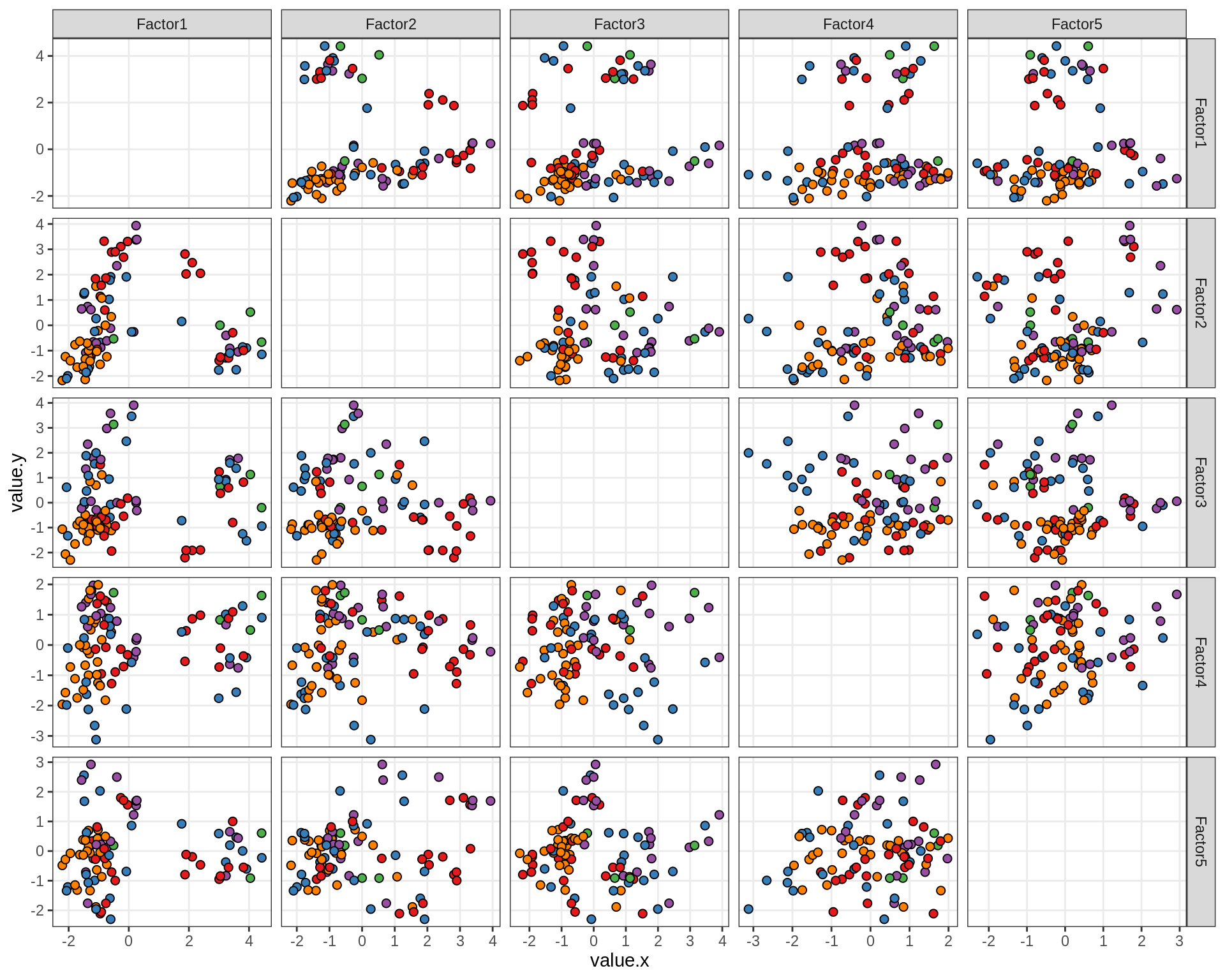
Factors over MDS layouts
for (cl in broad_ord) {
if (!(broad_short[[cl]] %in% views_names(model)))
next
cat('### ', cl, '\n', sep = '')
print(plot_factors_over_mds_samples(model, mds_sep_dt, cl = cl))
cat('\n\n')
}OPCs / COPs
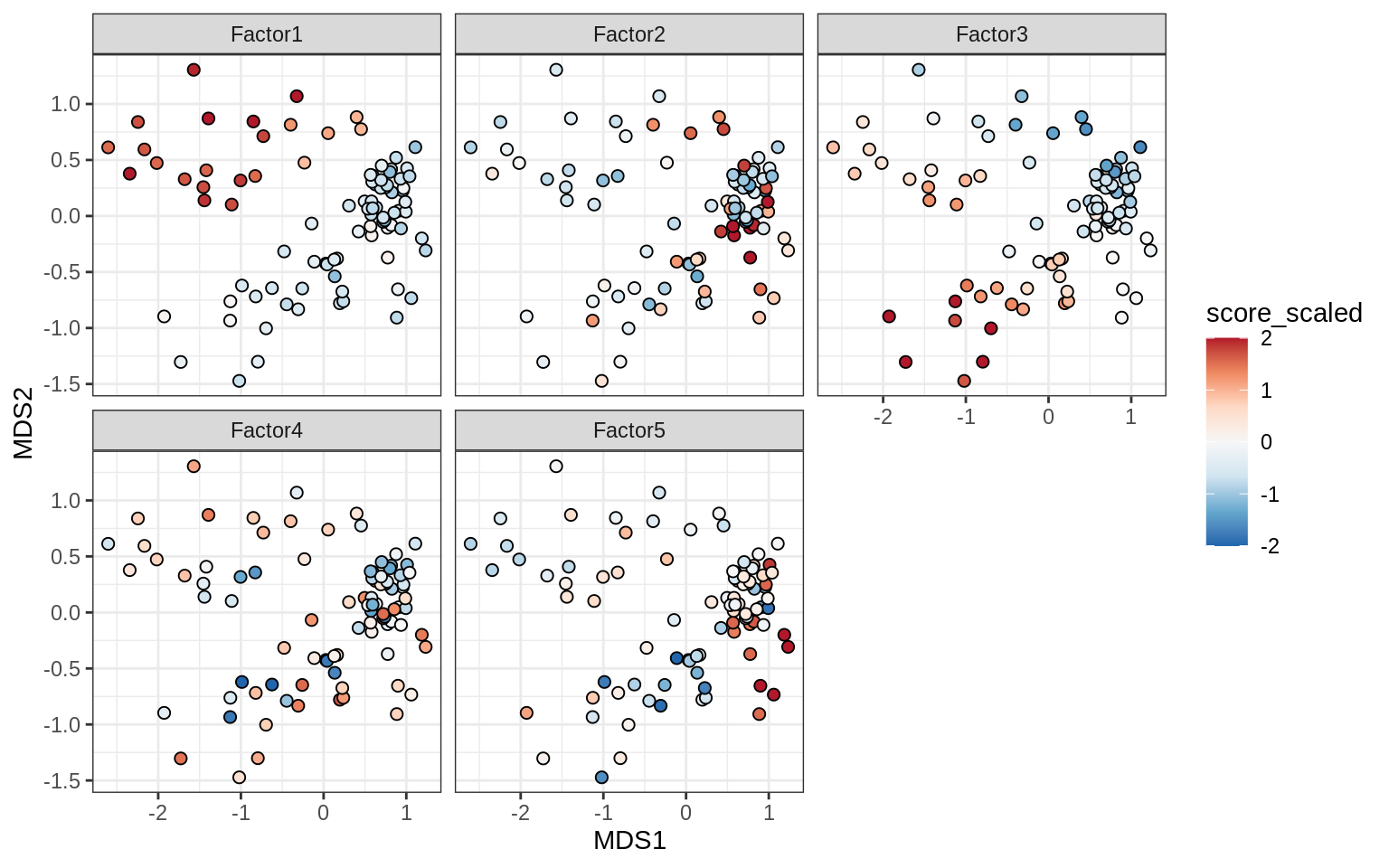
Oligodendrocytes
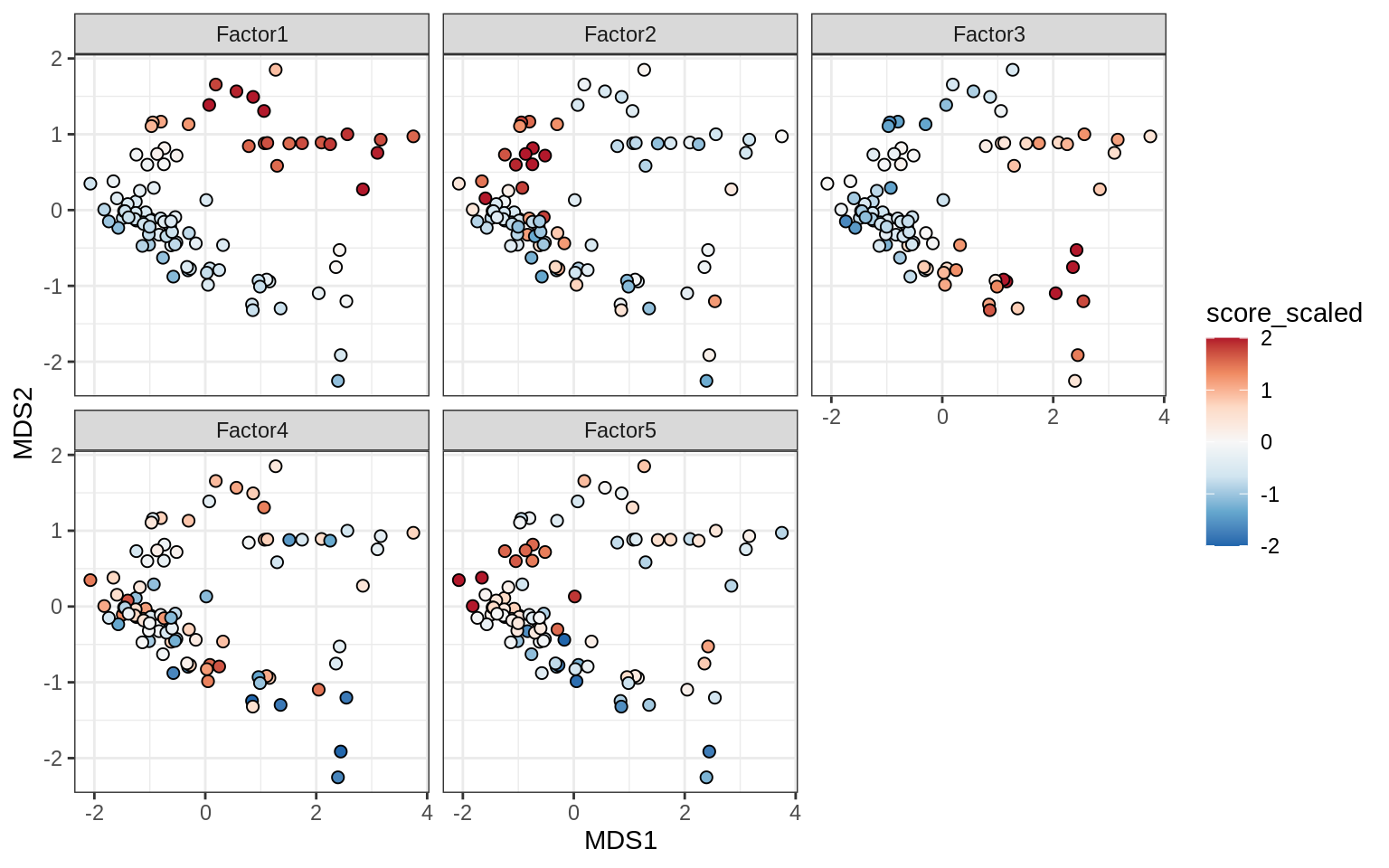
Astrocytes
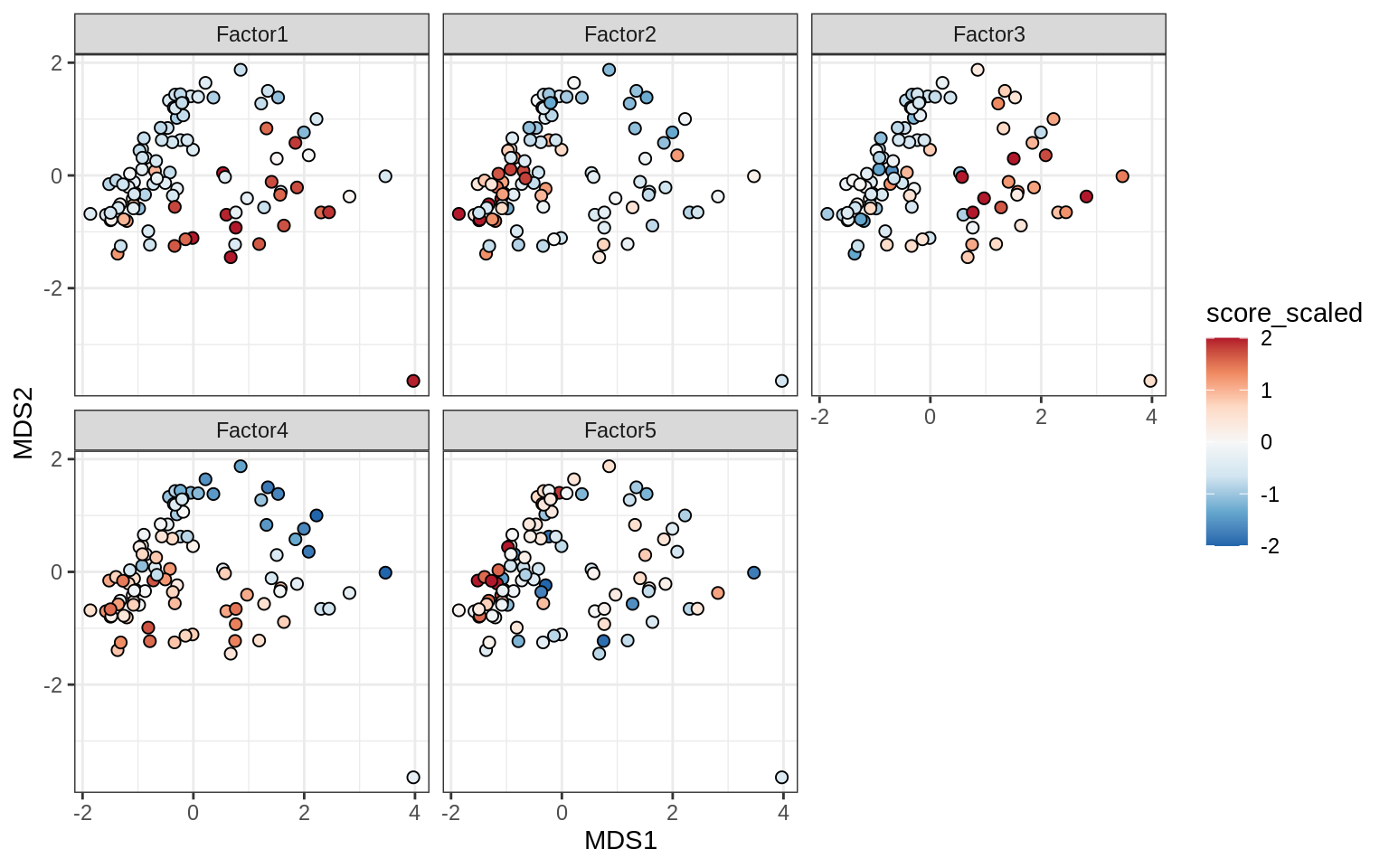
Microglia
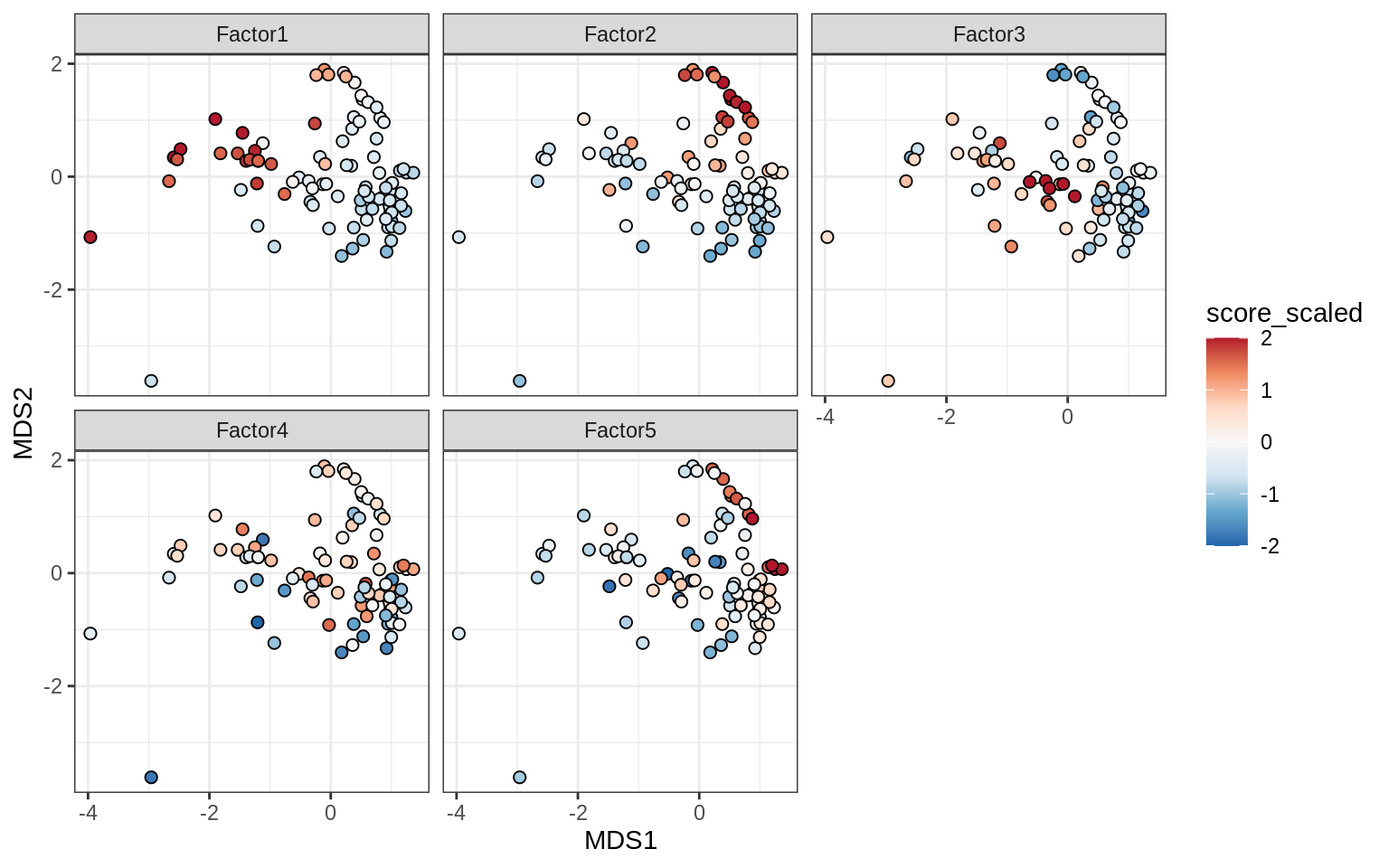
Endothelial cells
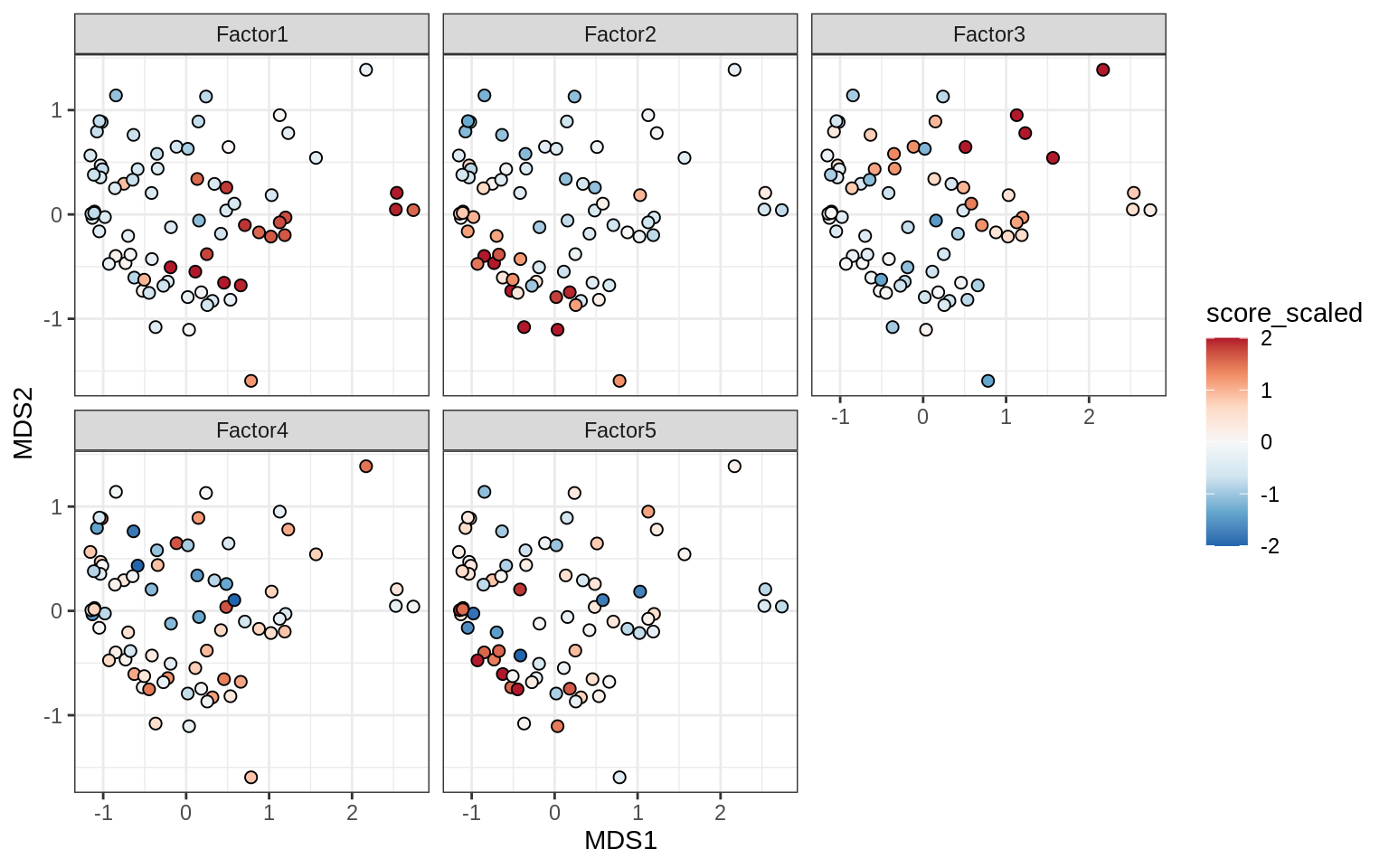
Pericytes
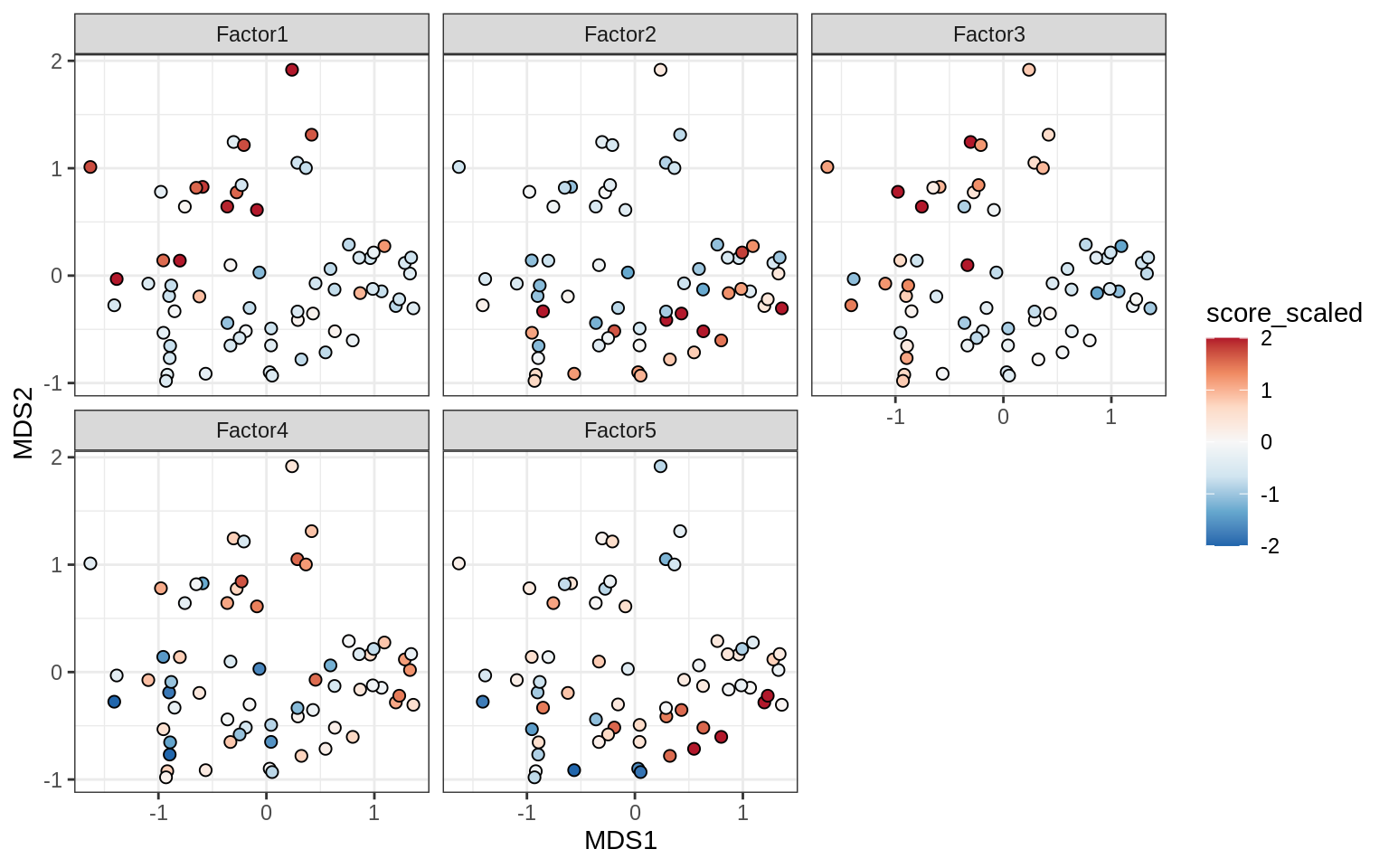
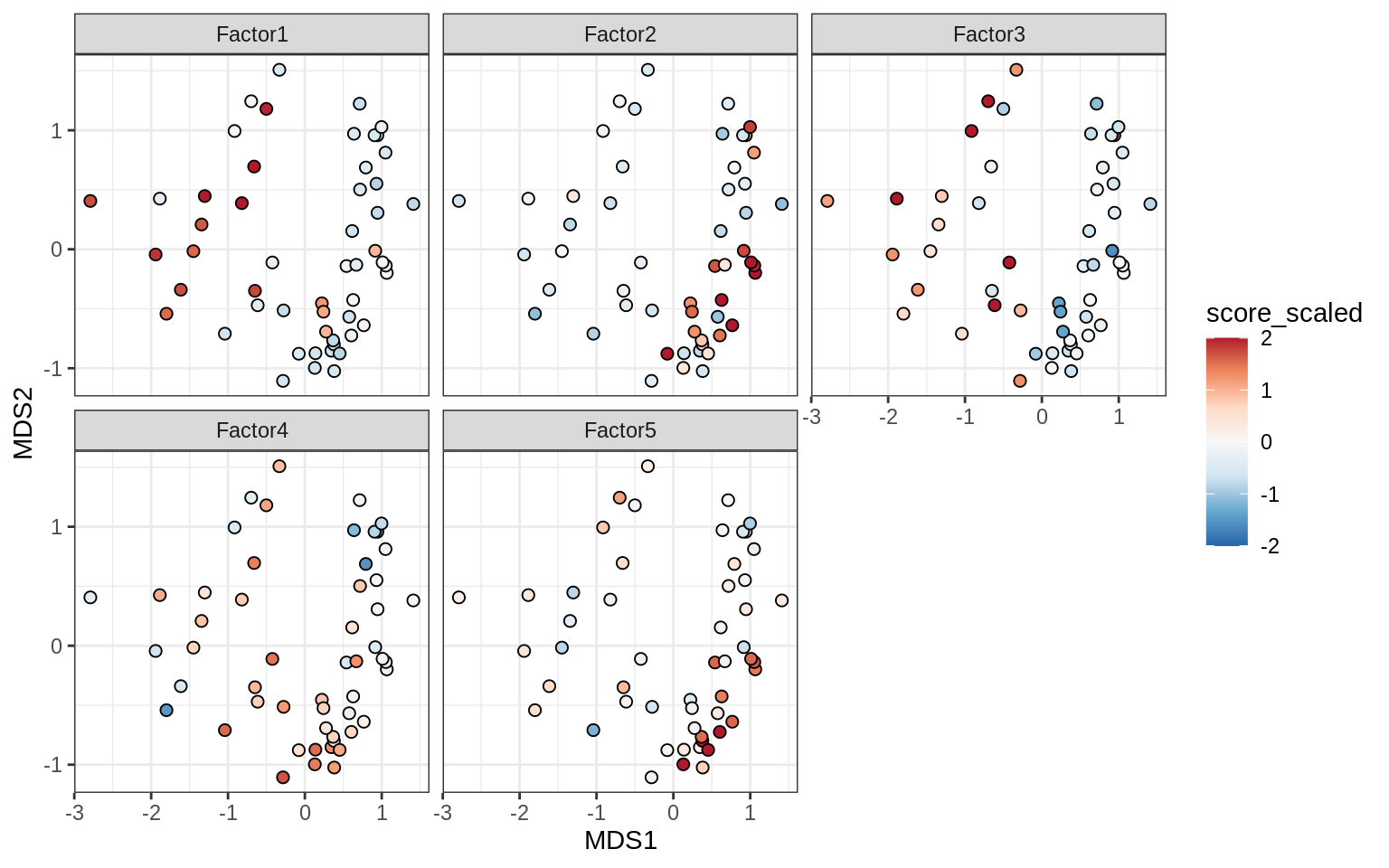
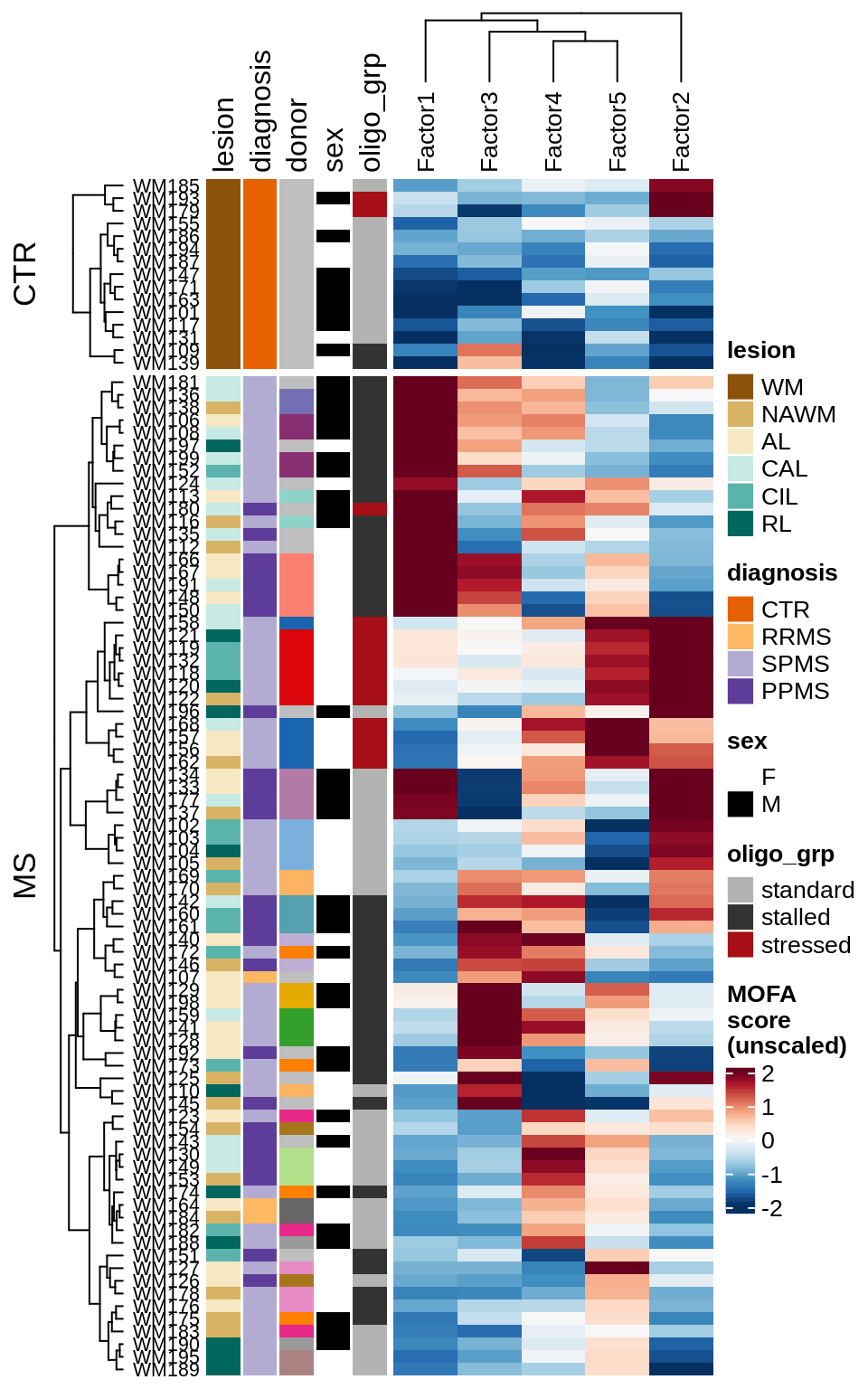
Factor distributions with patient annotations - few
for (v in c('score', 'score_scaled')) {
cat('### ', v, '\n', sep = '')
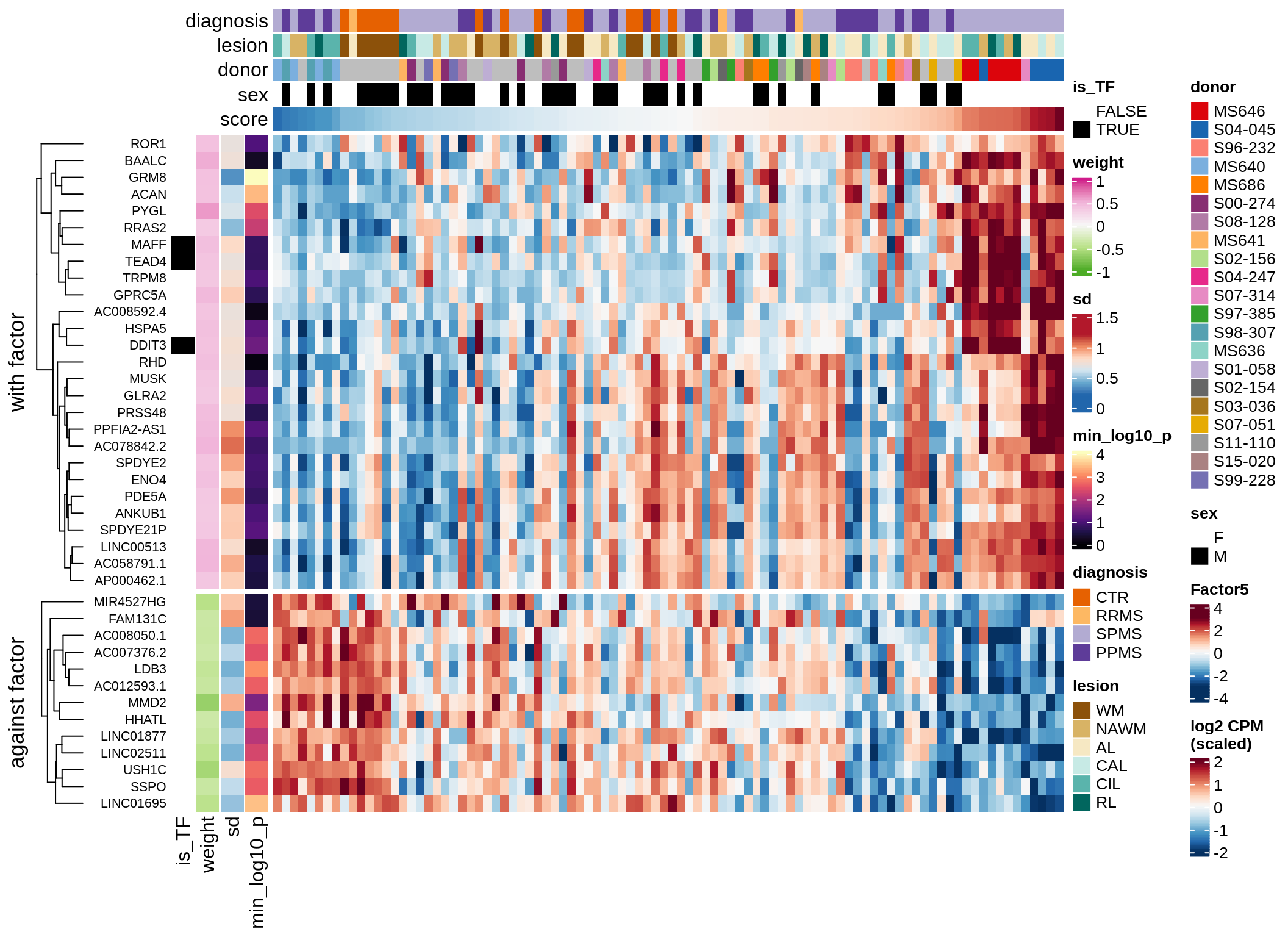
draw(plot_factors_heatmap(model, annots_dt, pb, what = 'few', plot_var = v))
cat('\n\n')
}
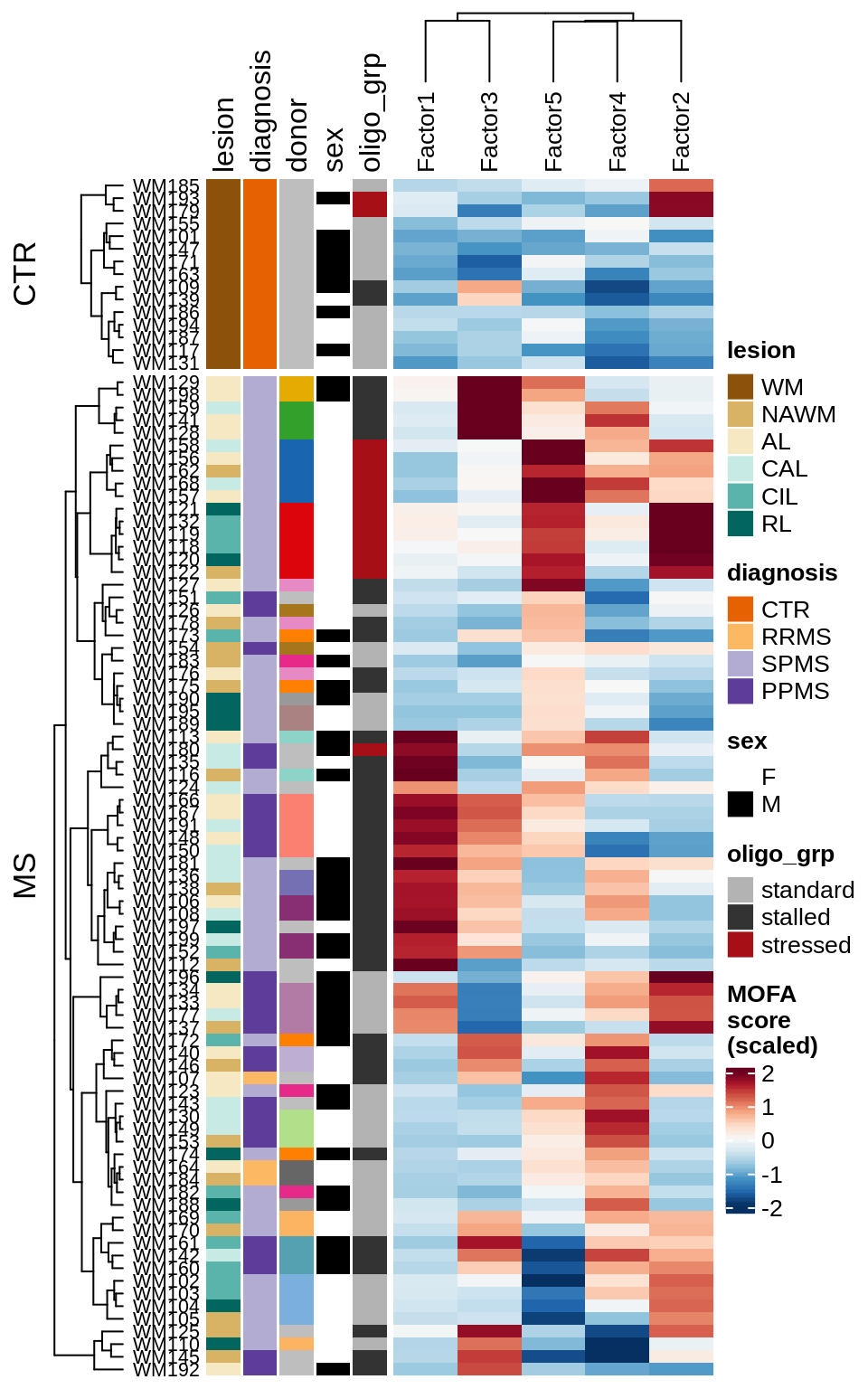
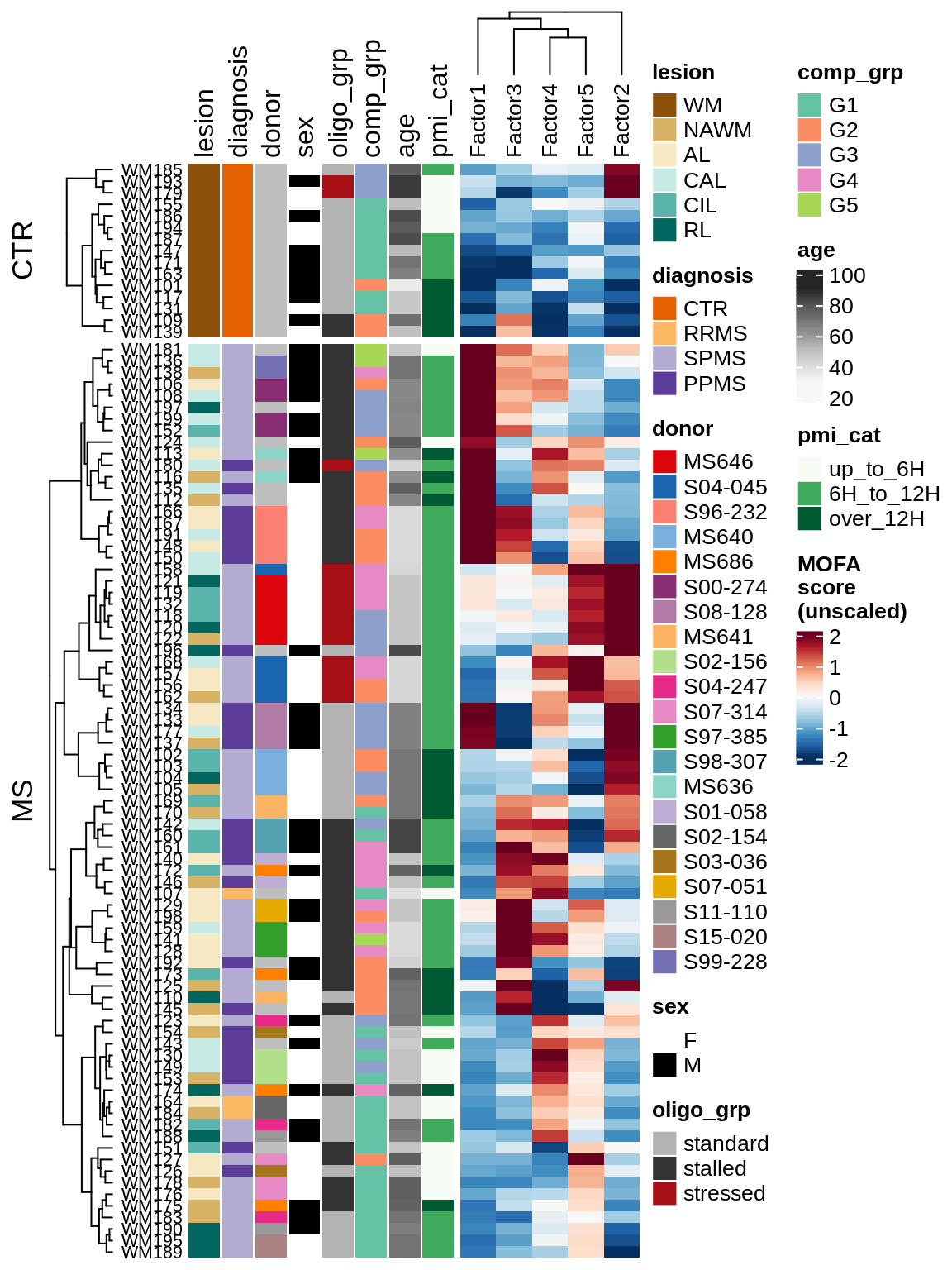
Factor distributions with patient annotations - all
for (v in c('score', 'score_scaled')) {
cat('### ', v, '\n', sep = '')
draw(plot_factors_heatmap(model, annots_dt, pb, what = 'all', plot_var = v))
cat('\n\n')
}
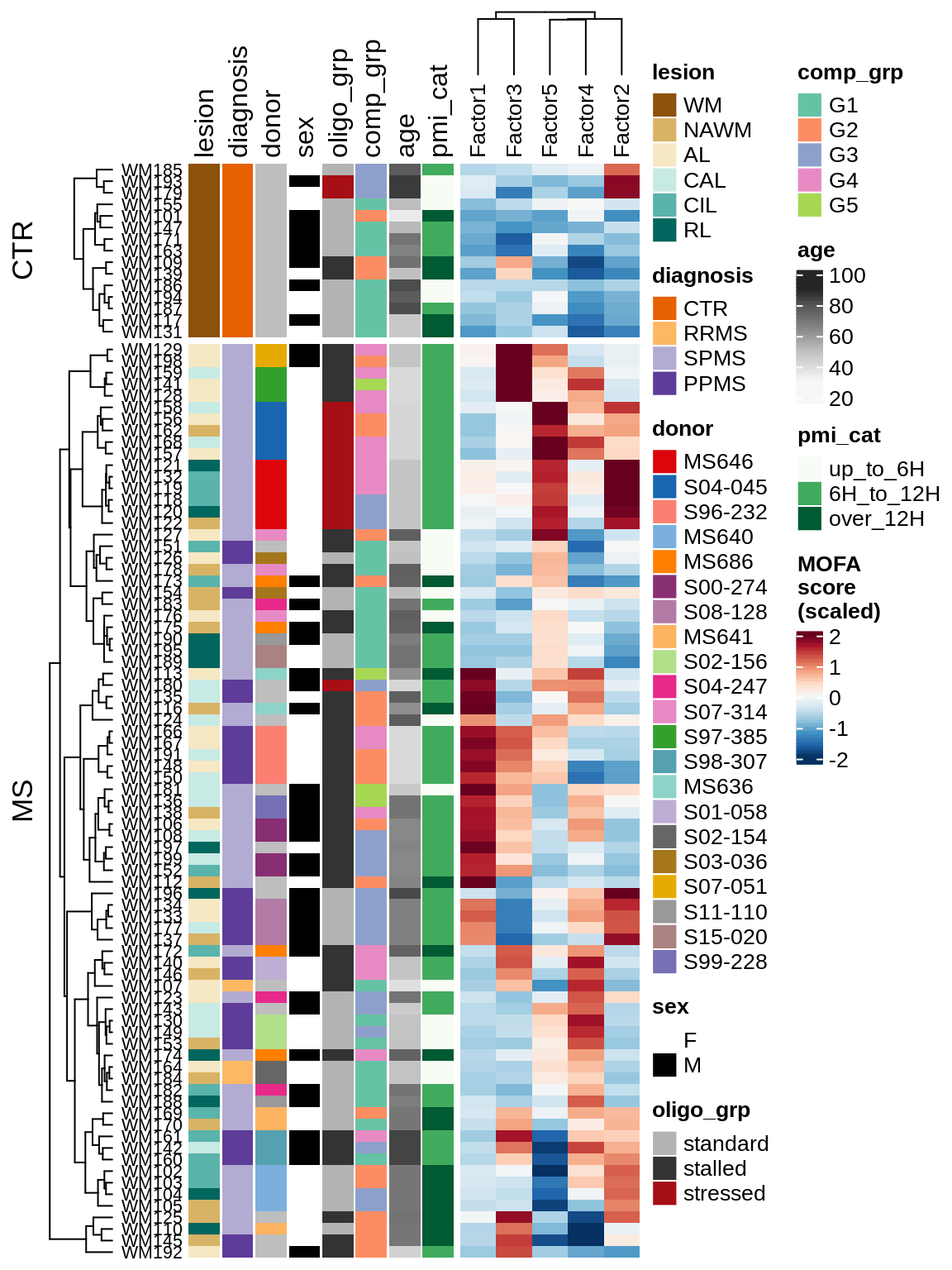
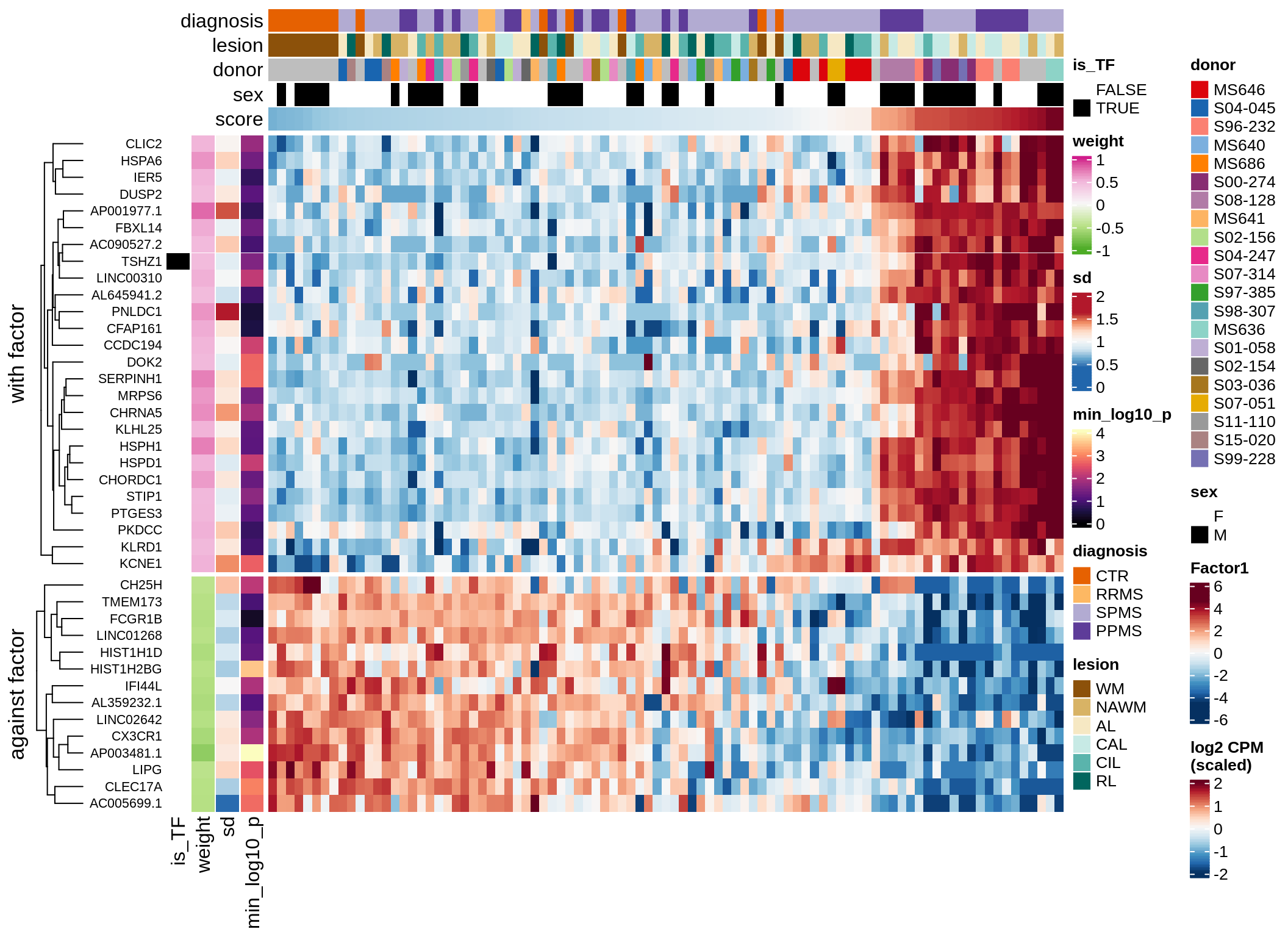
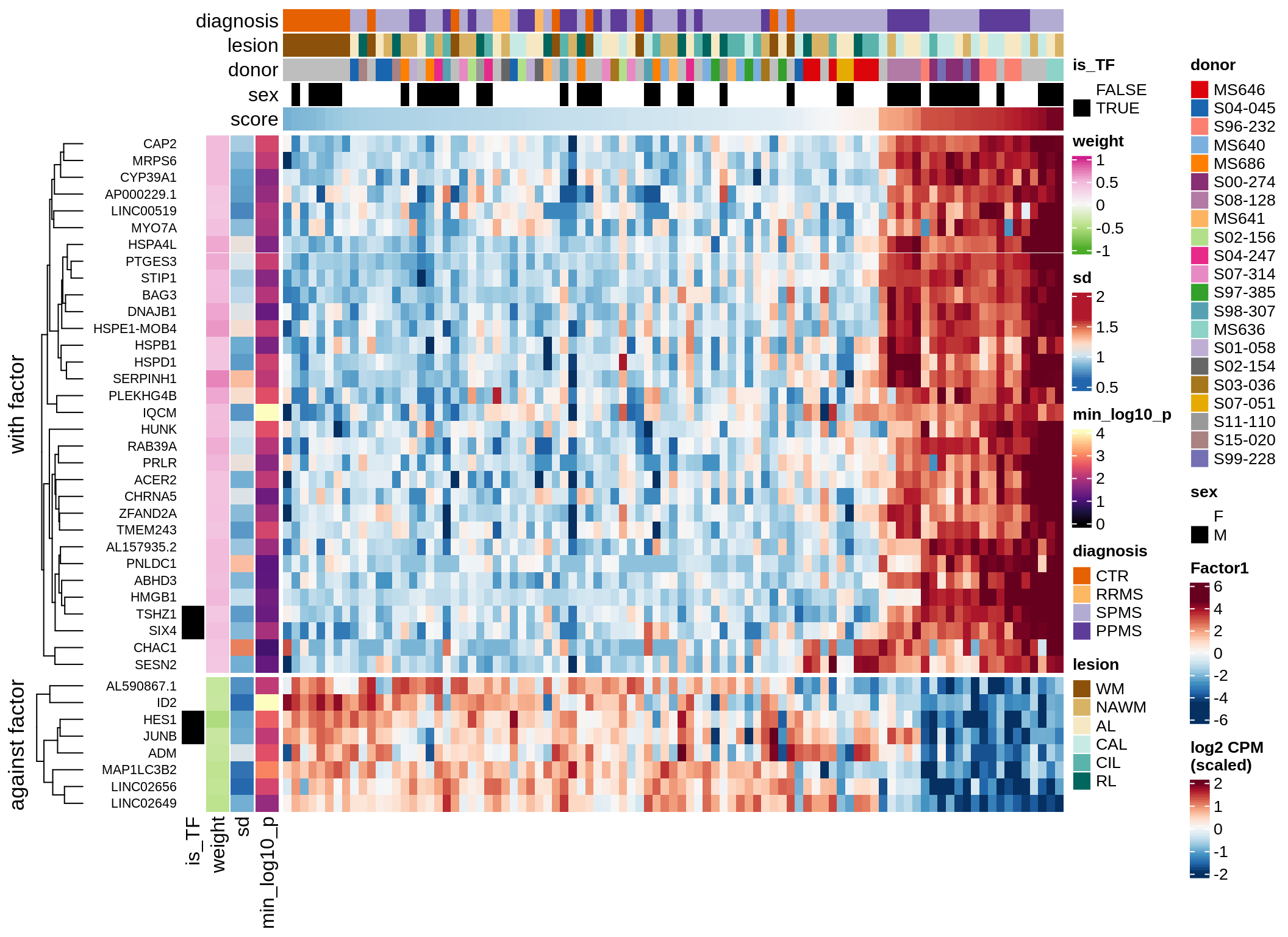
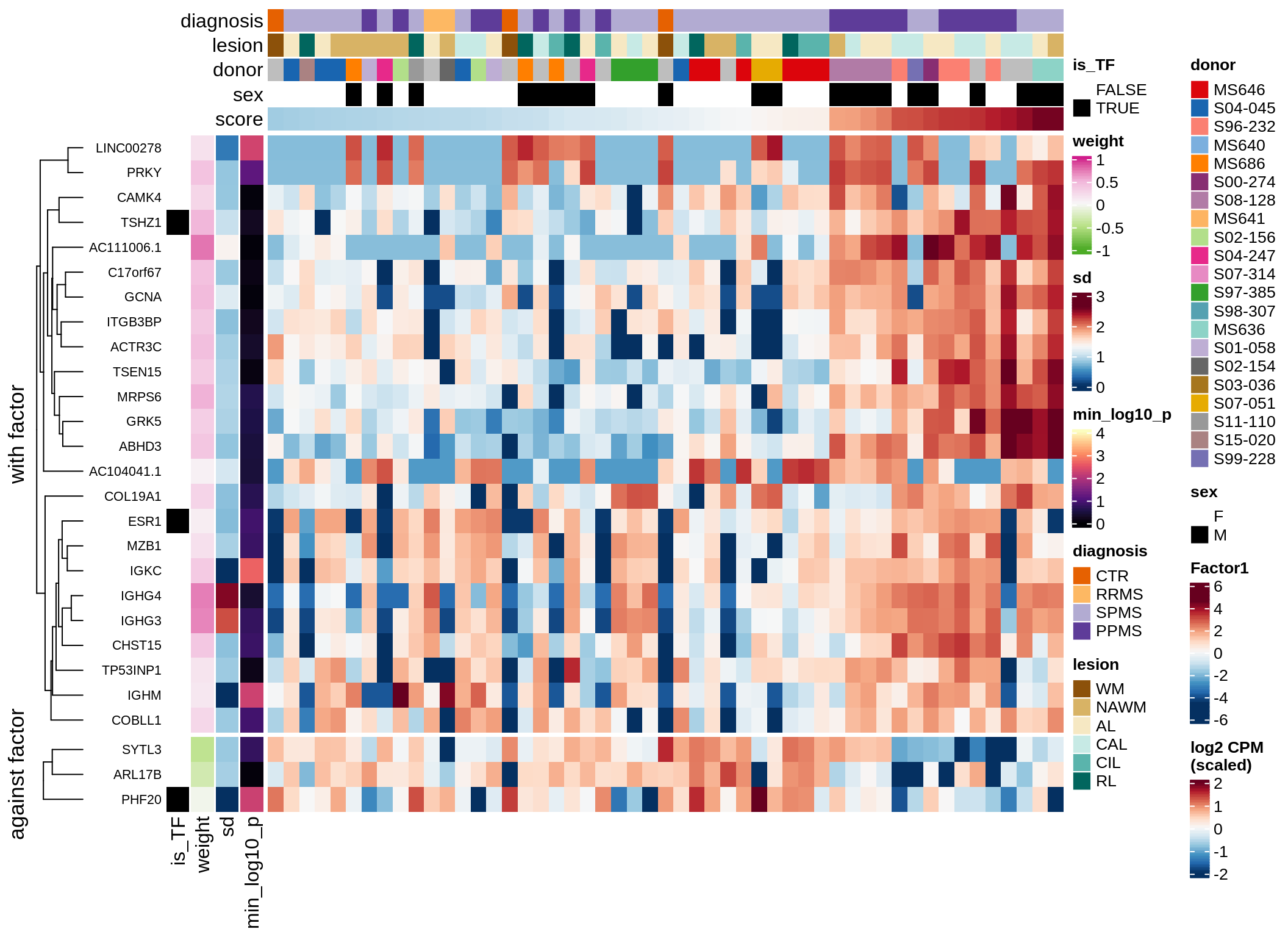
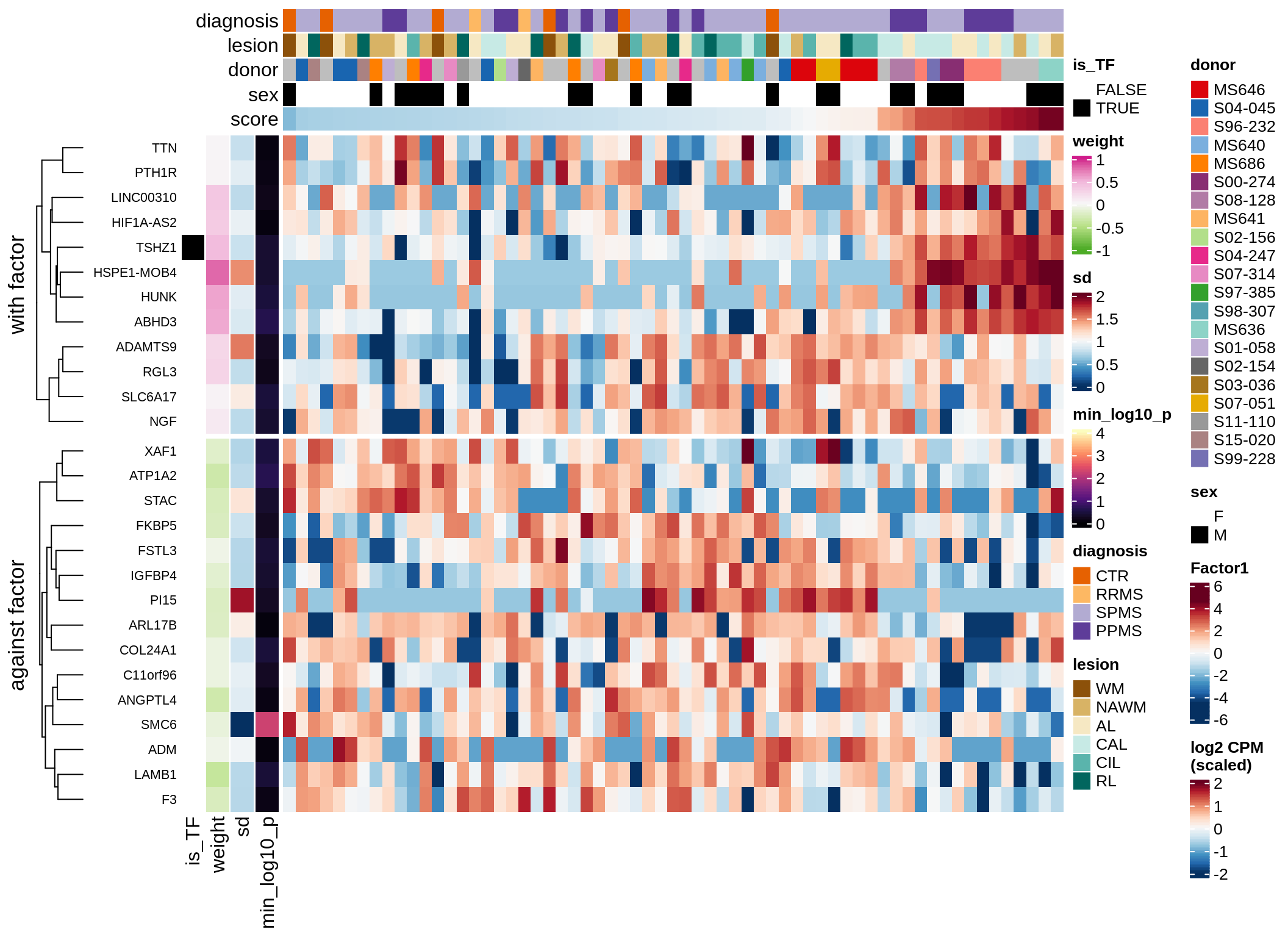
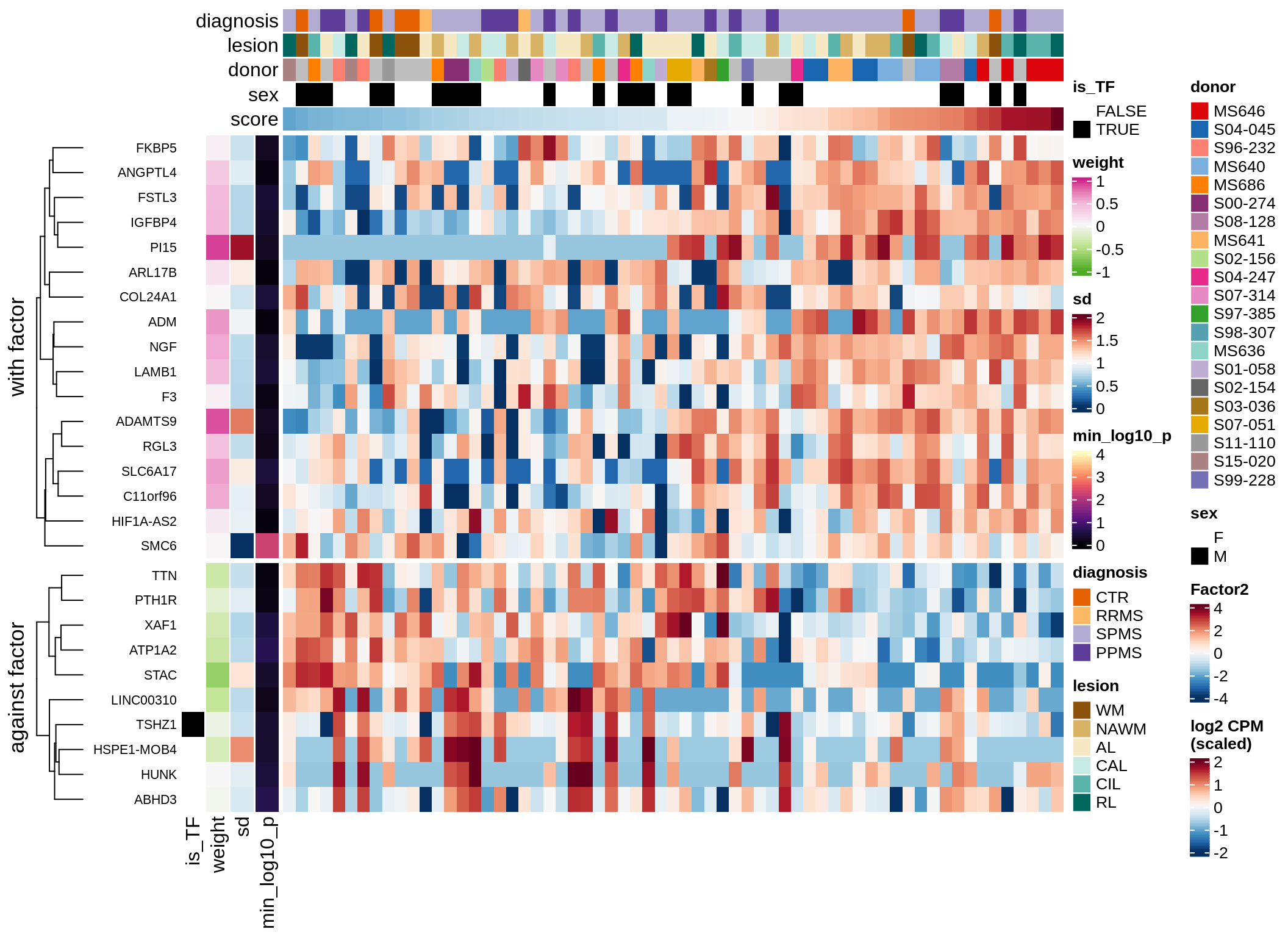
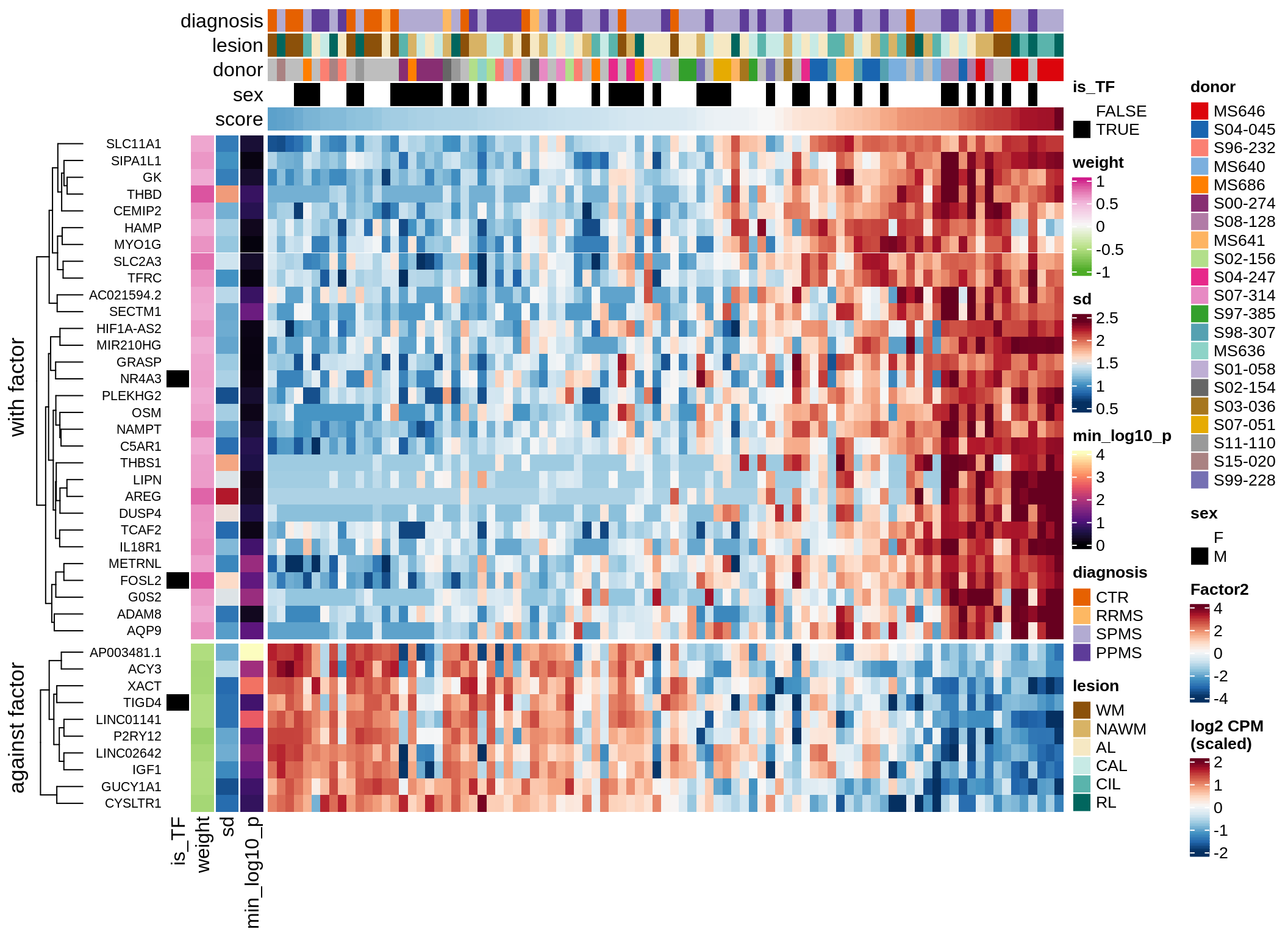
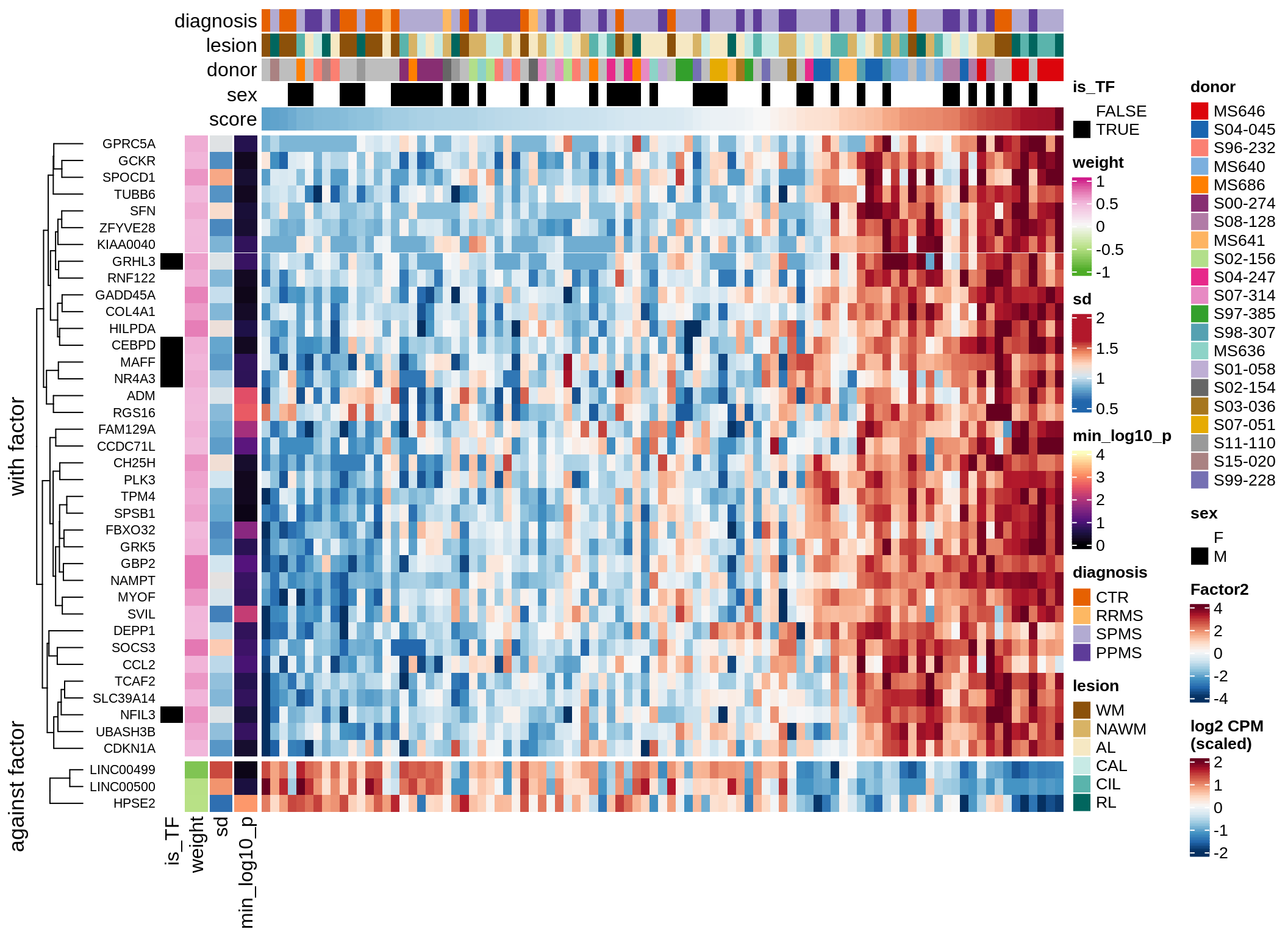
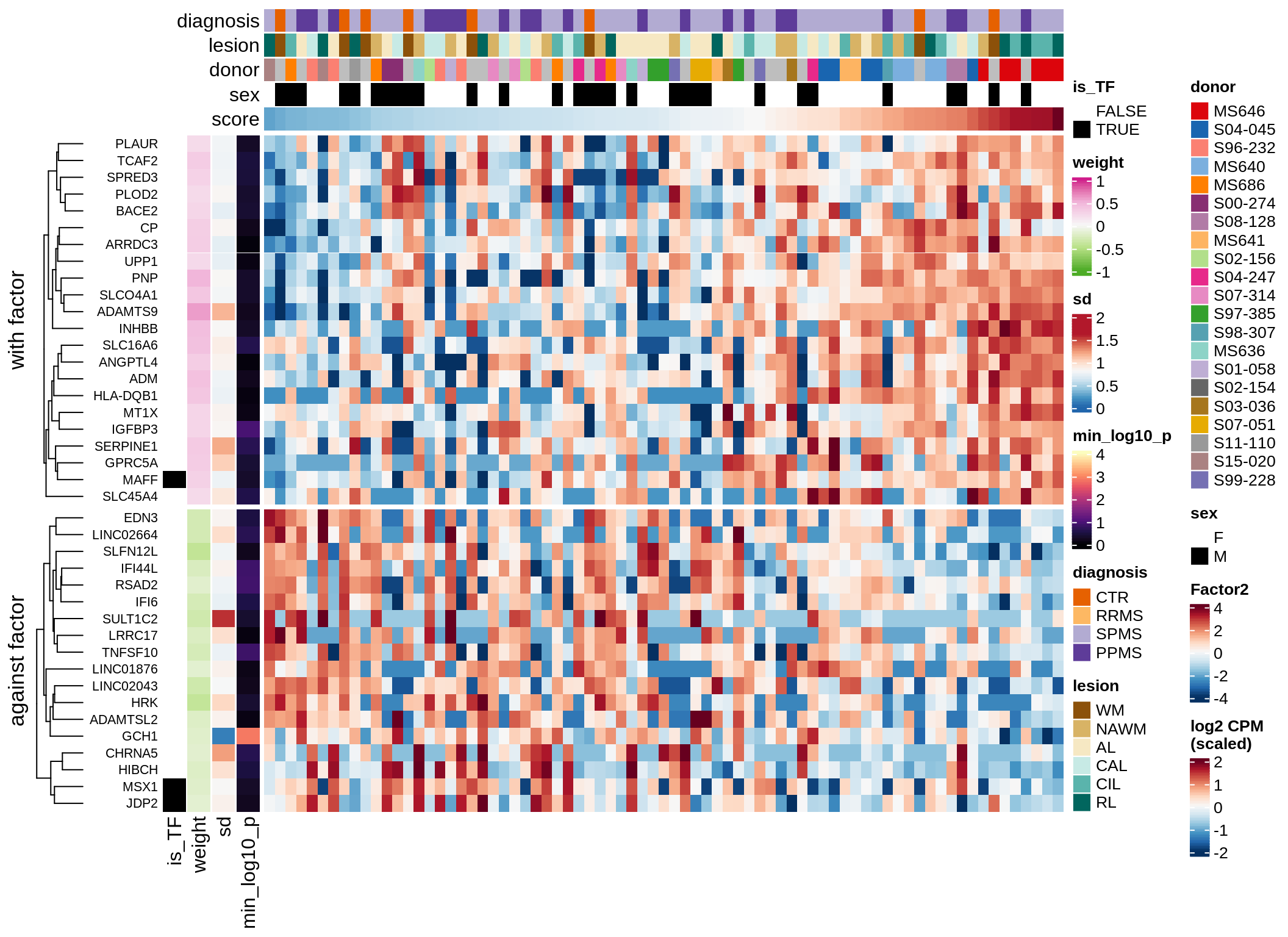
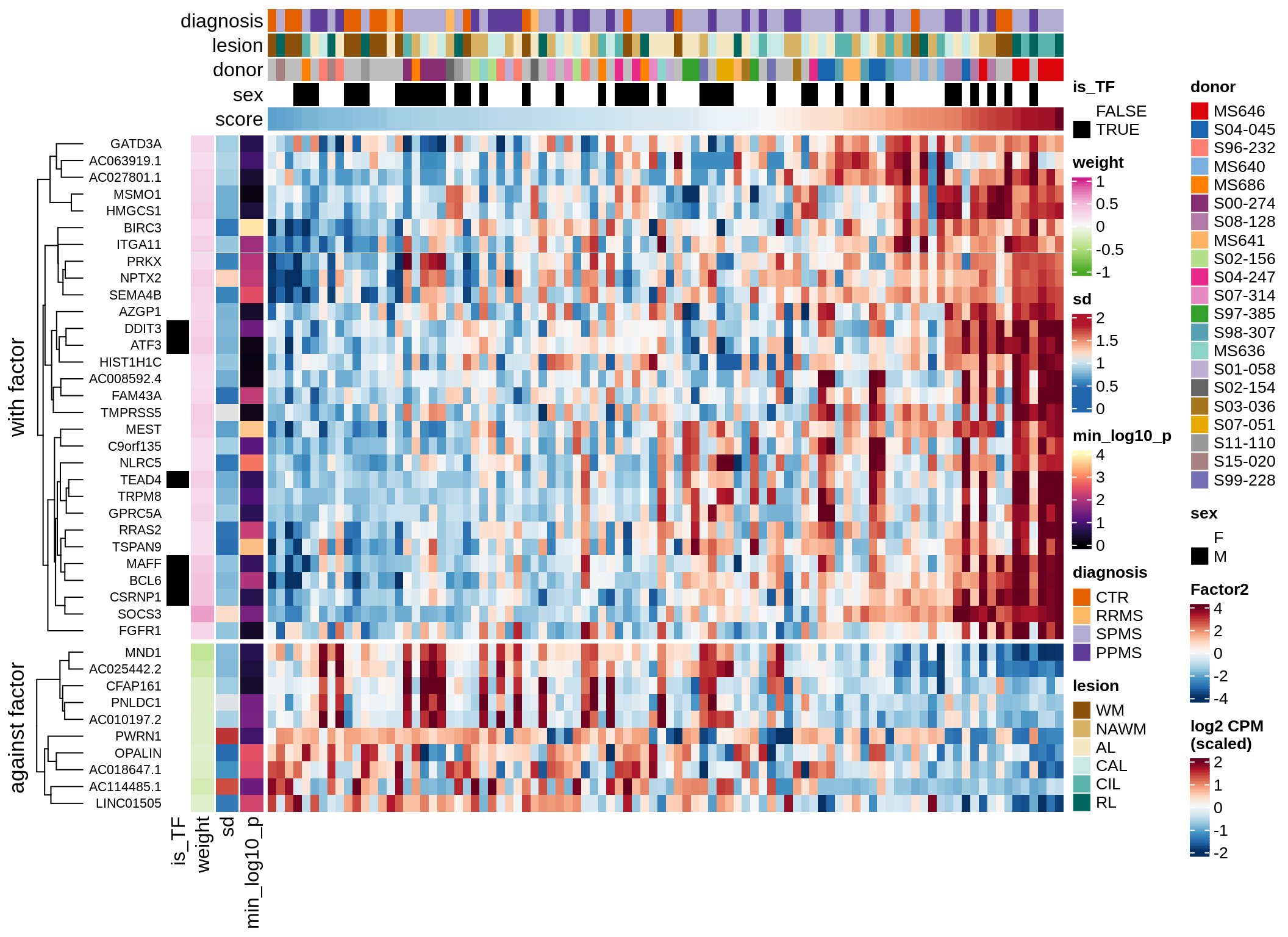
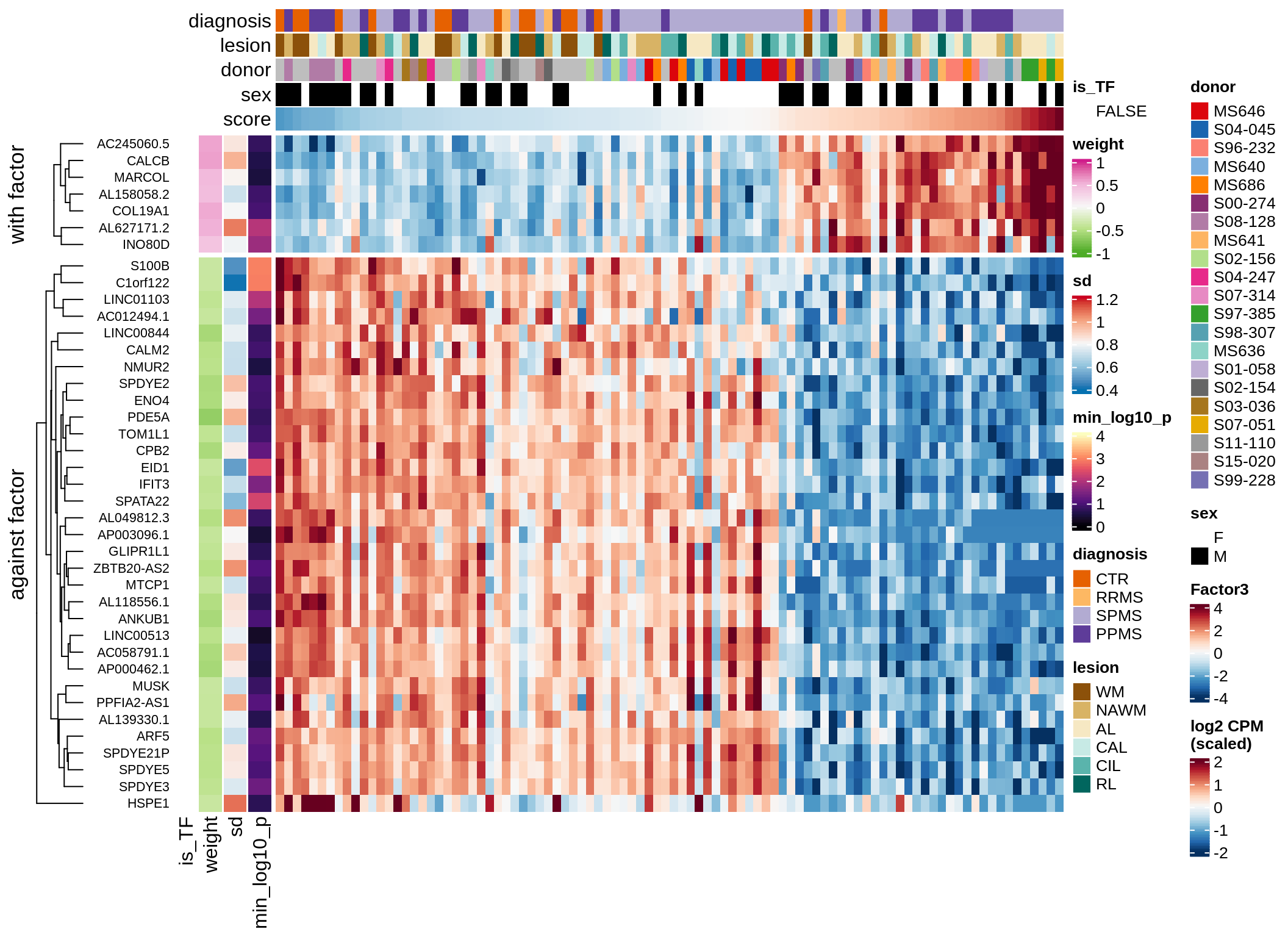
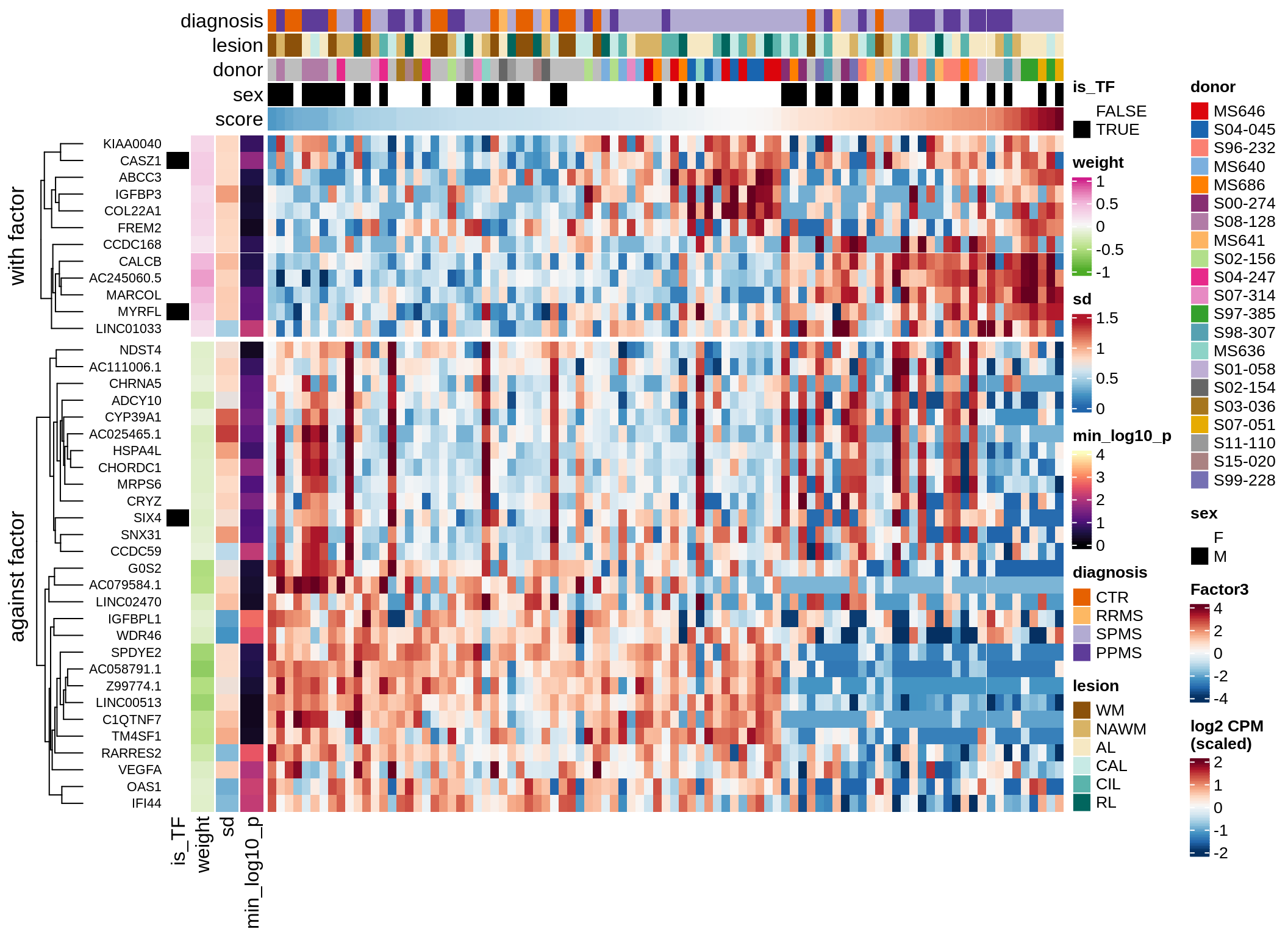
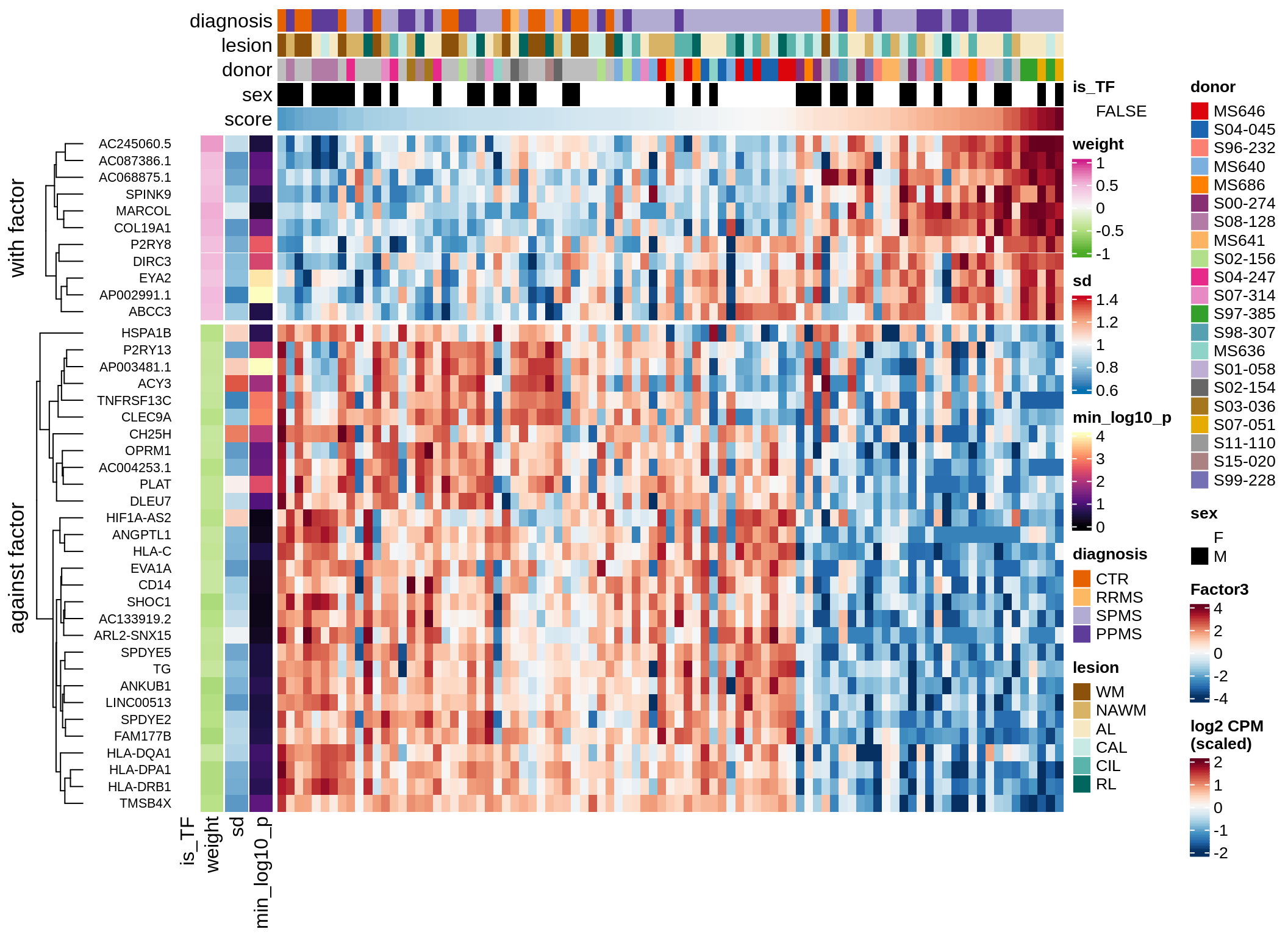
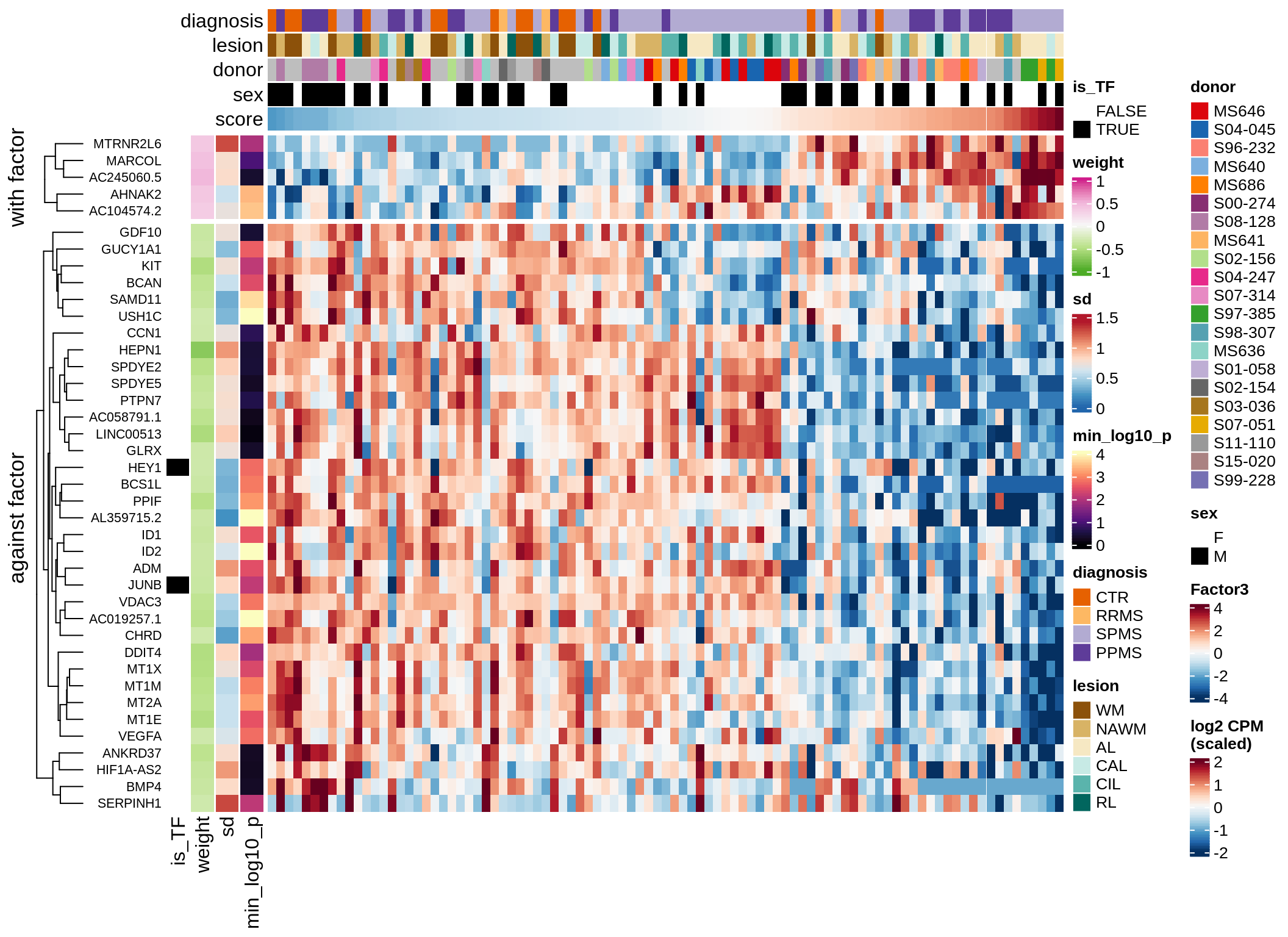
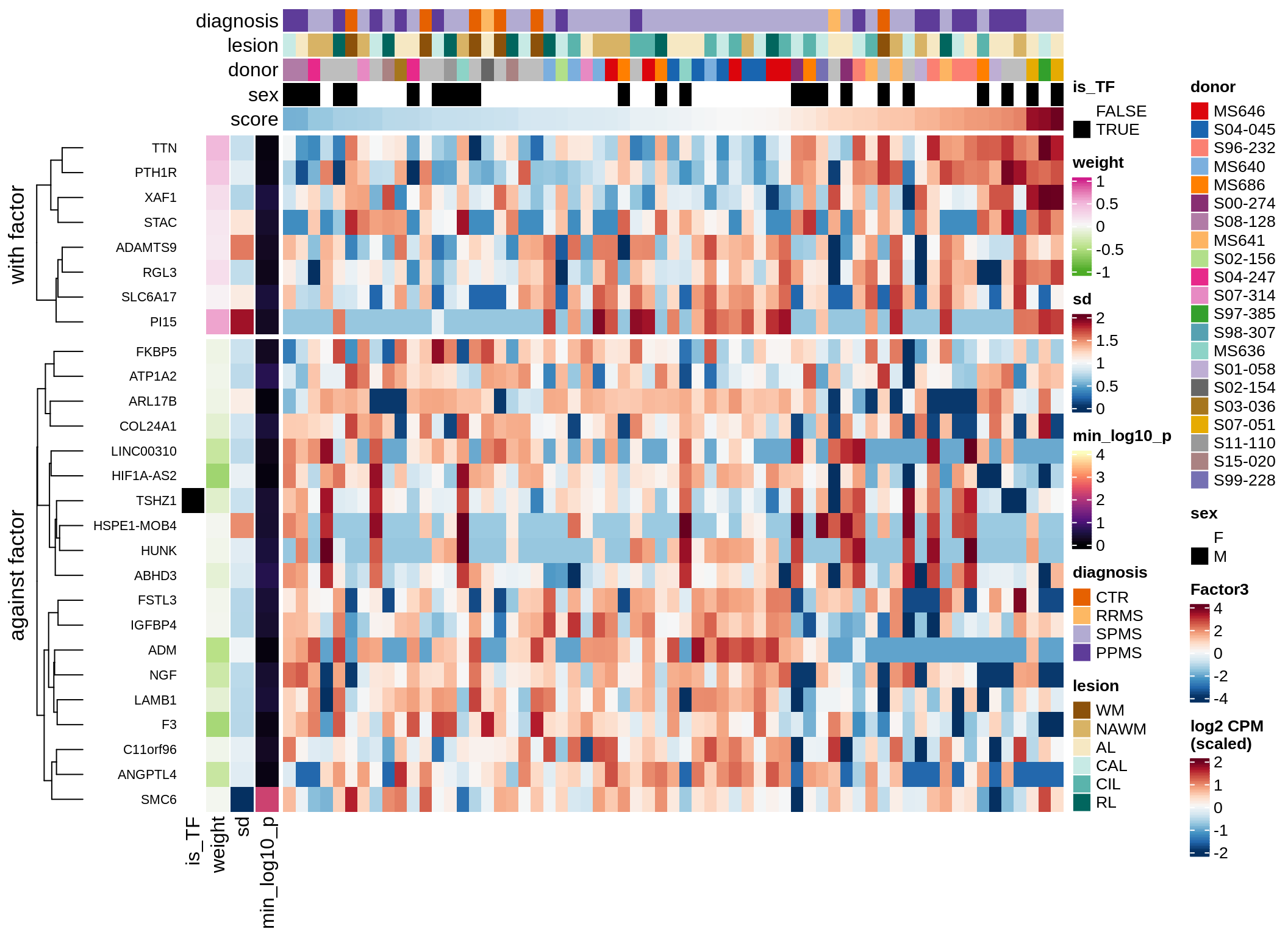
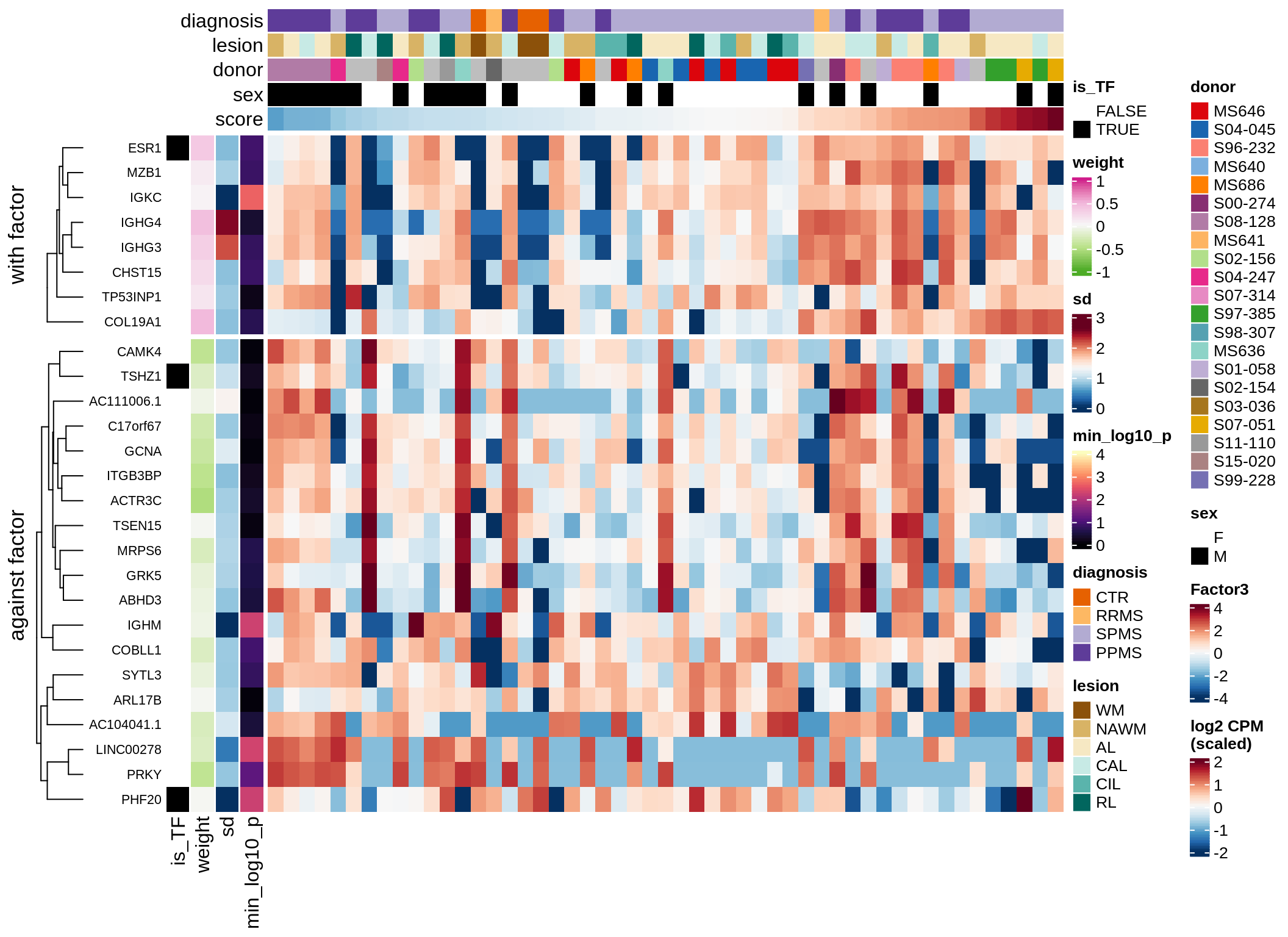
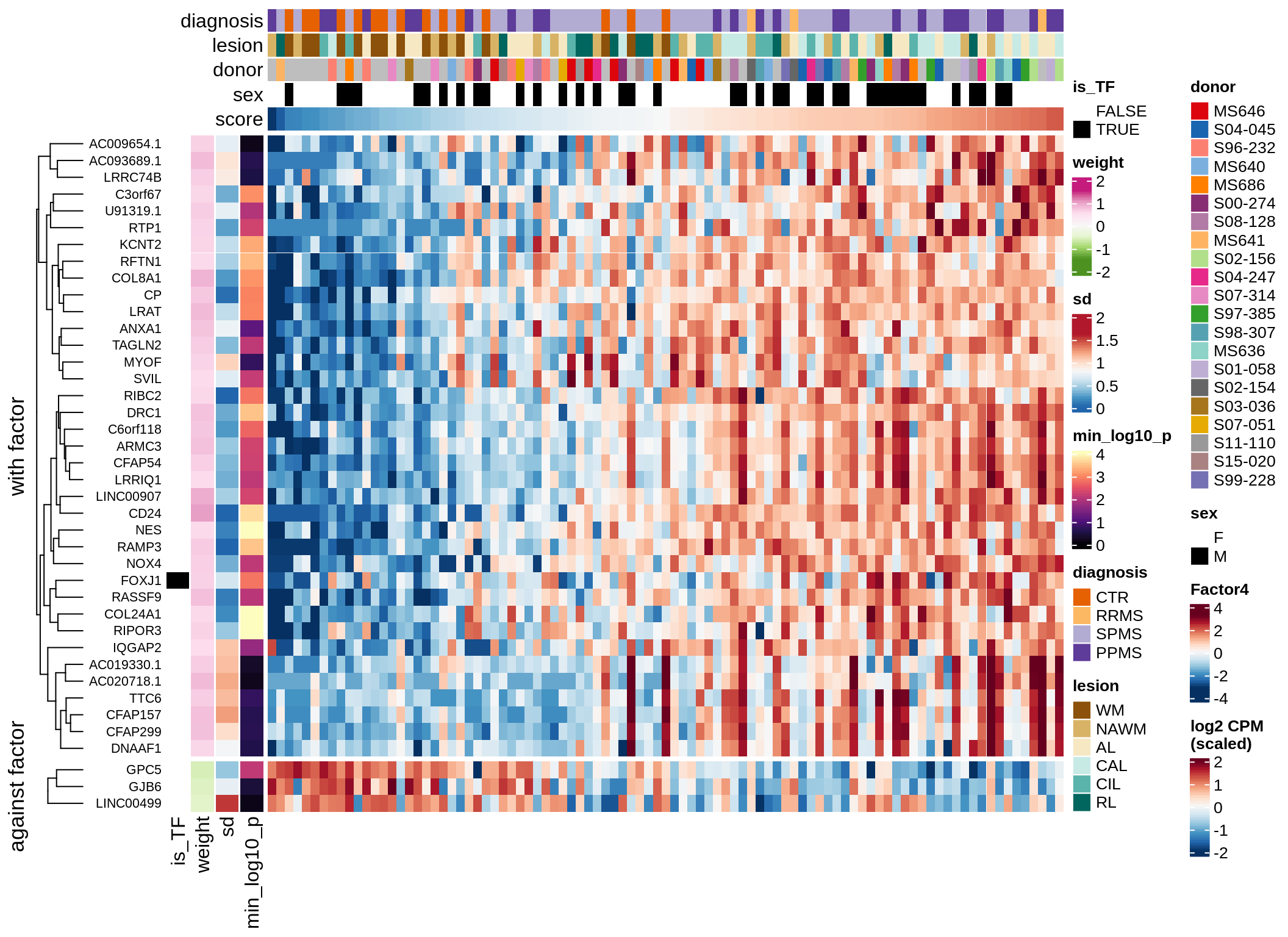
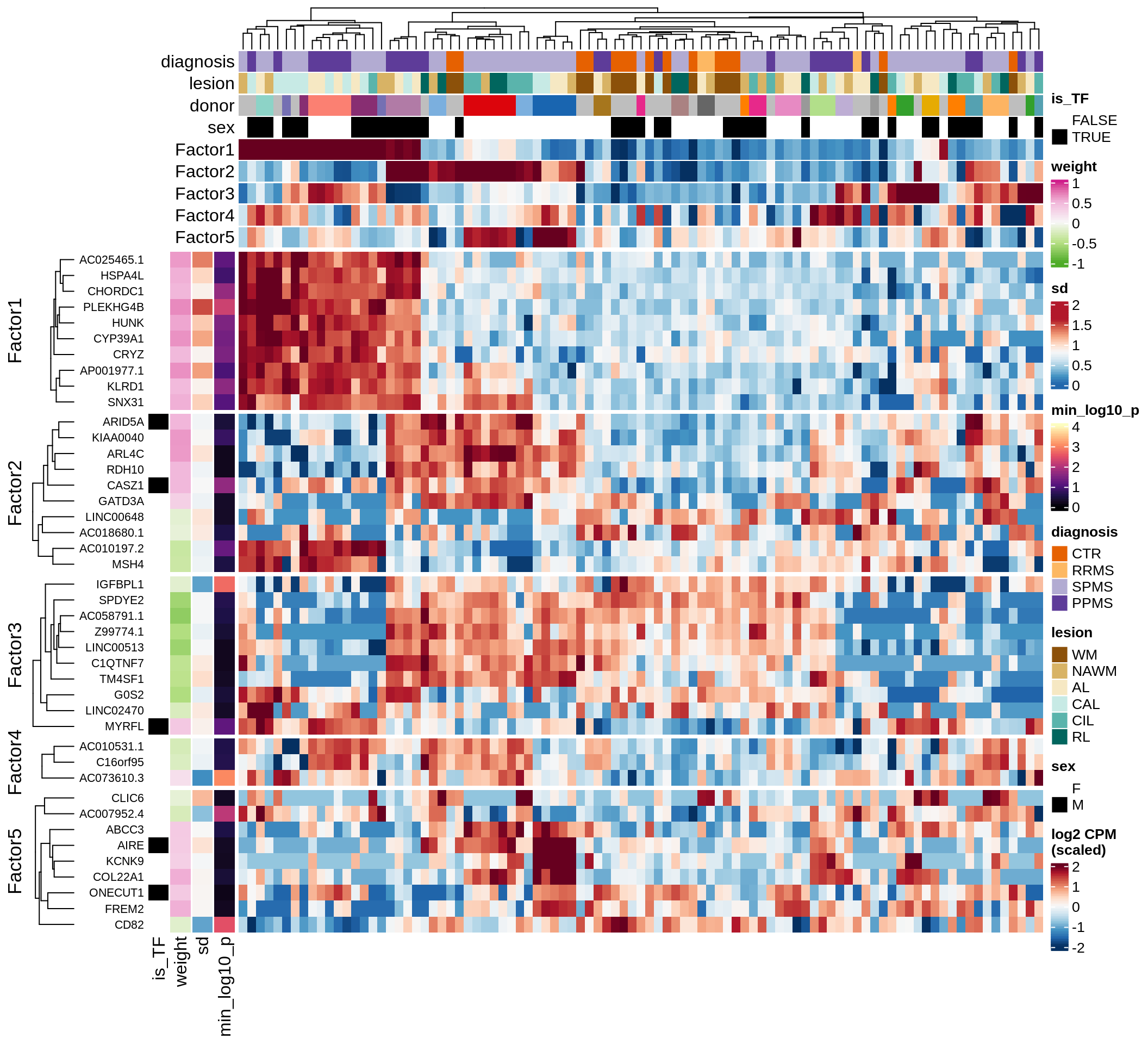
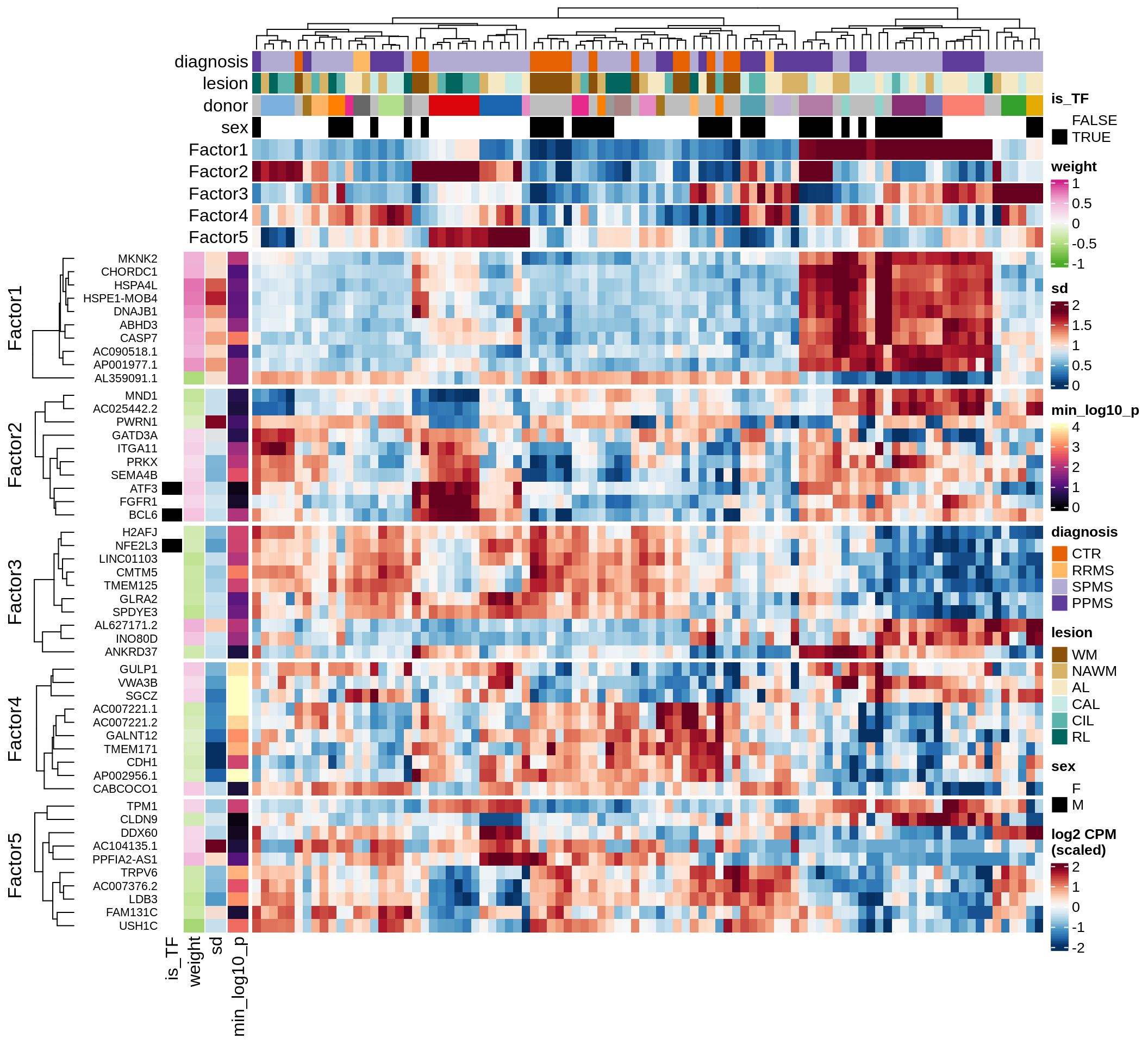
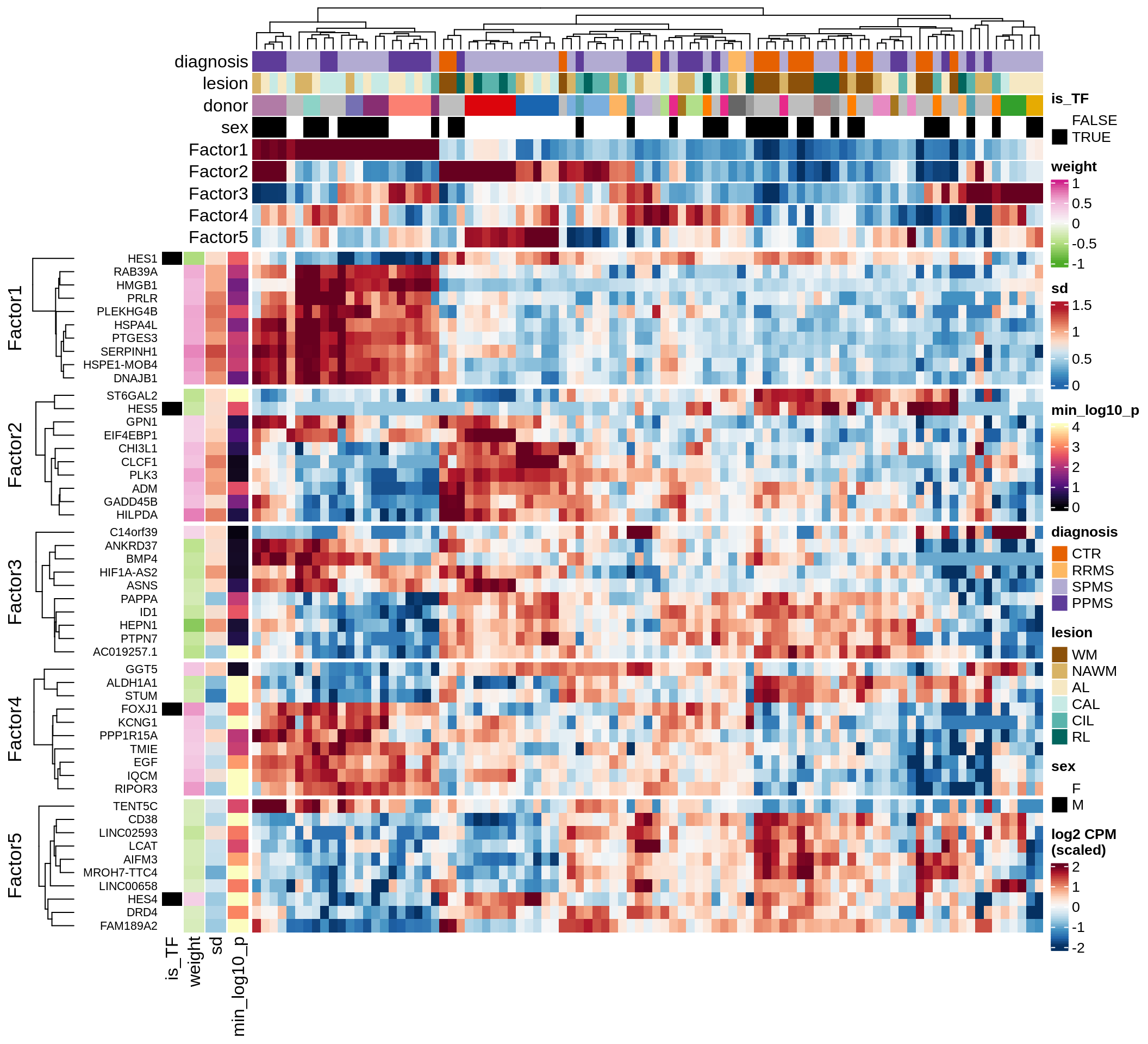
Coefficients of top genes (by factor)
for (f in factors_names(model)) {
cat('### ', f, '\n', sep = '')
draw(plot_top_weights_heatmap_by_factor(model, var_exp_dt, sel_f = f))
cat('\n\n')
}Factor1
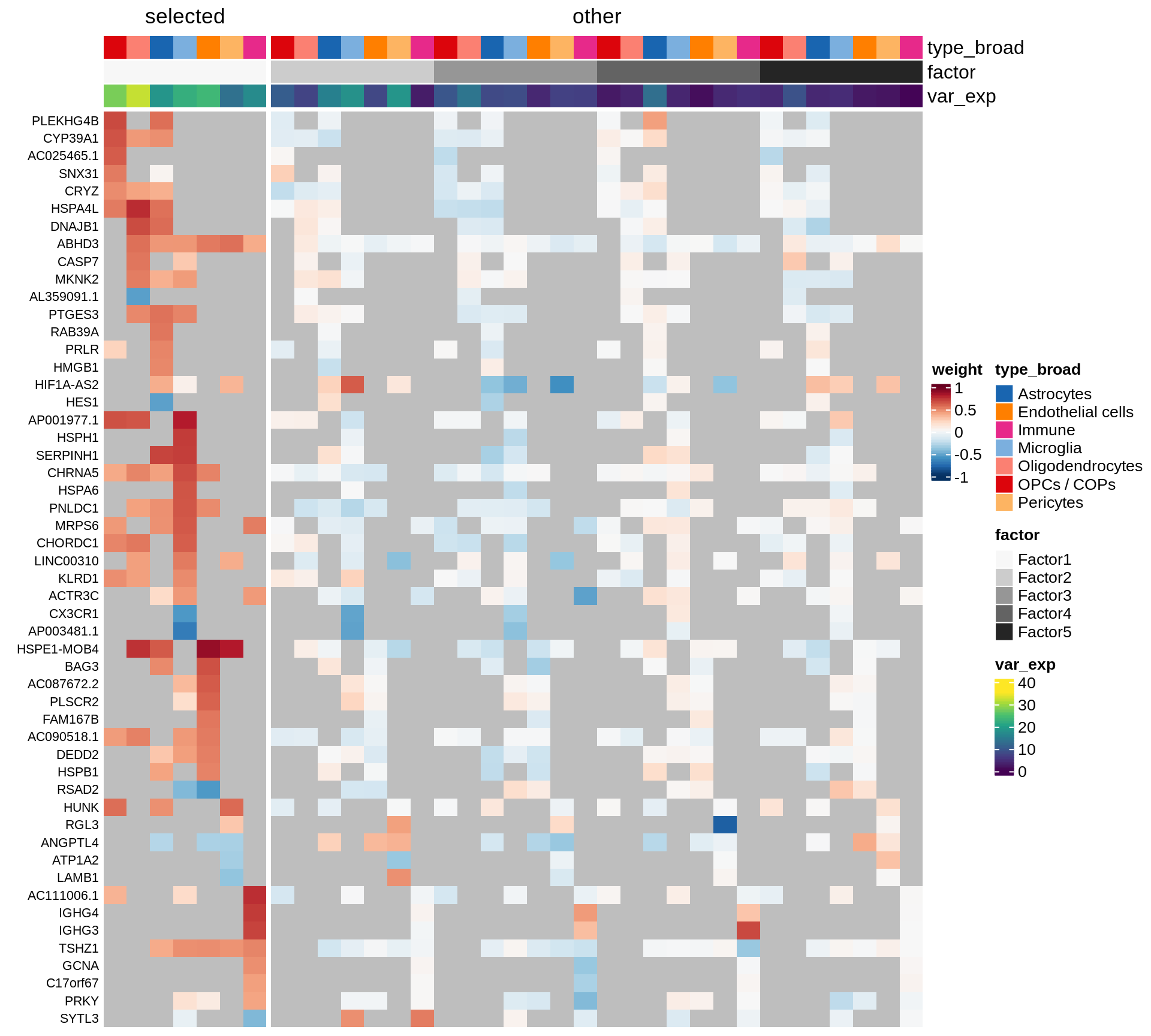
Factor2
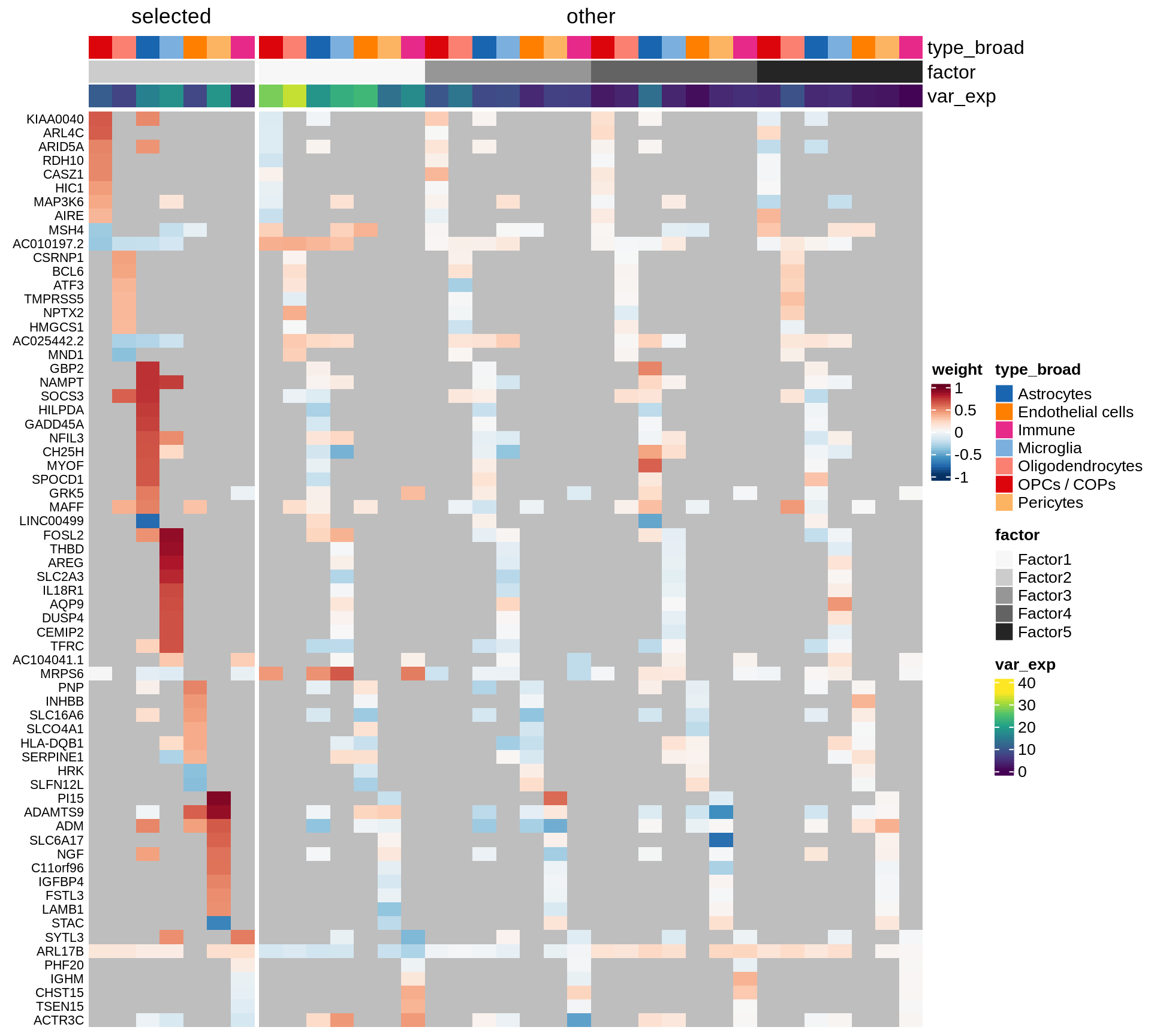
Factor3
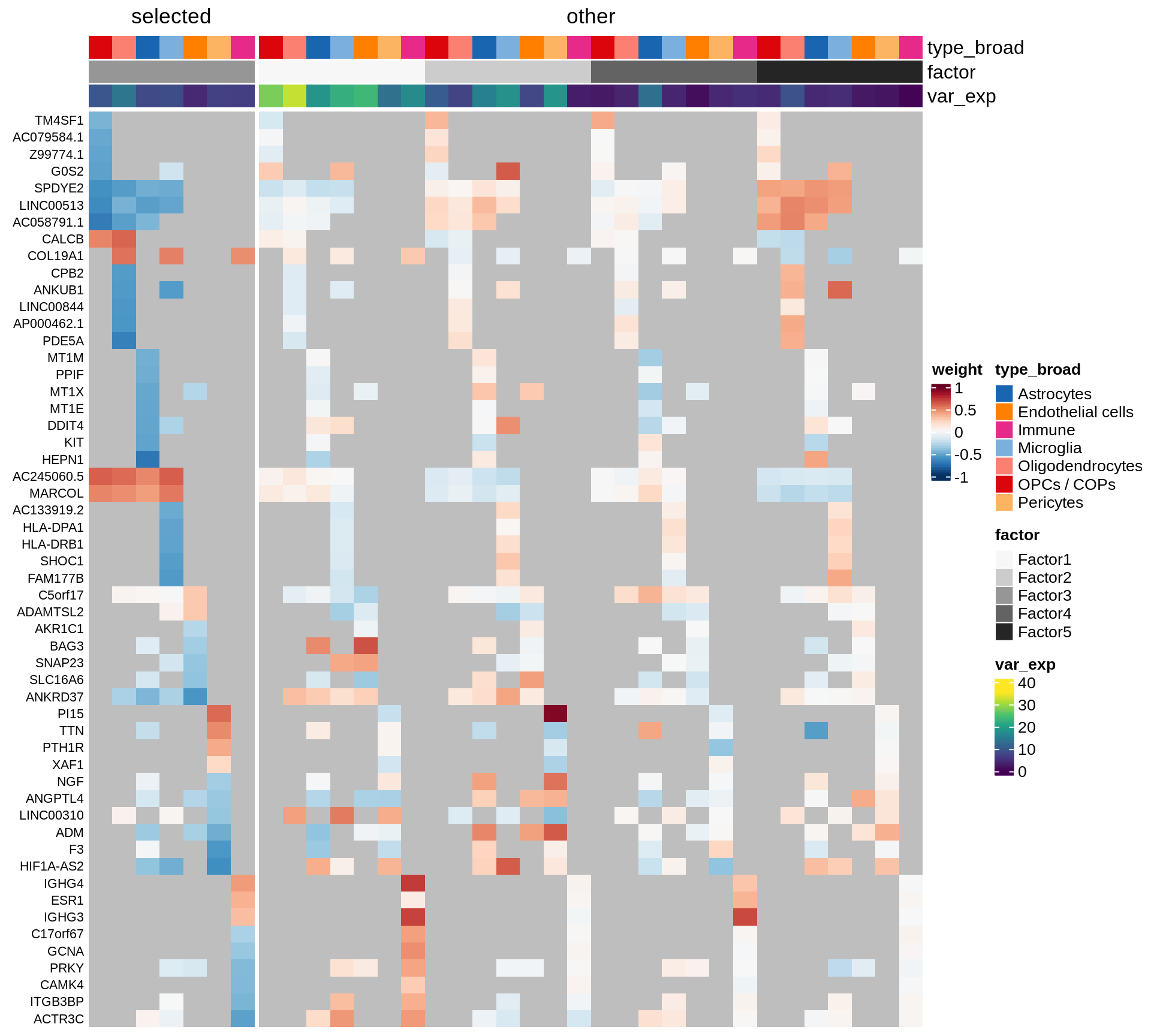
Factor4
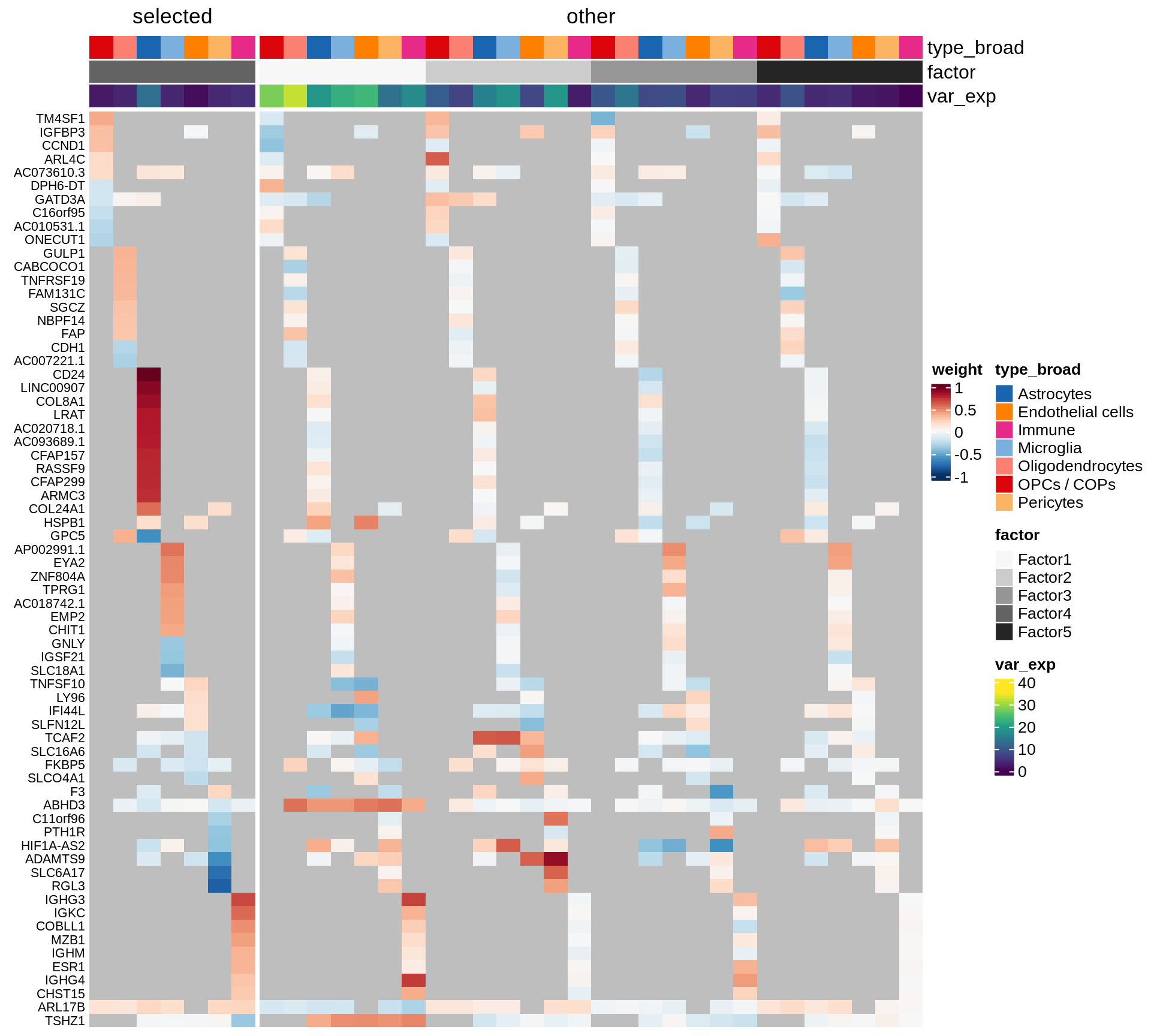
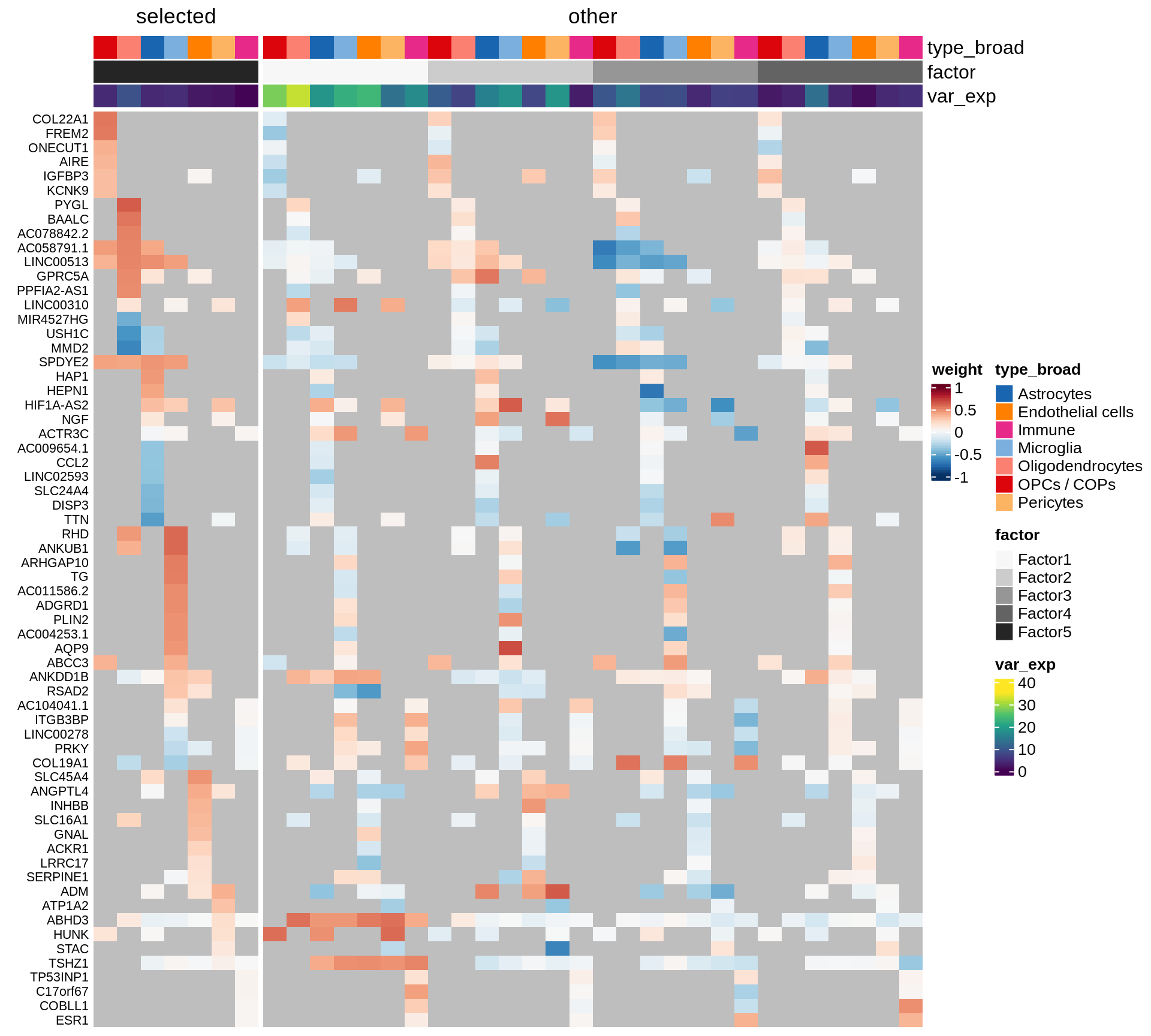
Expression of top genes per celltype
# iterate plots
for (i in seq.int(nrow(to_plot_dt))) {
sel_v = as.character(to_plot_dt[i]$view)
sel_f = to_plot_dt[i]$factor
this_r2 = to_plot_dt[i]$var_exp
cat('### ', sel_v, '-F', as.integer(sel_f),
' (', round(this_r2, 0), '%)', '\n', sep = '')
draw(plot_top_weights_expression_sample(model, pb, annots_dt, filter_dt,
tfs_dt, sel_v = sel_v, sel_f = sel_f, n_top = 40), merge_legend = TRUE )
cat('\n\n')
}

astro-F4 (13%)
Expression of top genes across all factors per celltype
# iterate plots
for (sel_v in broad_short[sel_cl]) {
cat('### ', sel_v, '\n', sep = '')
draw(plot_top_genes_expression_all_factors(model, pb, annots_dt, filter_dt,
tfs_dt, var_exp_dt, sel_v = sel_v, n_top = 10, min_var), merge_legend = TRUE )
cat('\n\n')
}
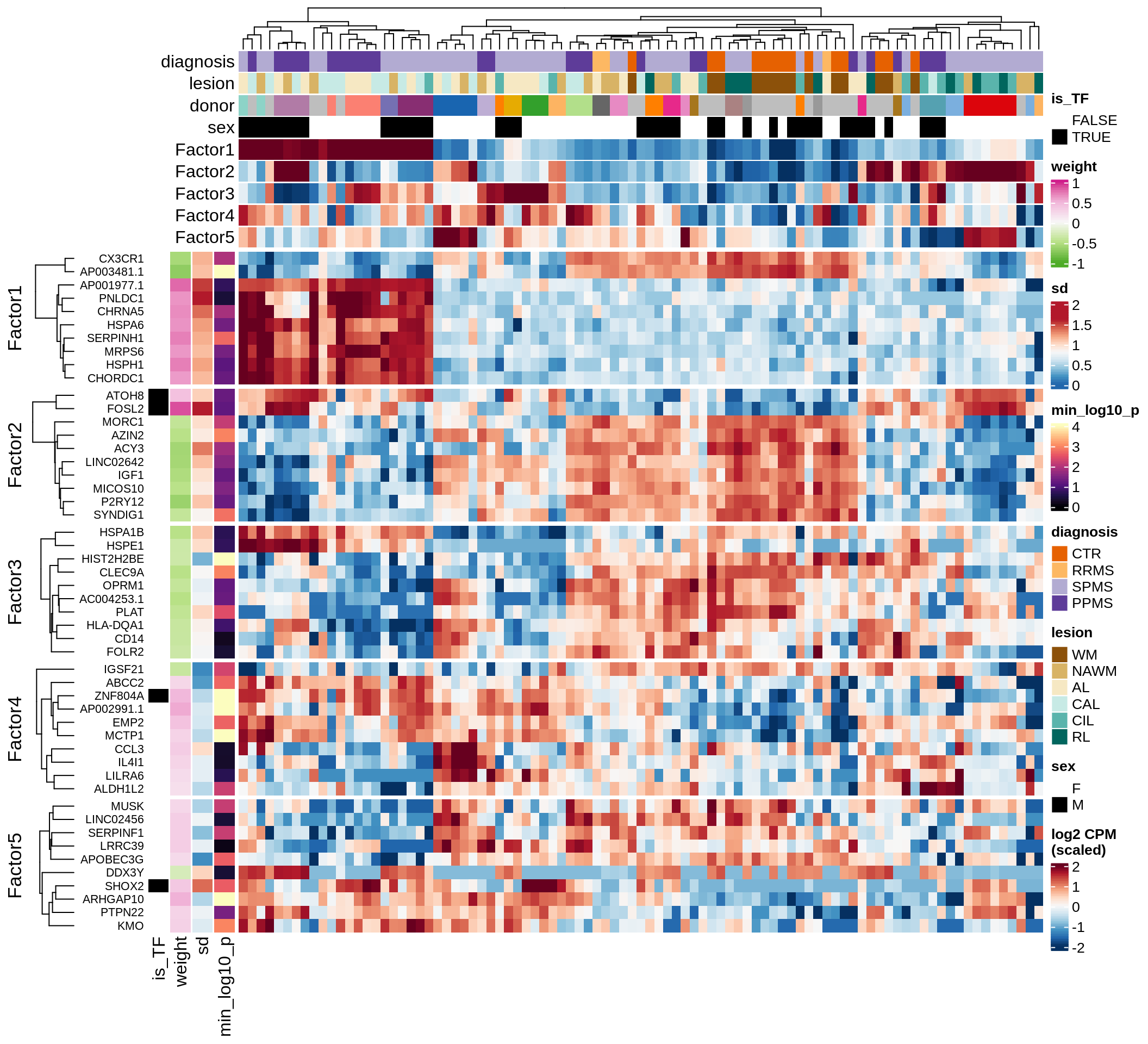
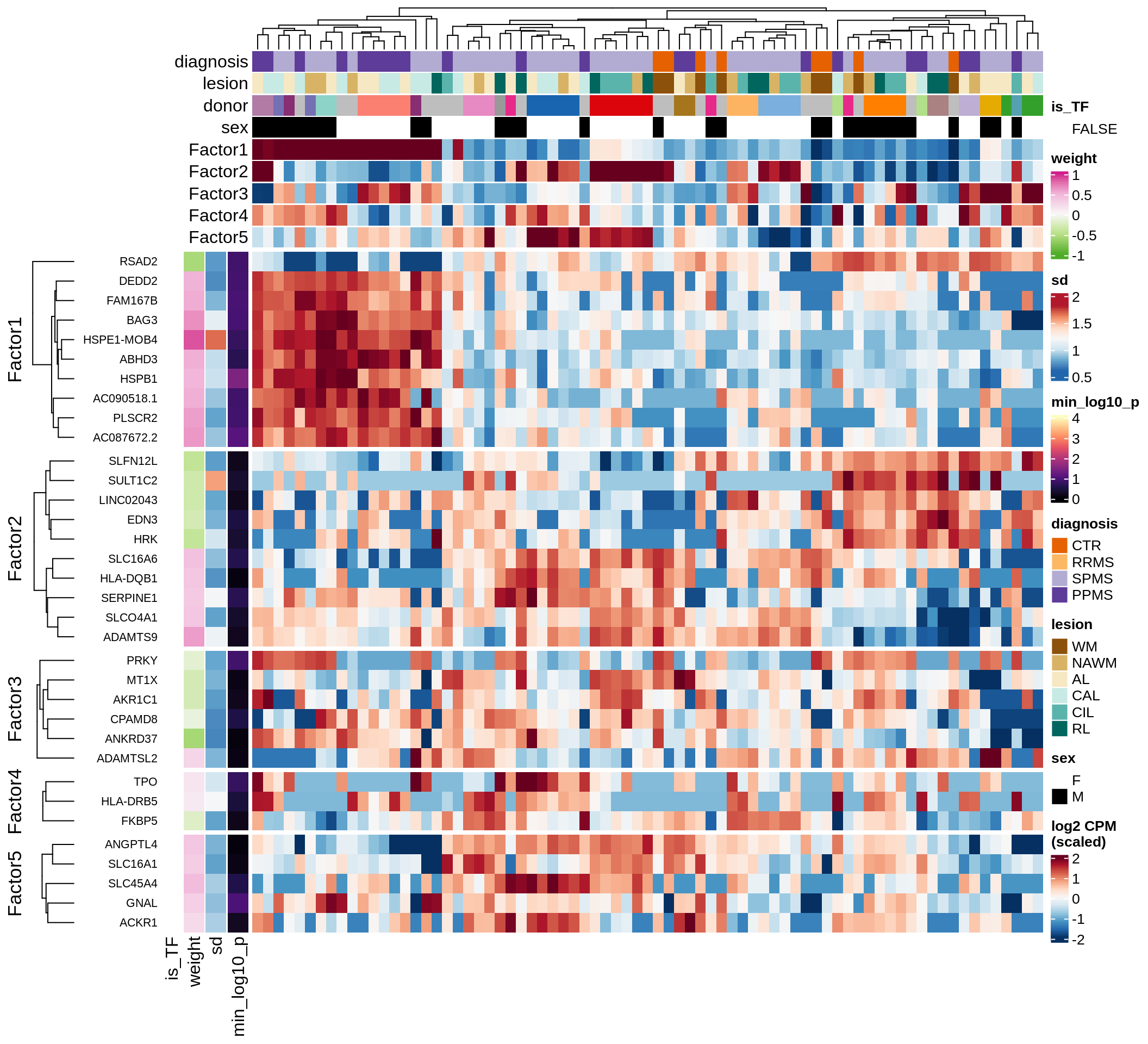
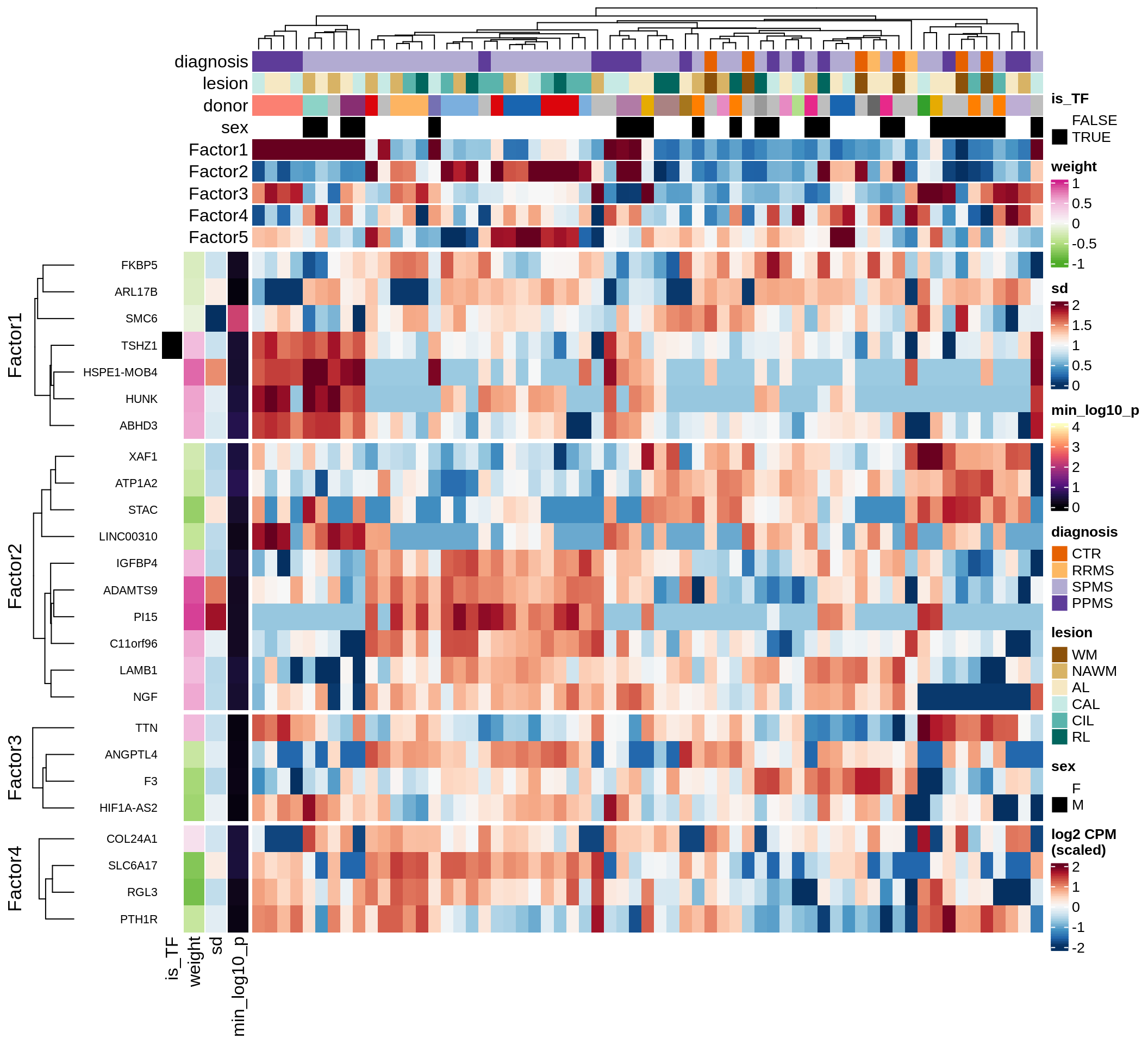
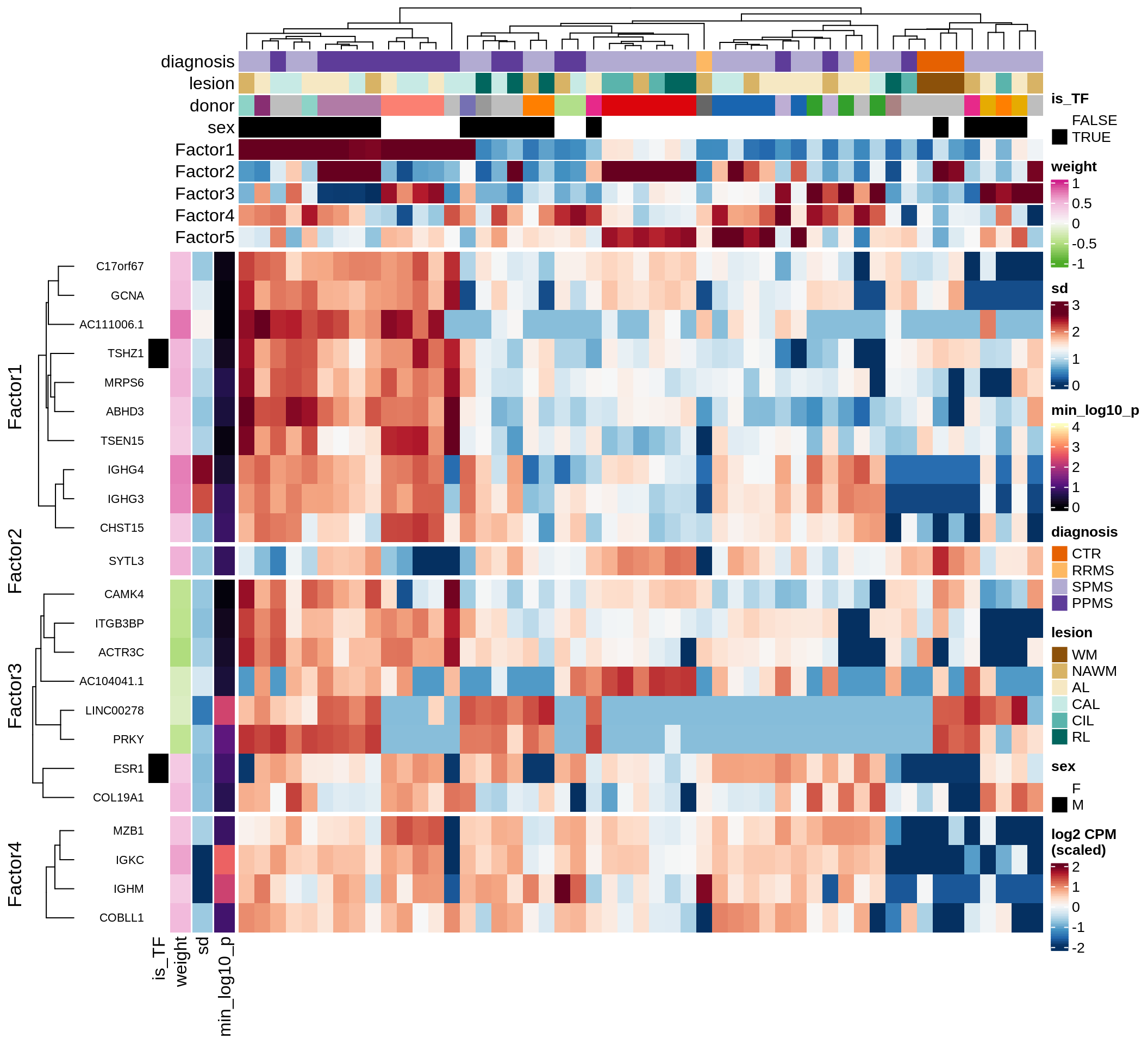
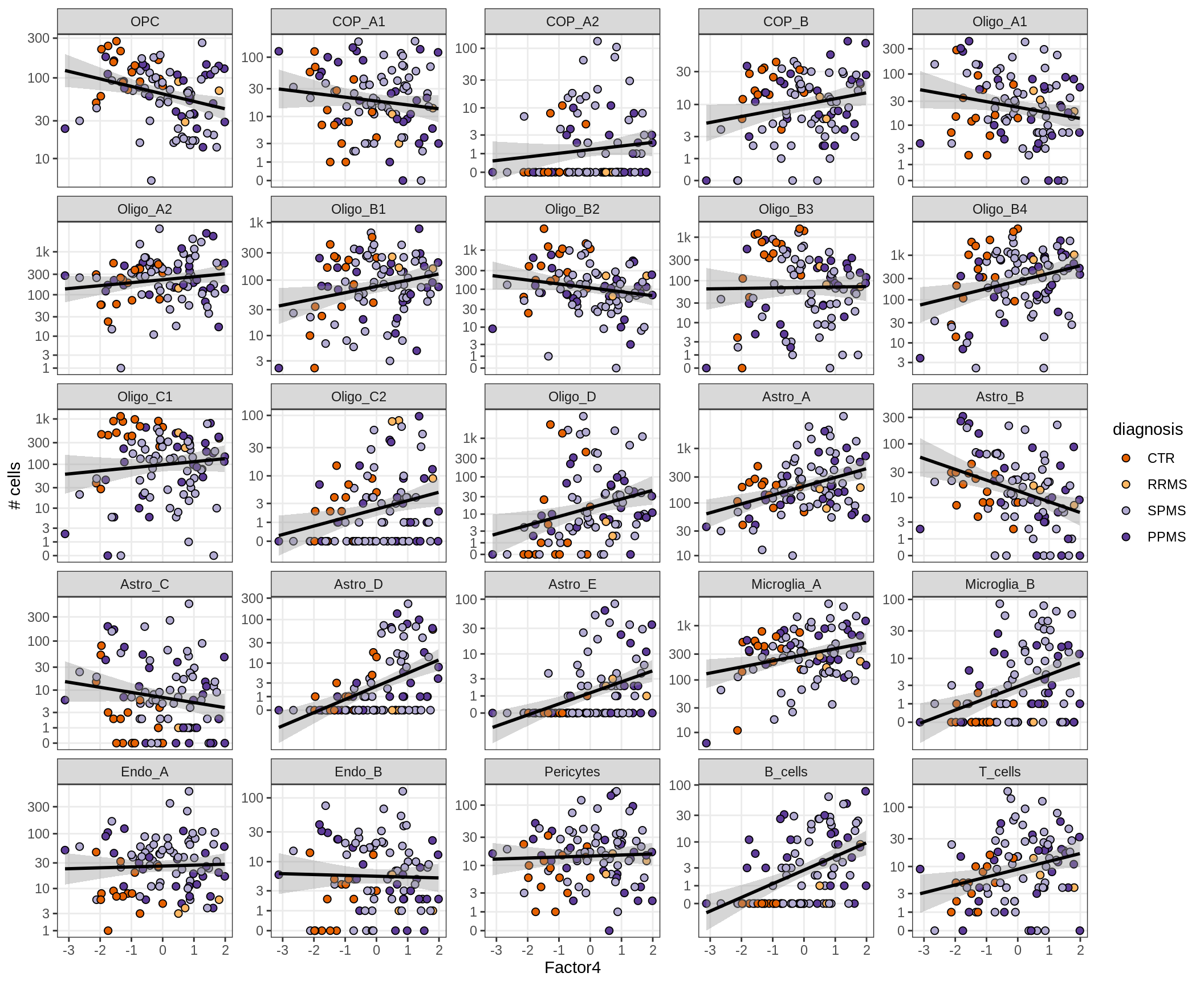
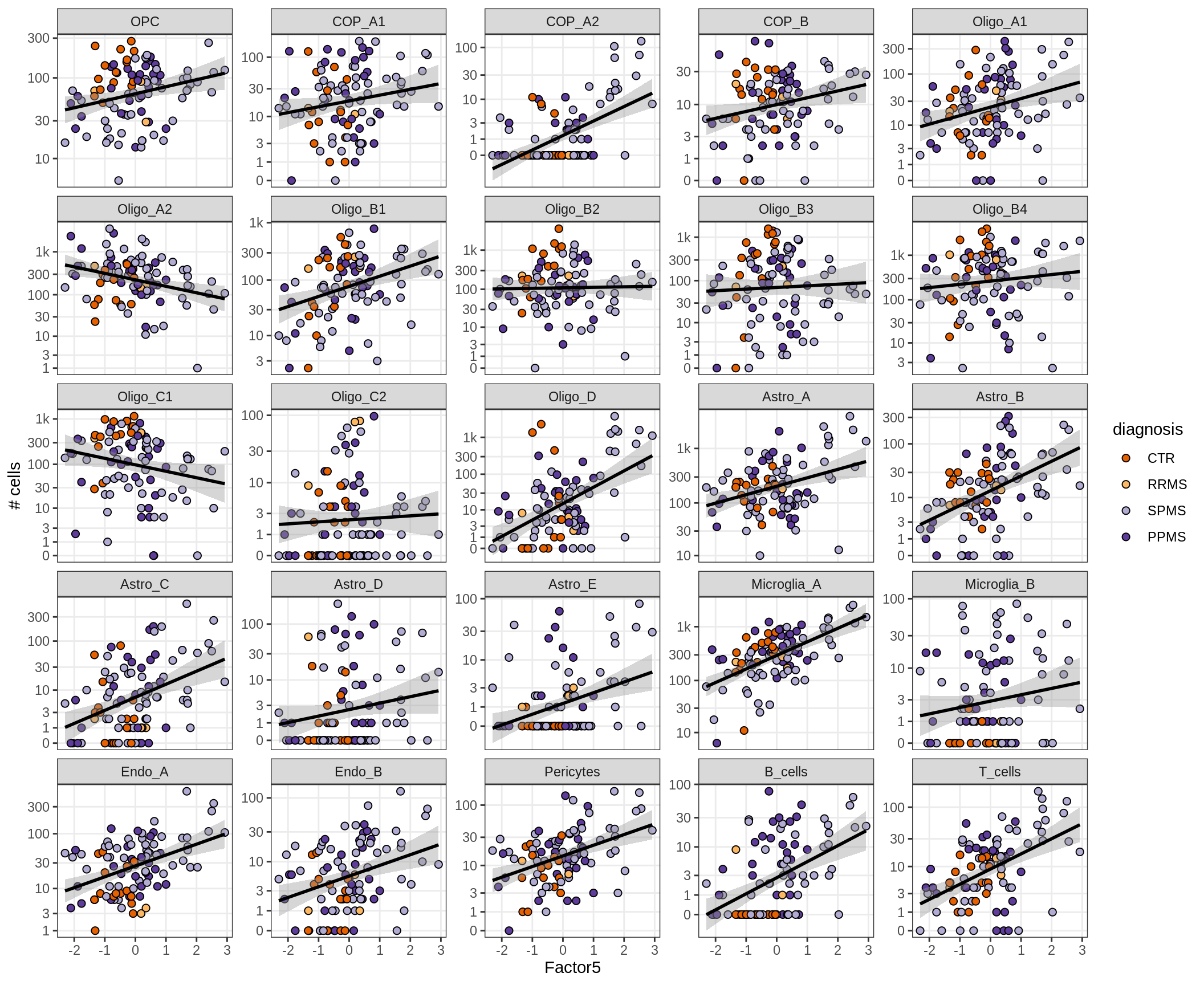
Factors vs number of cells
for (f in factors_names(model) ) {
cat('### ', f, '\n', sep = '')
print(plot_mofa_vs_n_cells(model, n_cells_dt, sel_f = f))
cat('\n\n')
}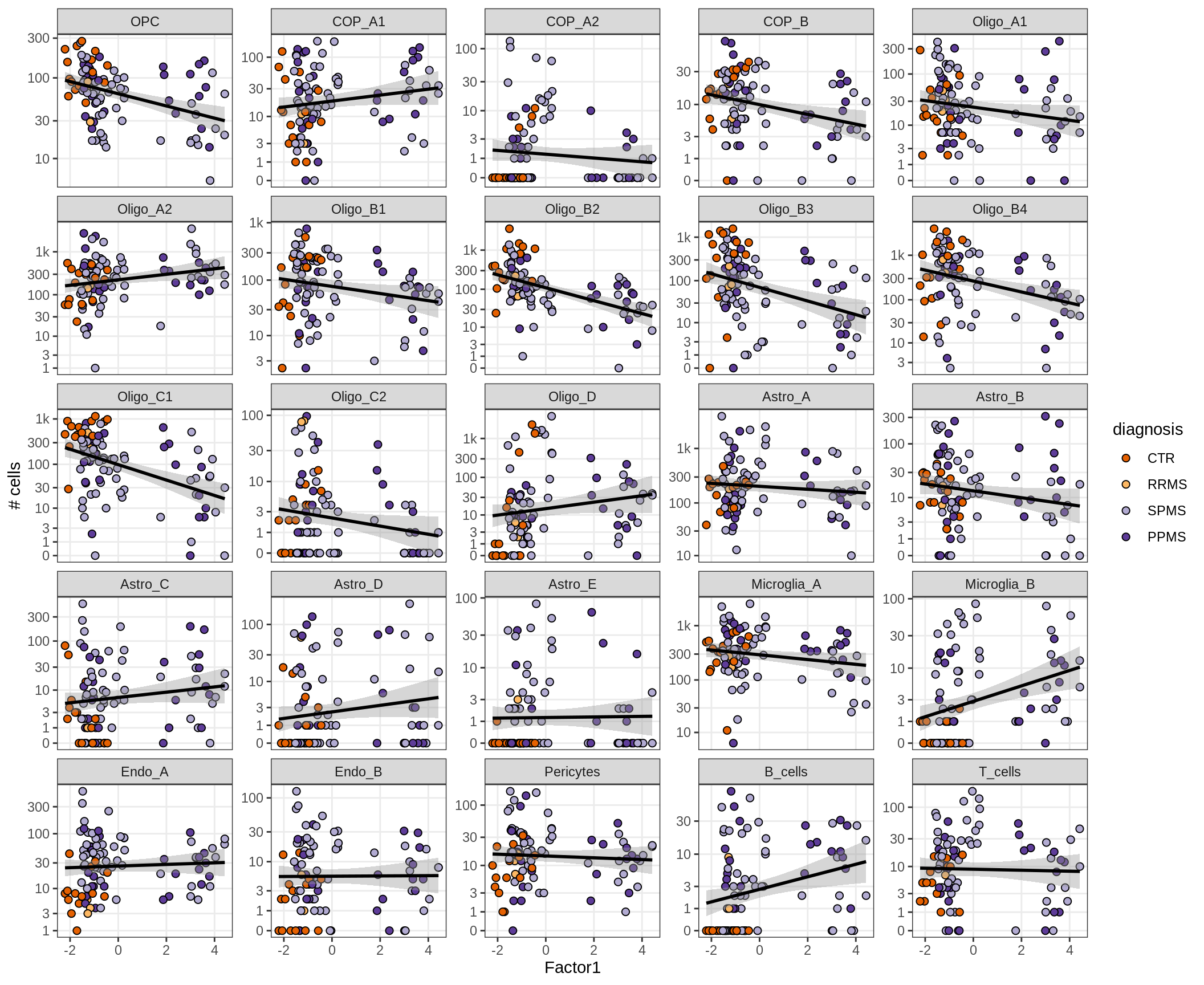
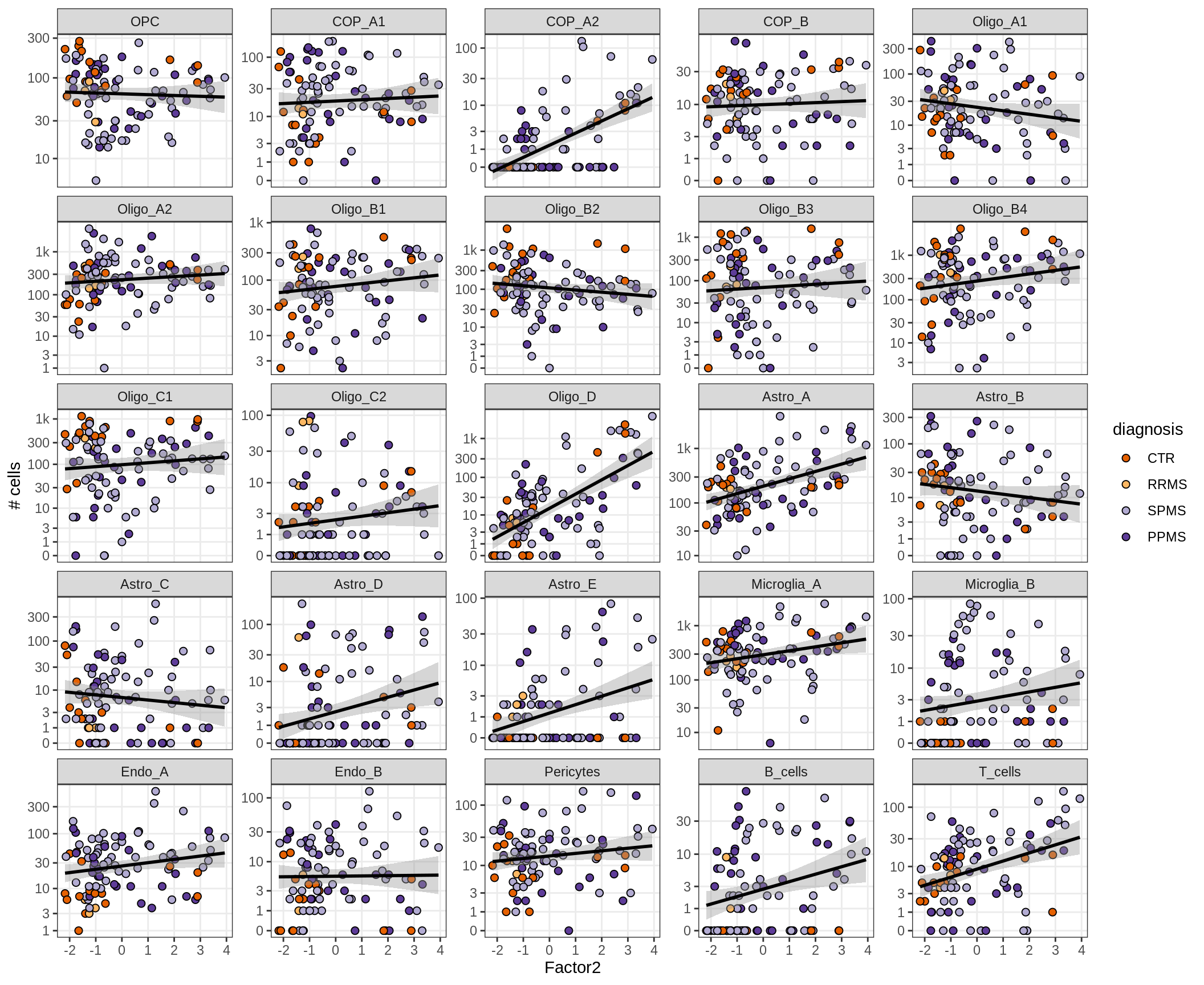
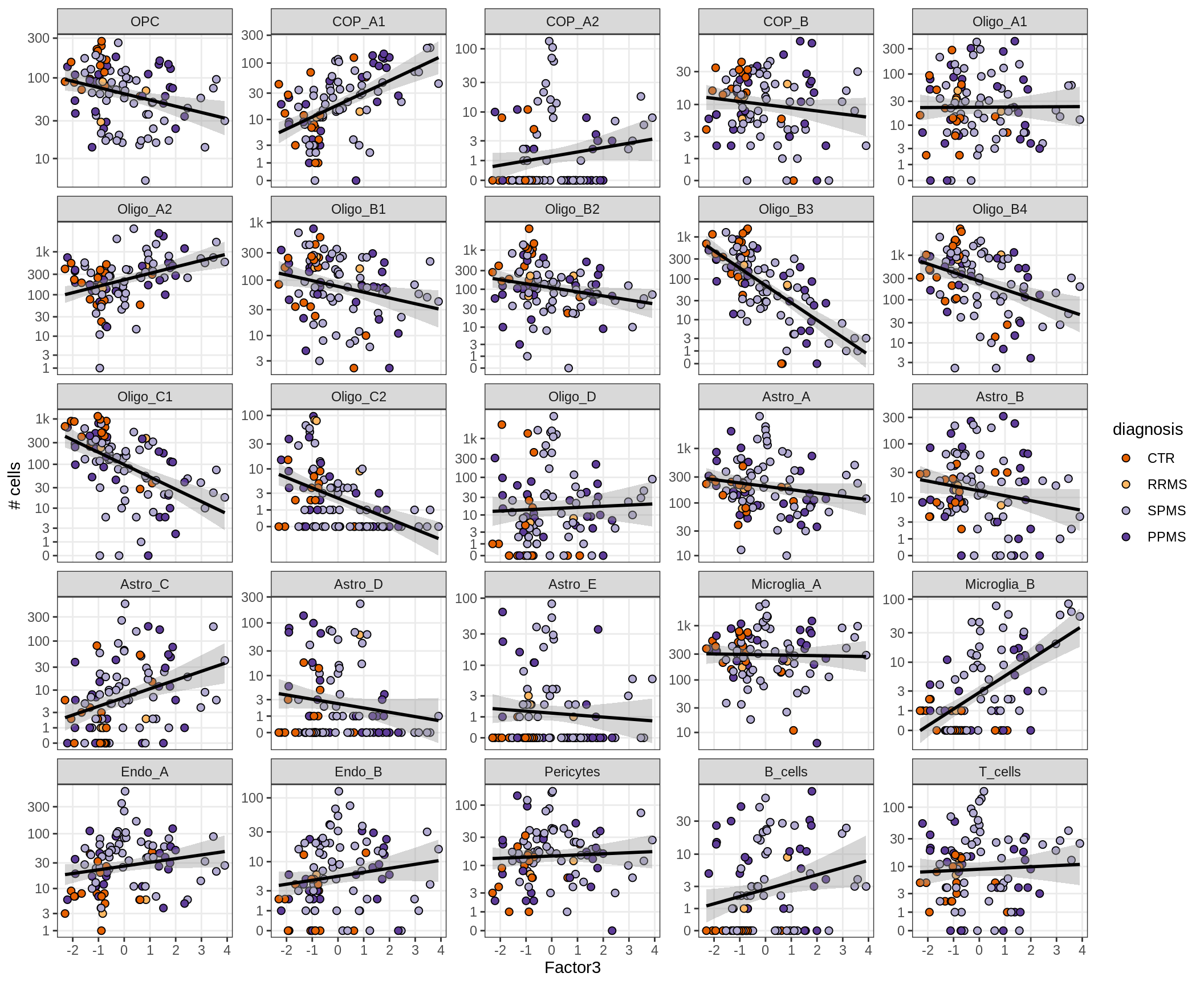
Factor4
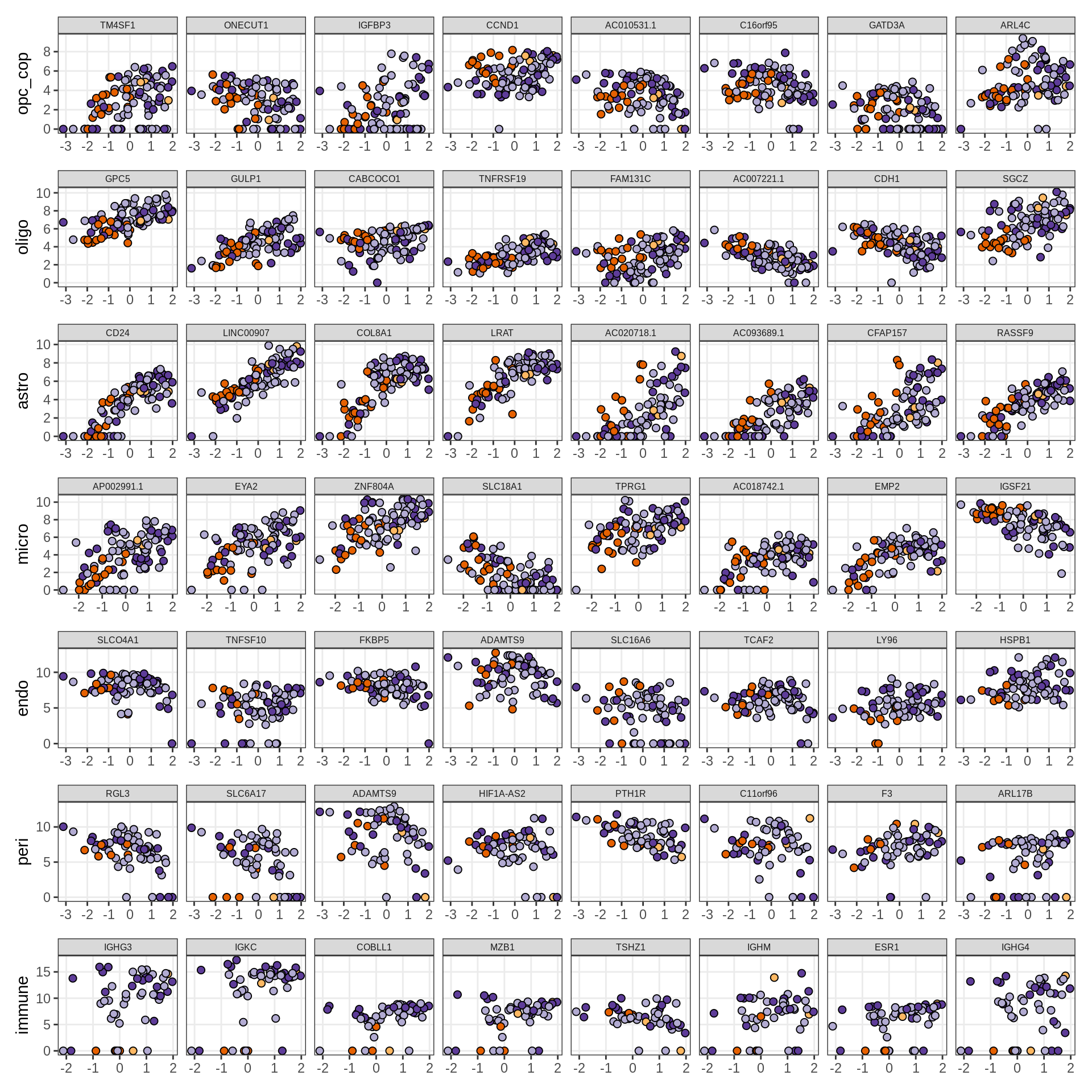
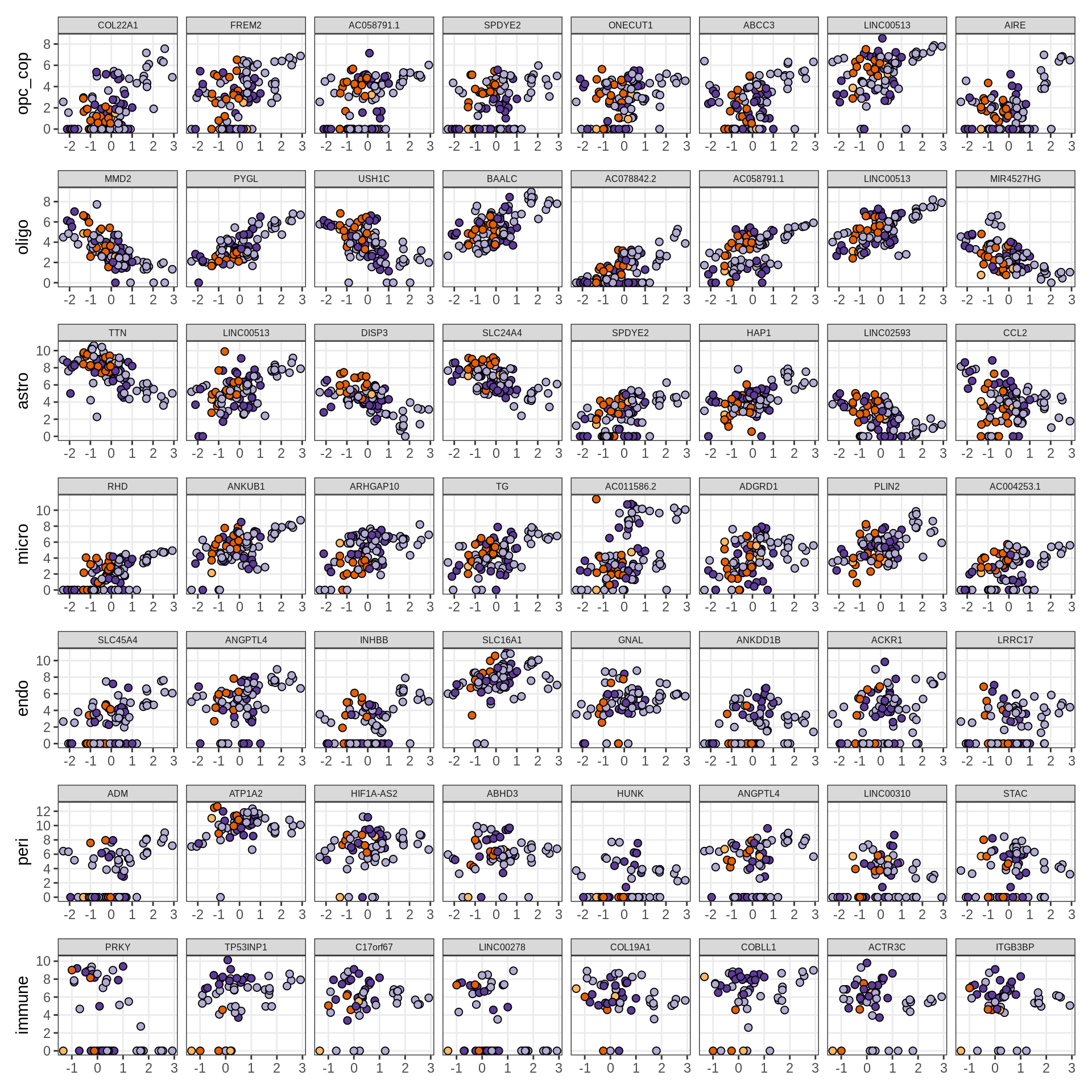
Factors vs top genes
for (f in factors_names(model)) {
cat('### ', f, '\n', sep = '')
print(plot_mofa_vs_logcpm(model, annots_dt, sel_f = f))
cat('\n\n')
}


Factor4
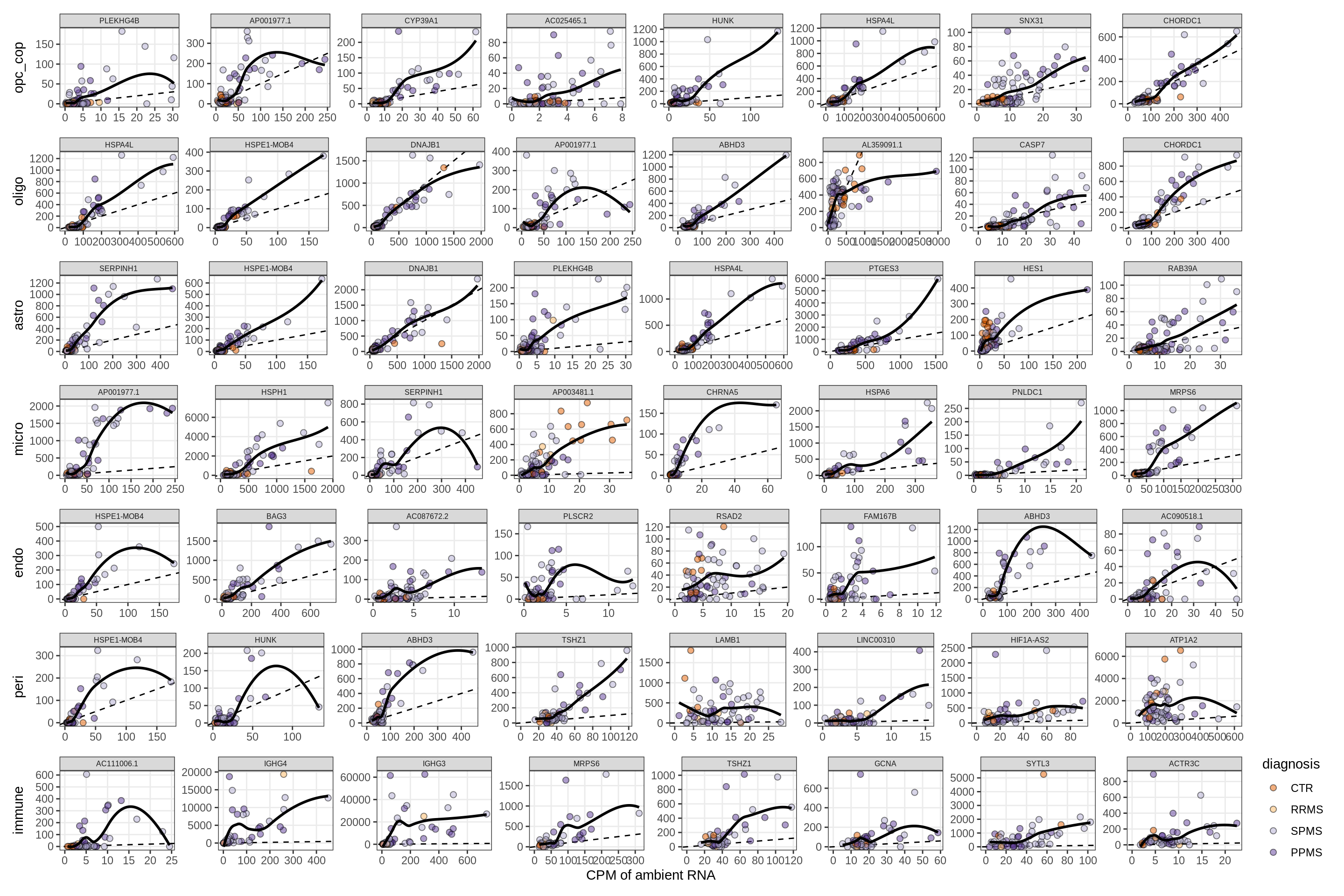
Factors vs top genes - soup
for (f in factors_names(model) ) {
cat('### ', f, '\n', sep = '')
print(plot_mofa_vs_soup_logcpm(model, annots_dt, soup_dt,
sel_f = f, trans = 'linear'))
cat('\n\n')
}
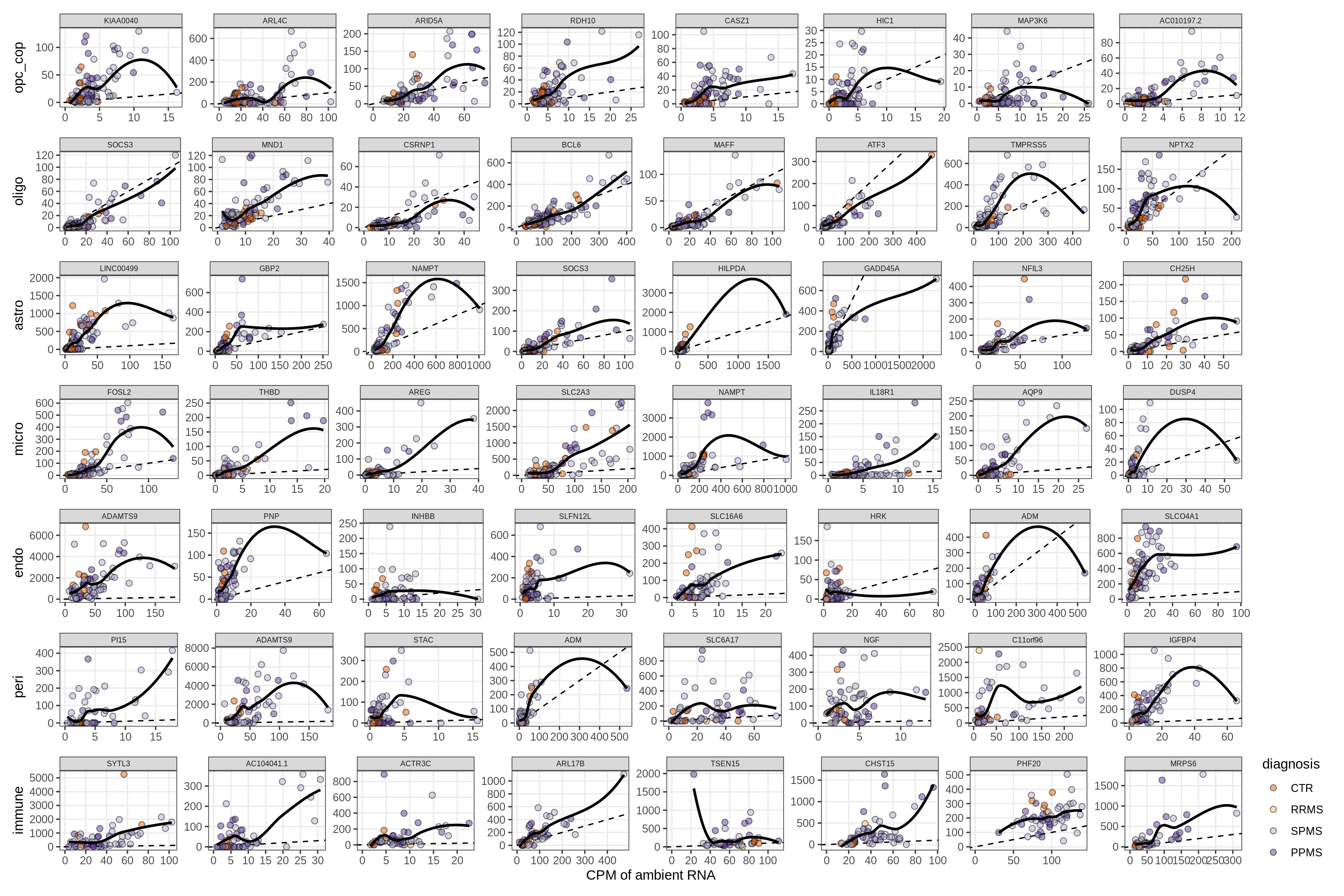

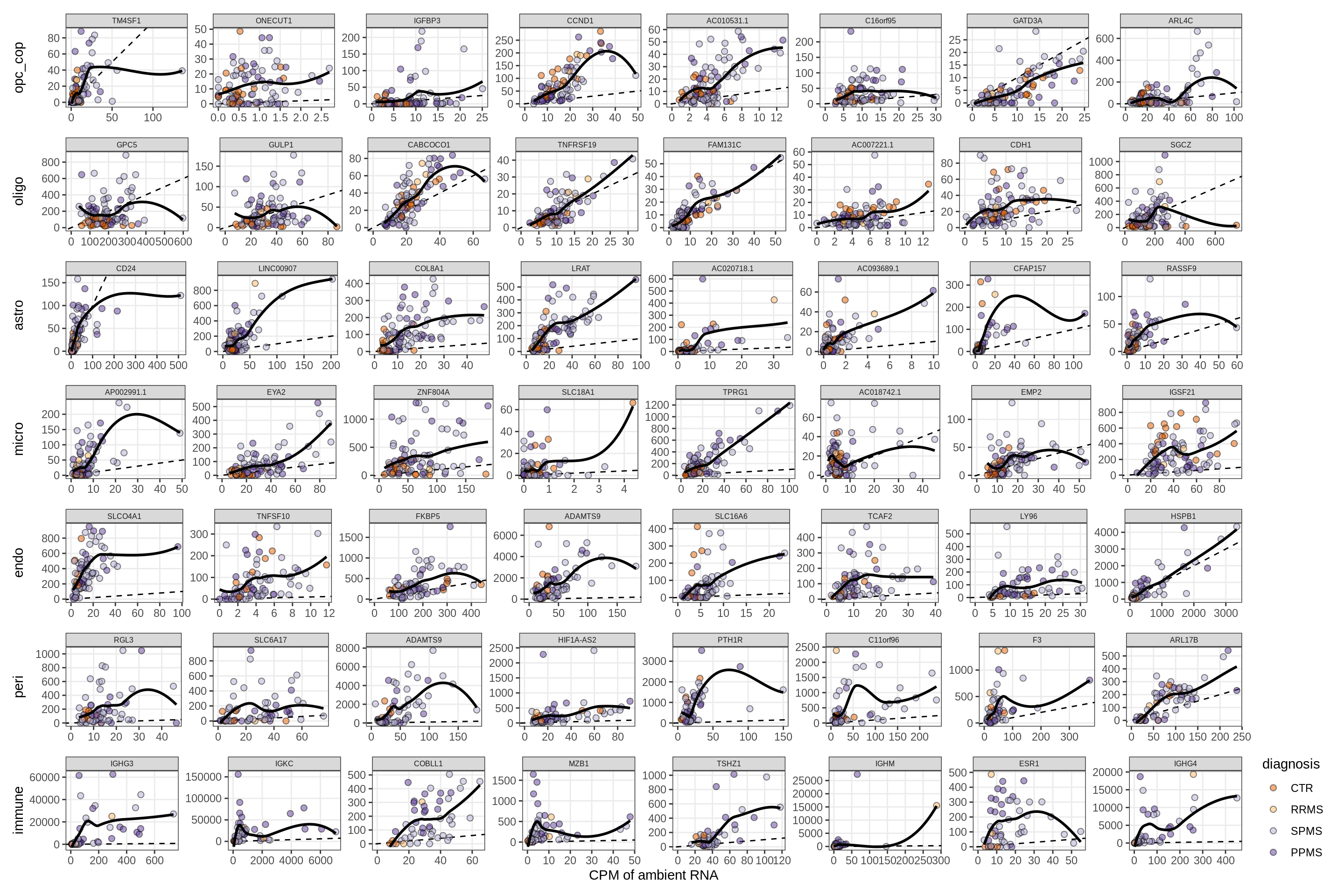

Distributions of factor weights
(plot_mofa_weights(model))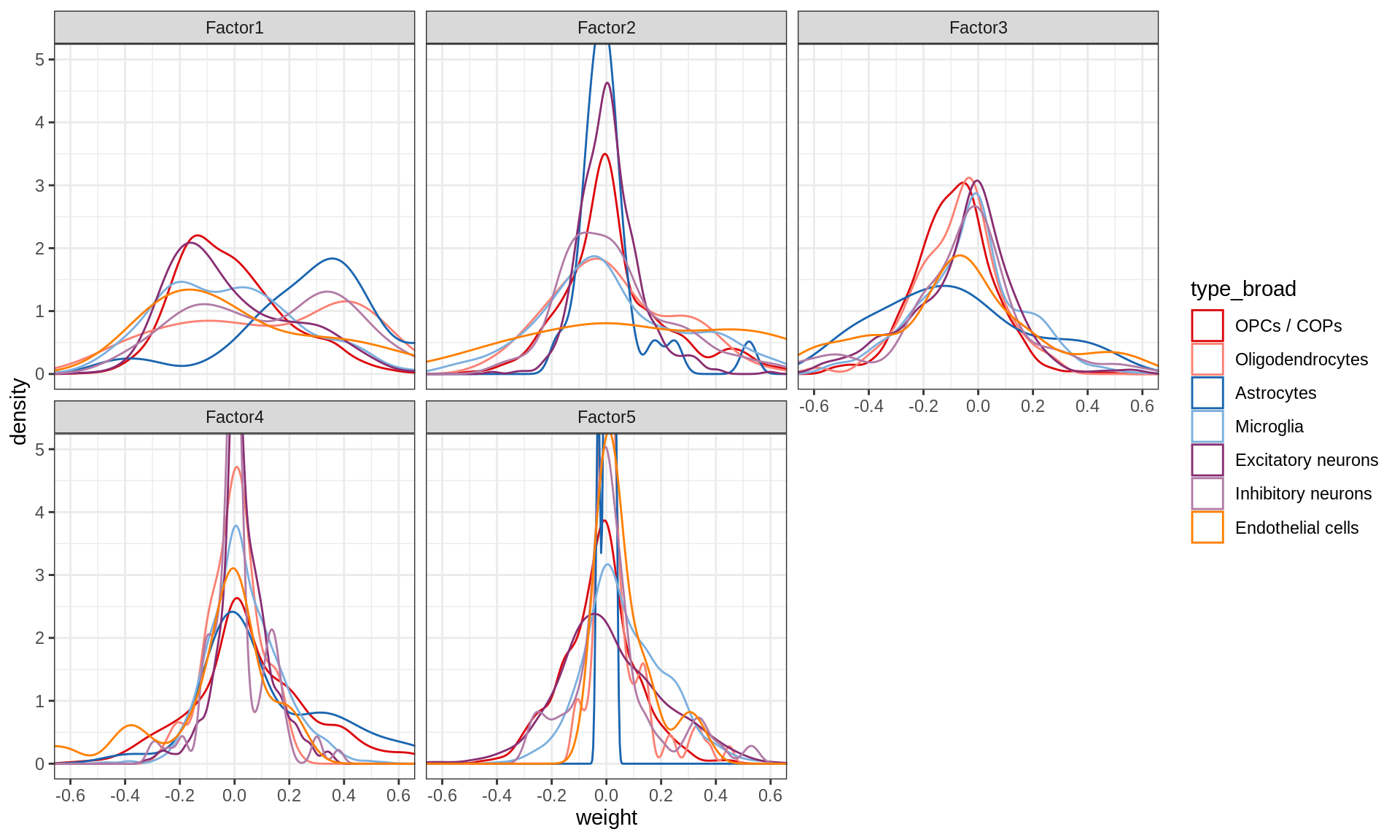
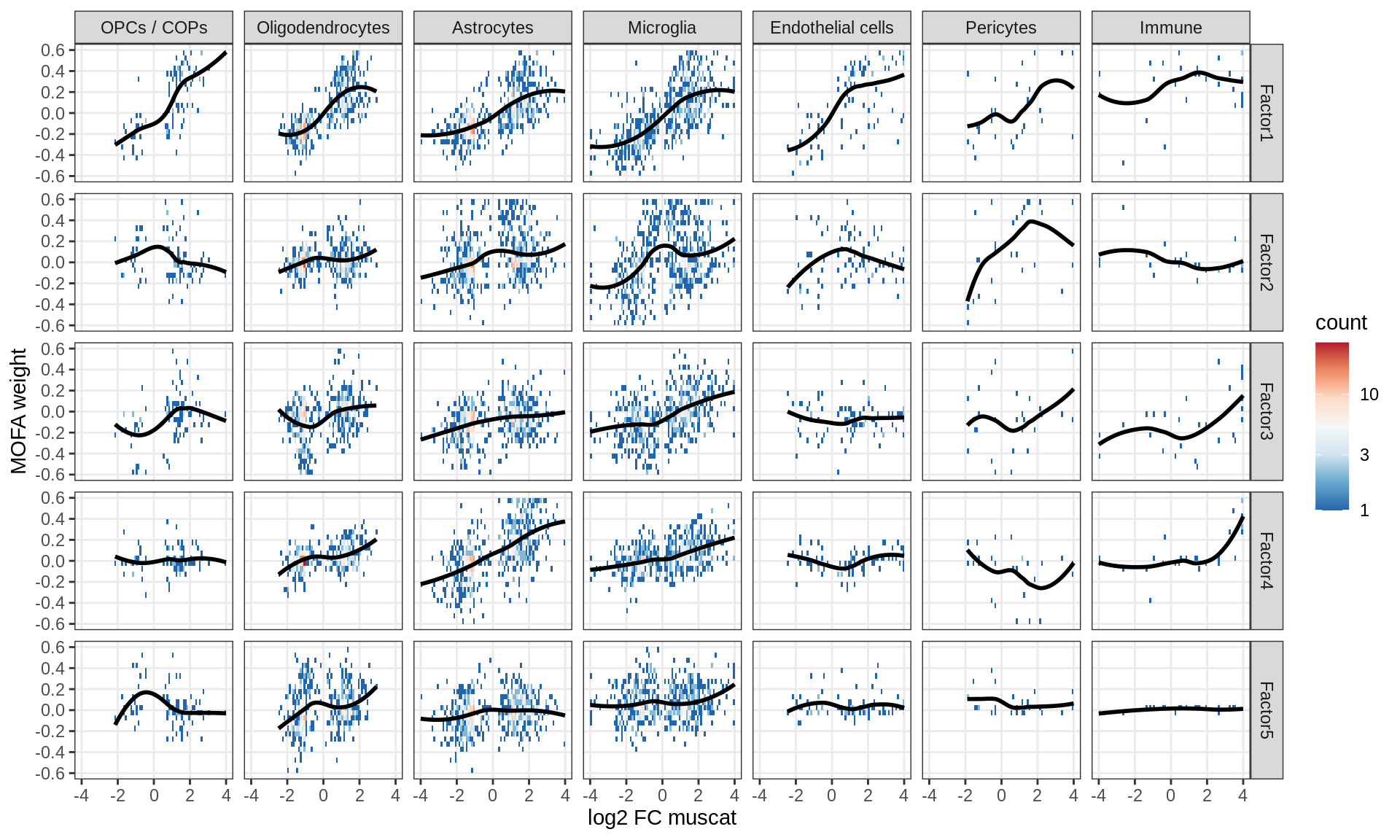
Factor weights vs muscat results
for (what in c('log10_padj', 'log2FC')) {
cat('### ', what, '\n', sep = '')
print(plot_muscat_vs_mofa(model, filter_dt, what = what))
cat('\n\n')
}log10_padj
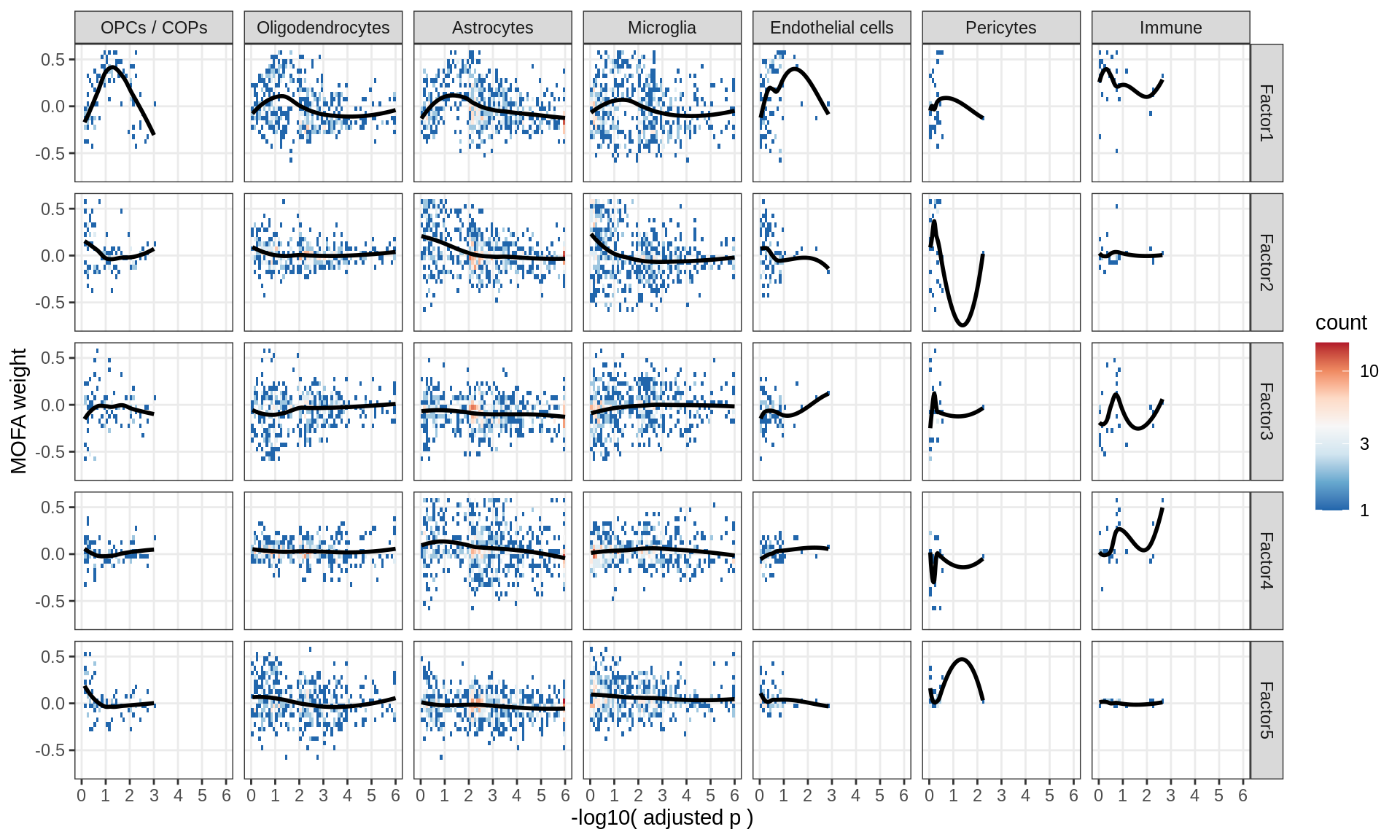
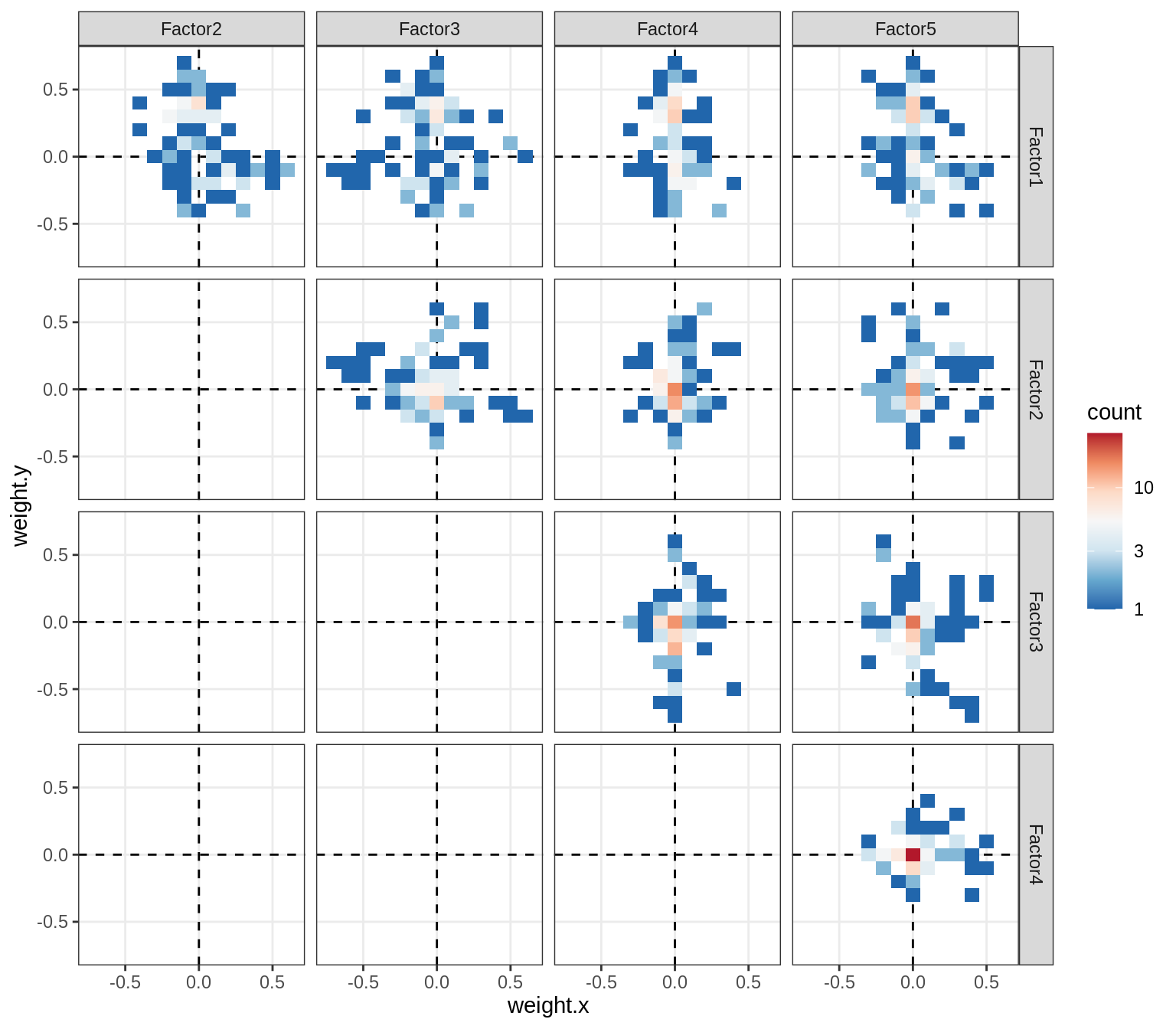
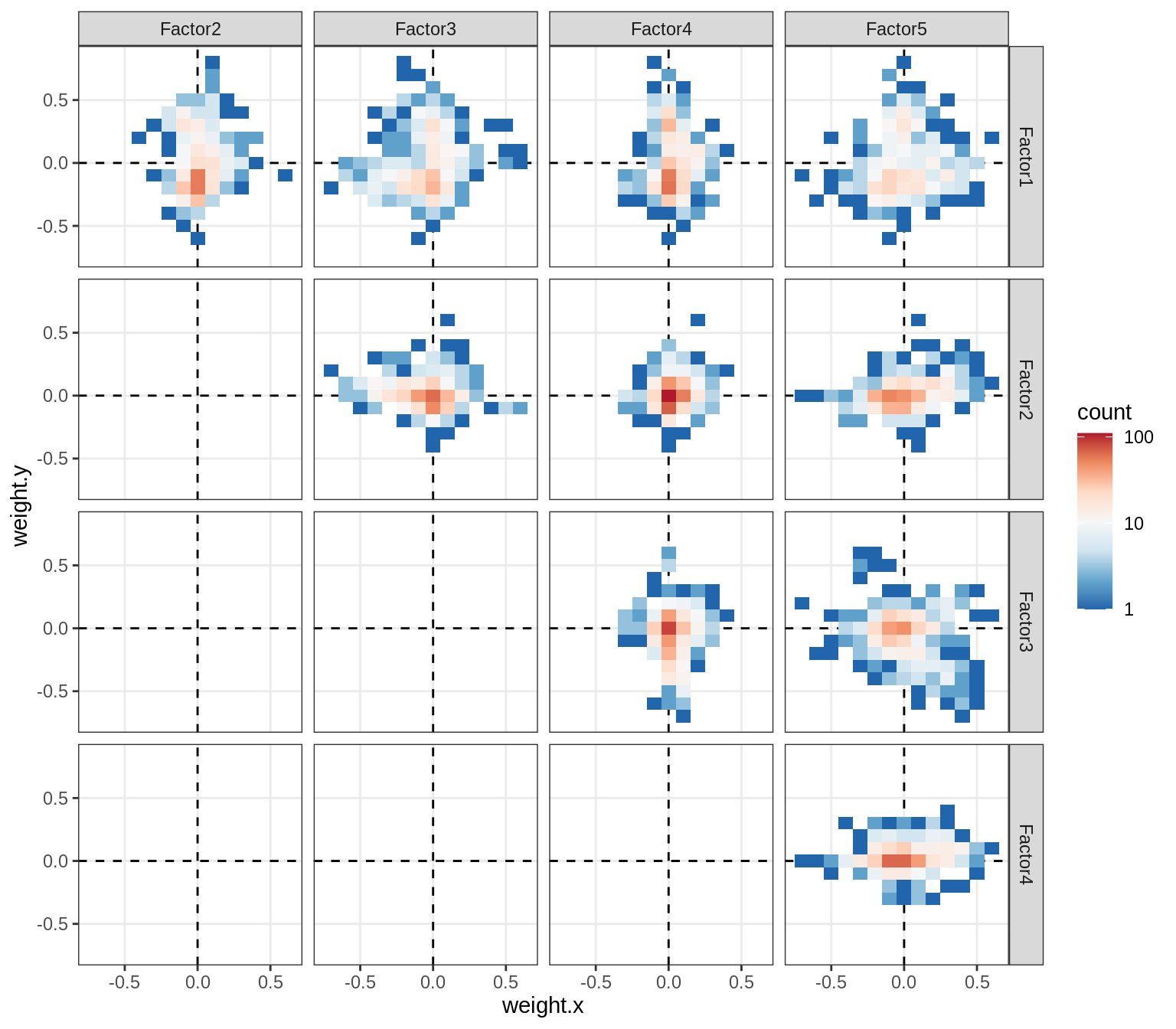
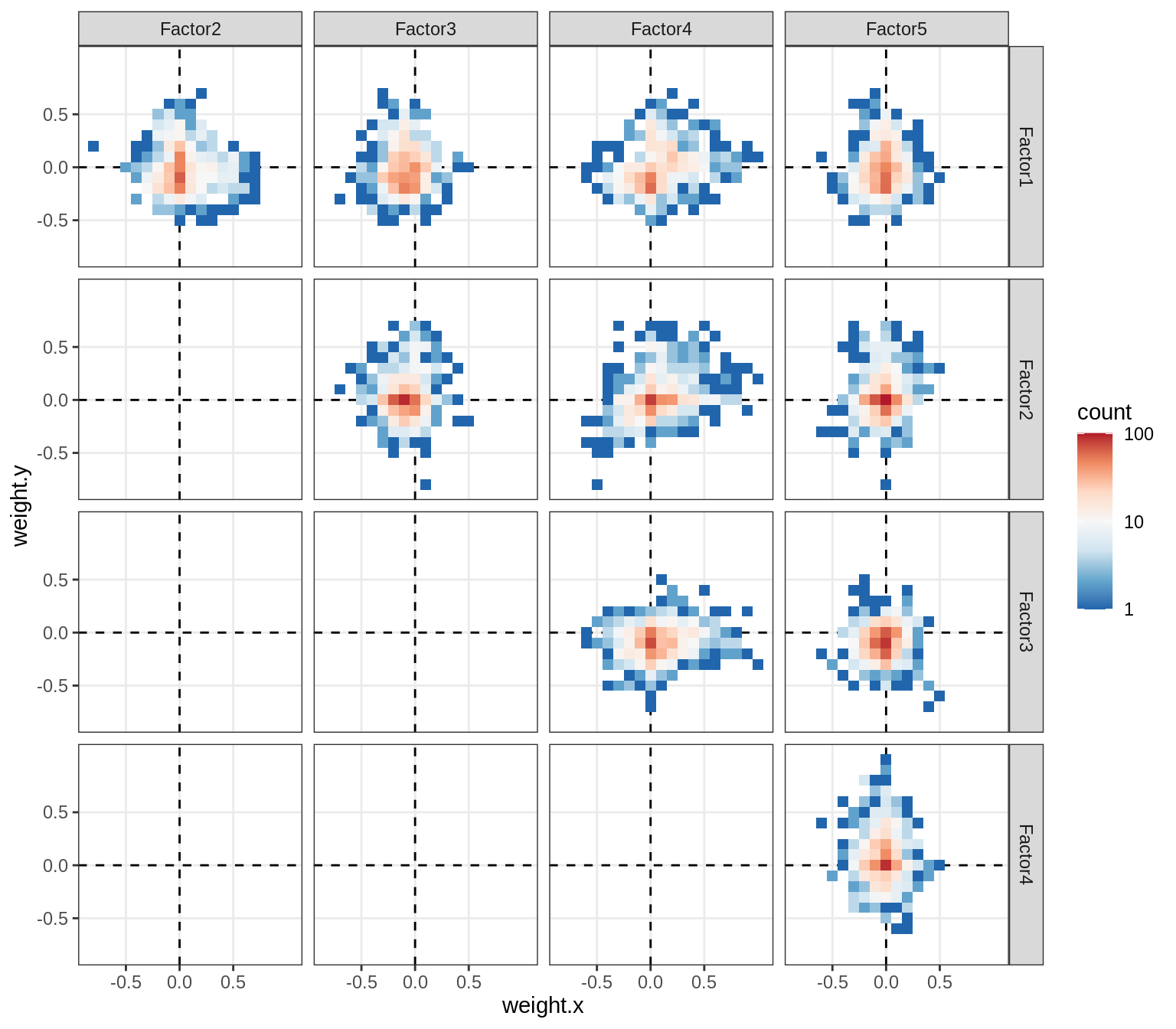
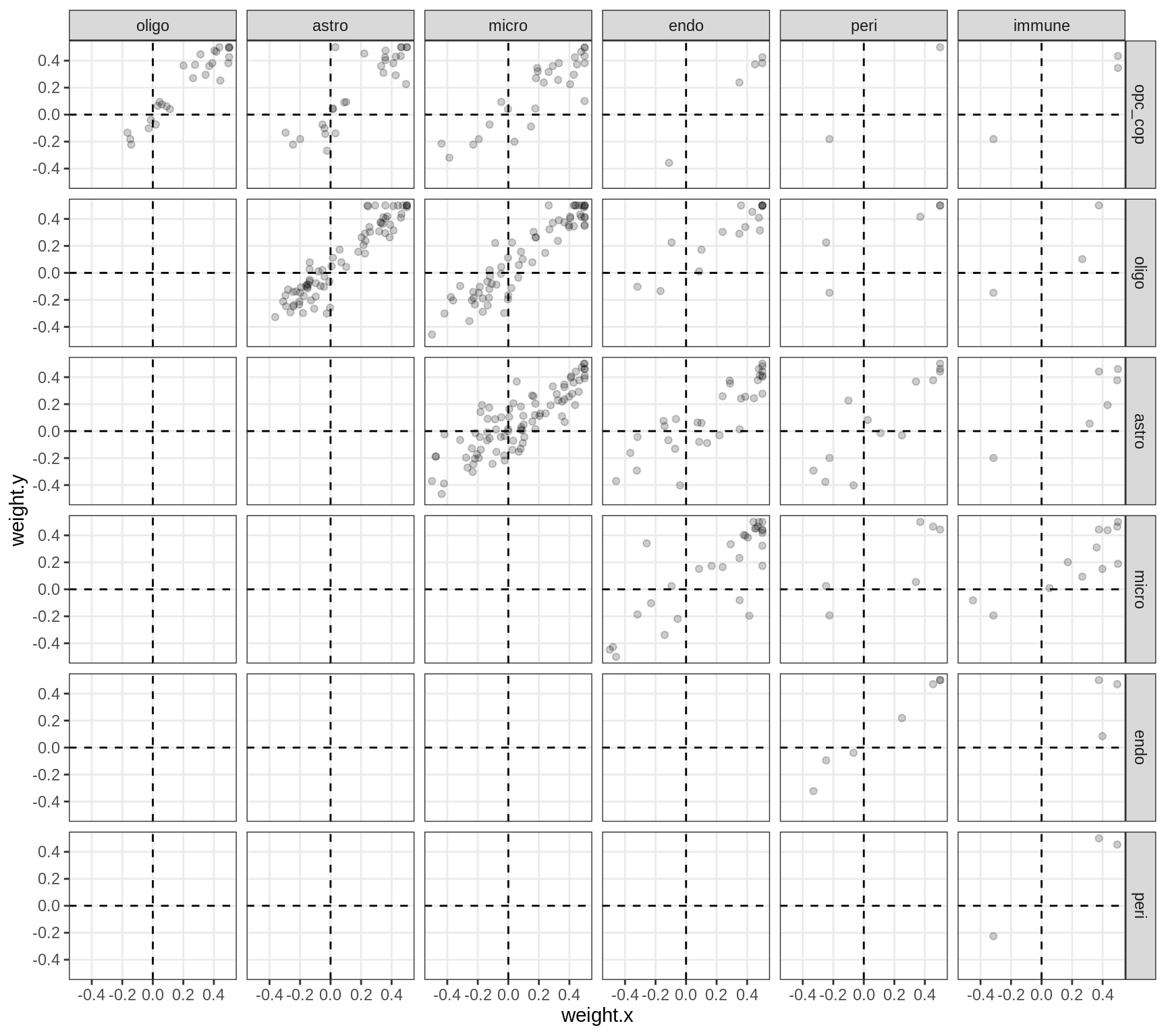
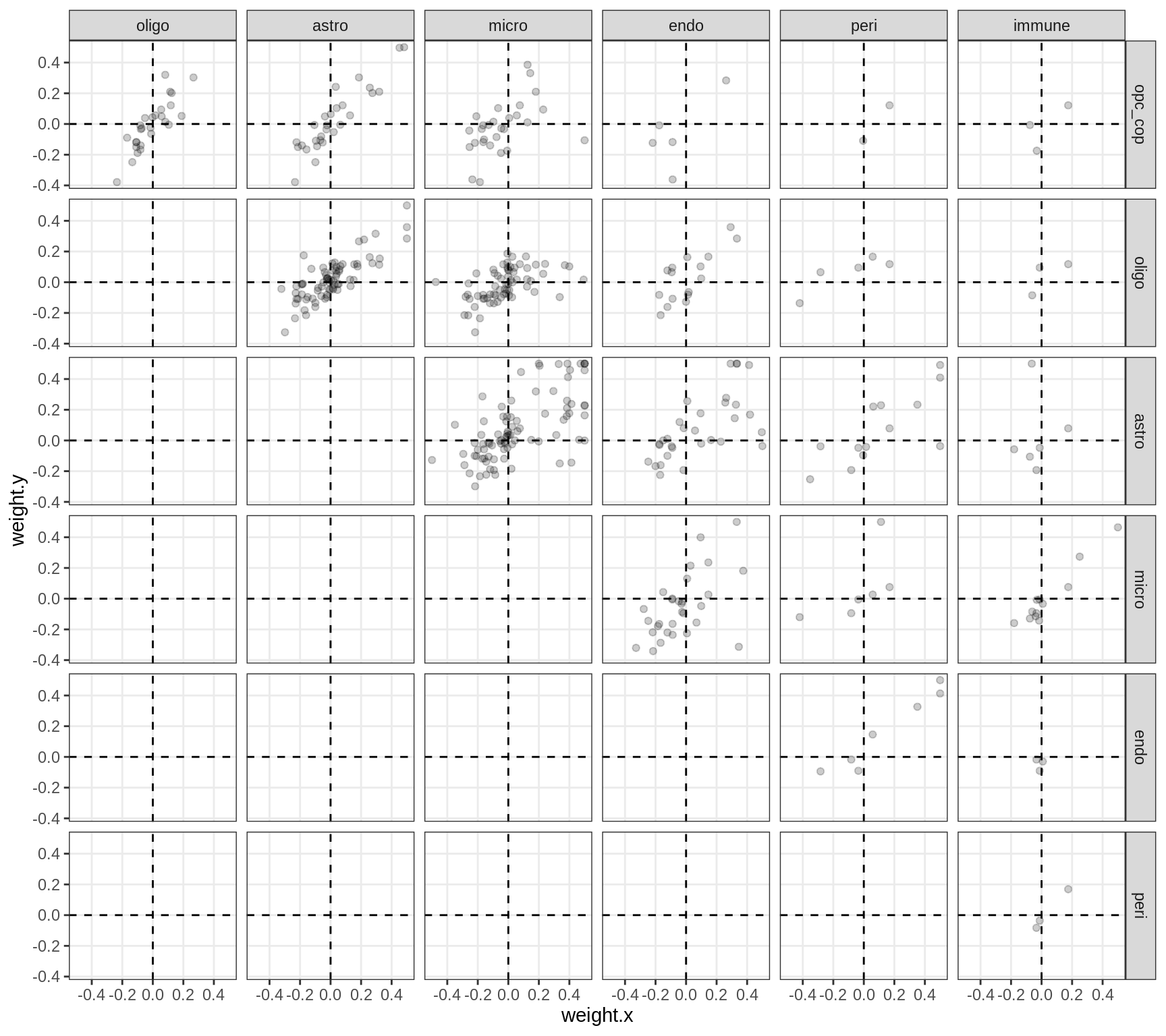
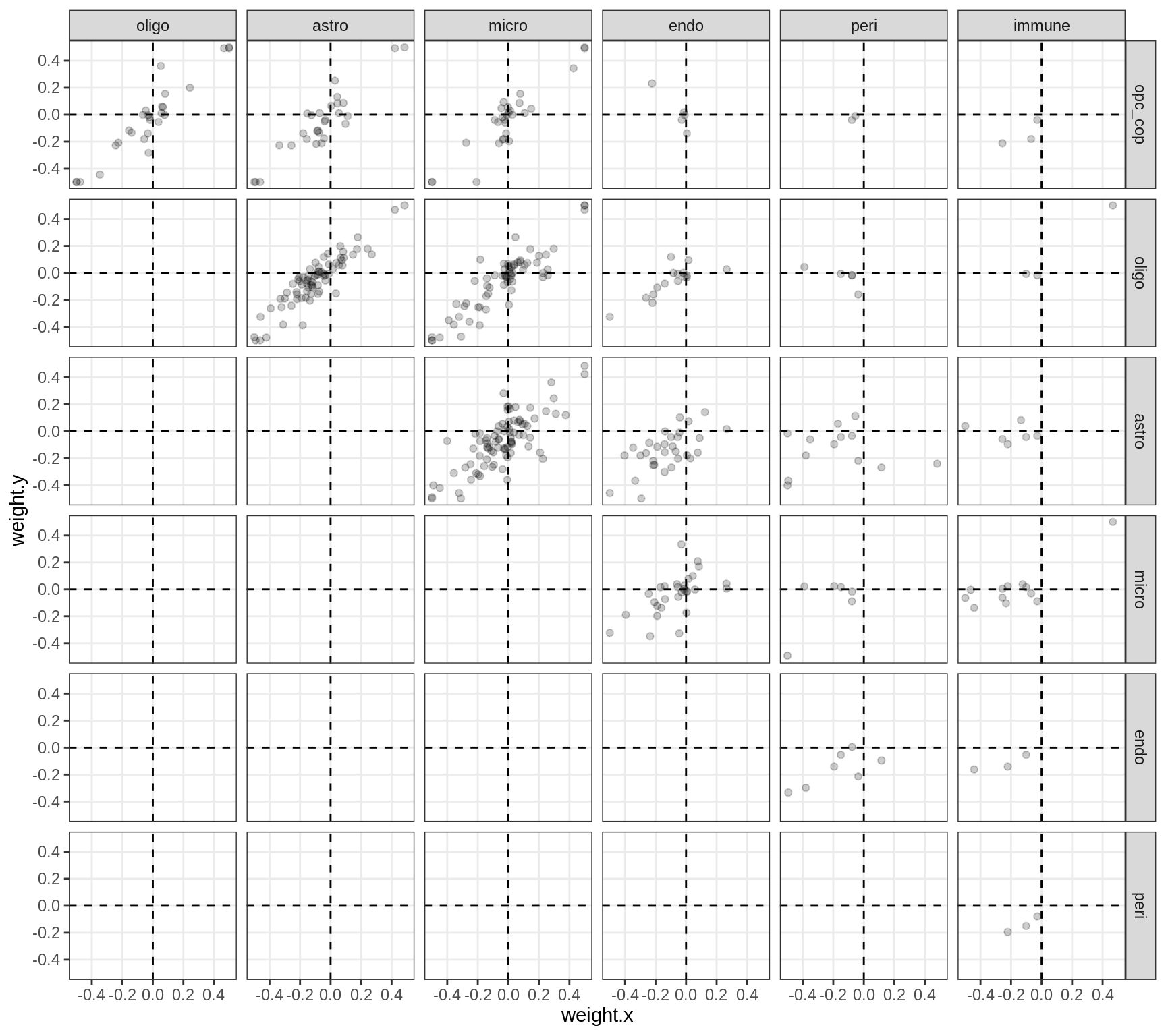
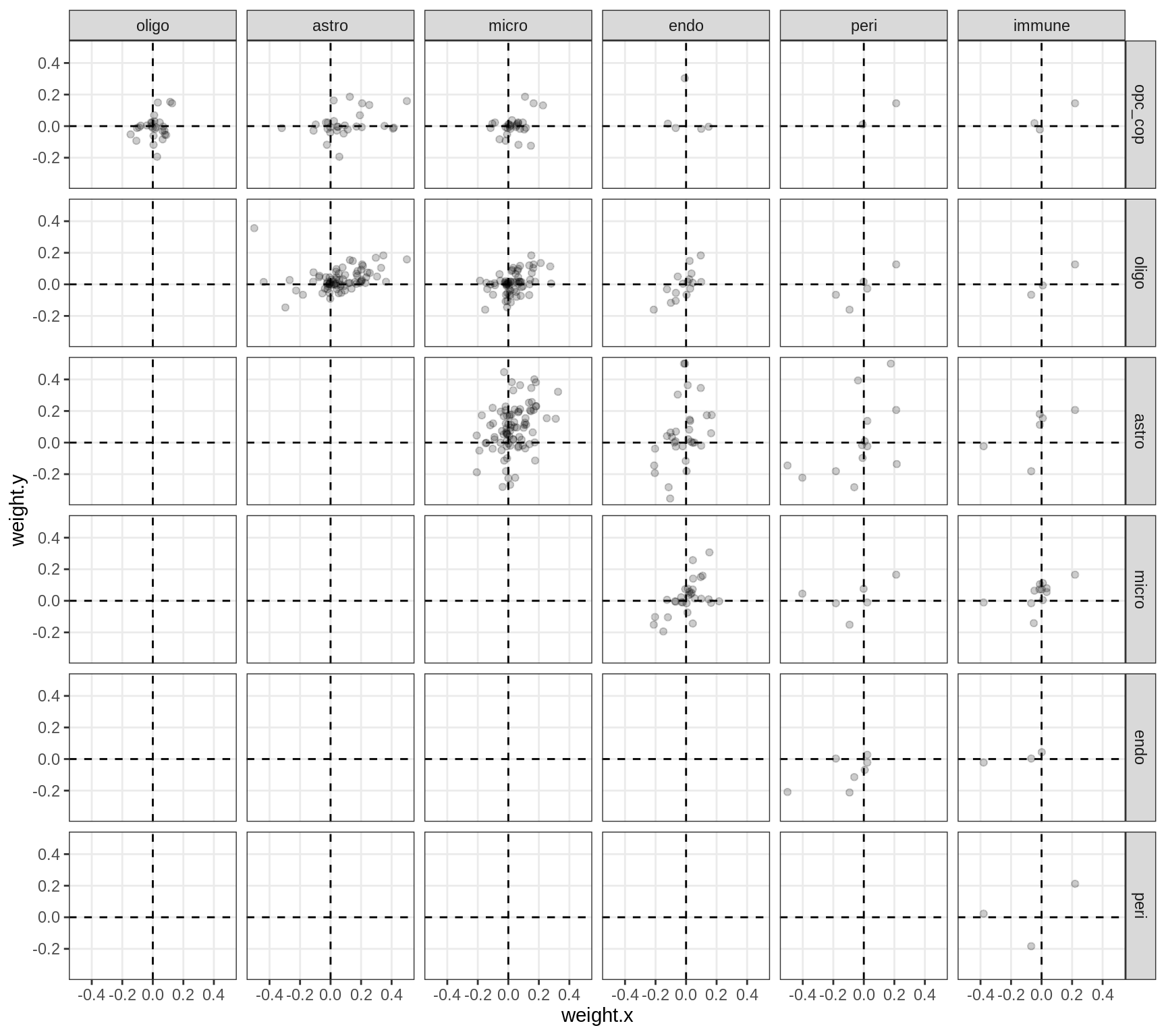
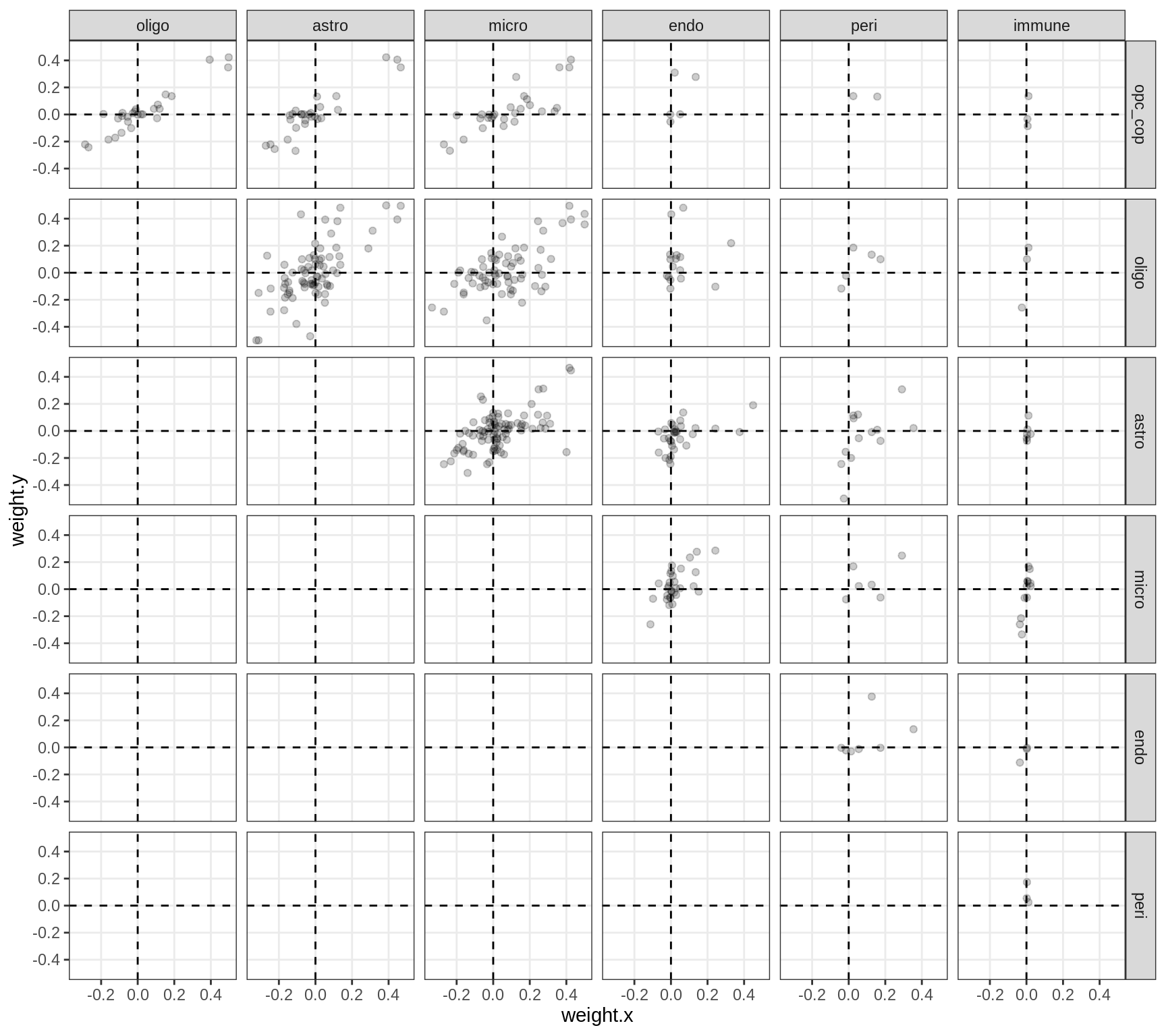
Correlations between factor weights - split by celltype
for (v in broad_short[sel_cl]) {
cat('### ', v, '\n', sep = '')
print(plot_factor_weight_corrs(model, v, by = 'type', how = 'bin'))
cat('\n\n')
}opc_cop
oligo
astro
micro
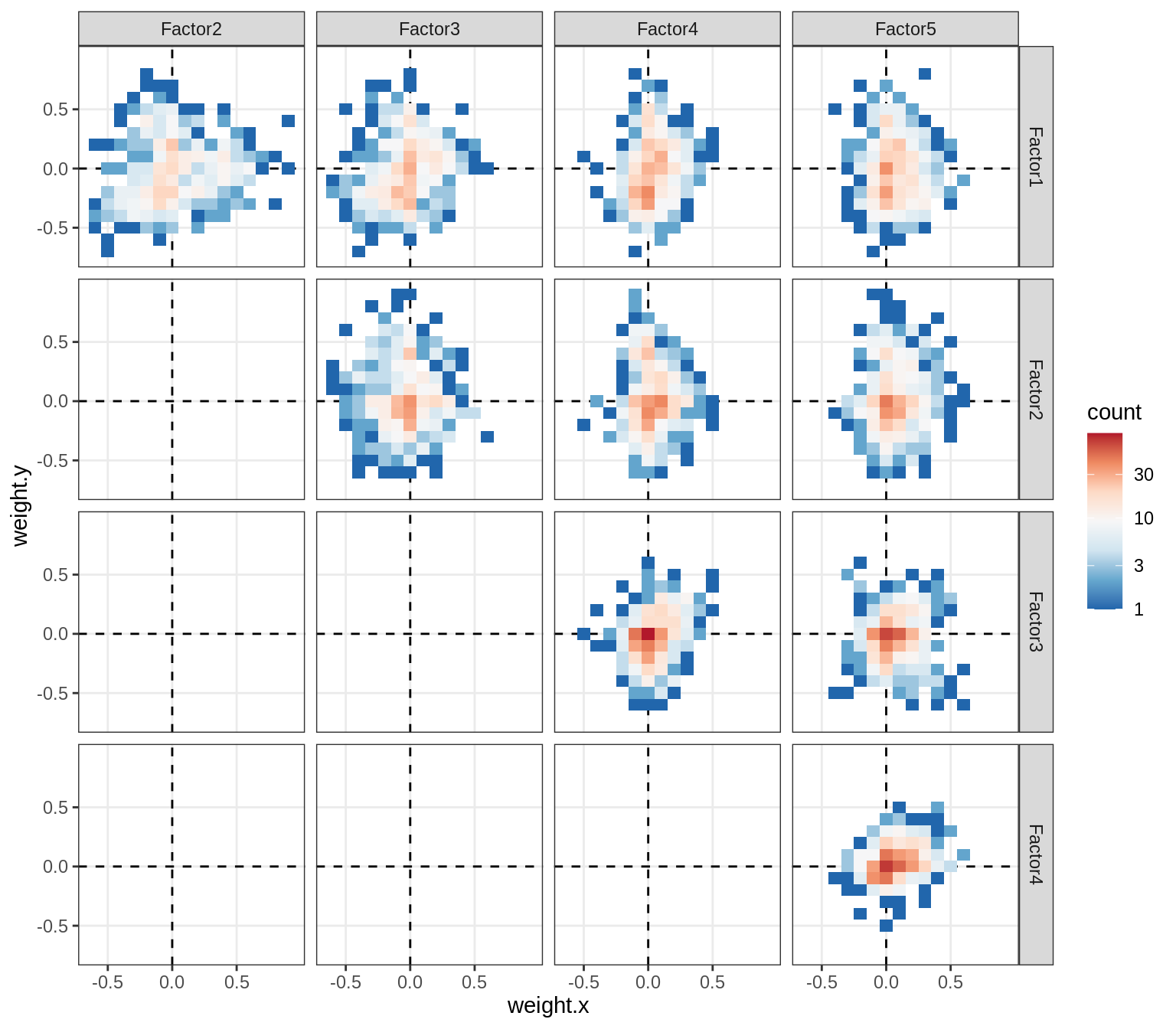
endo
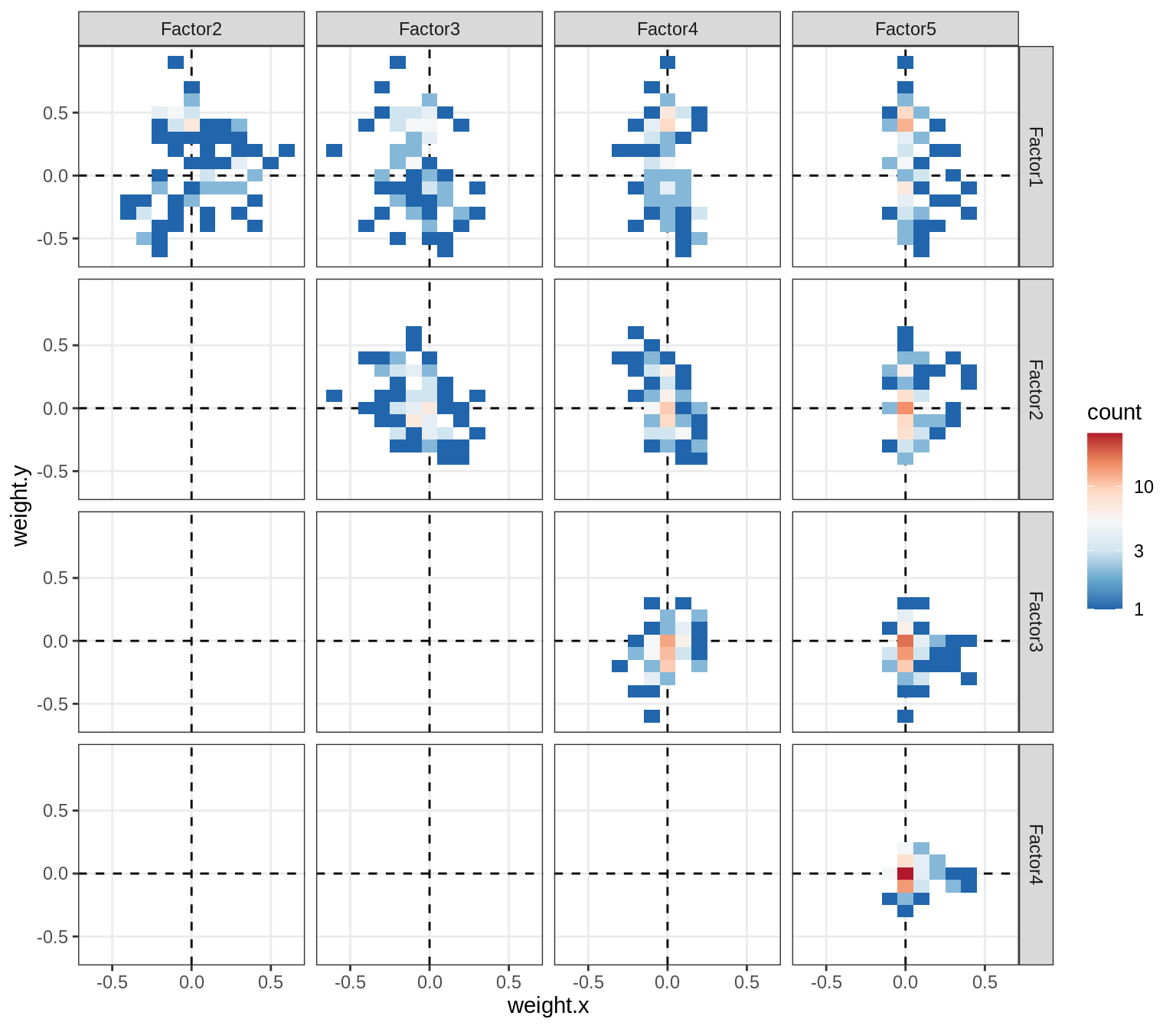
peri
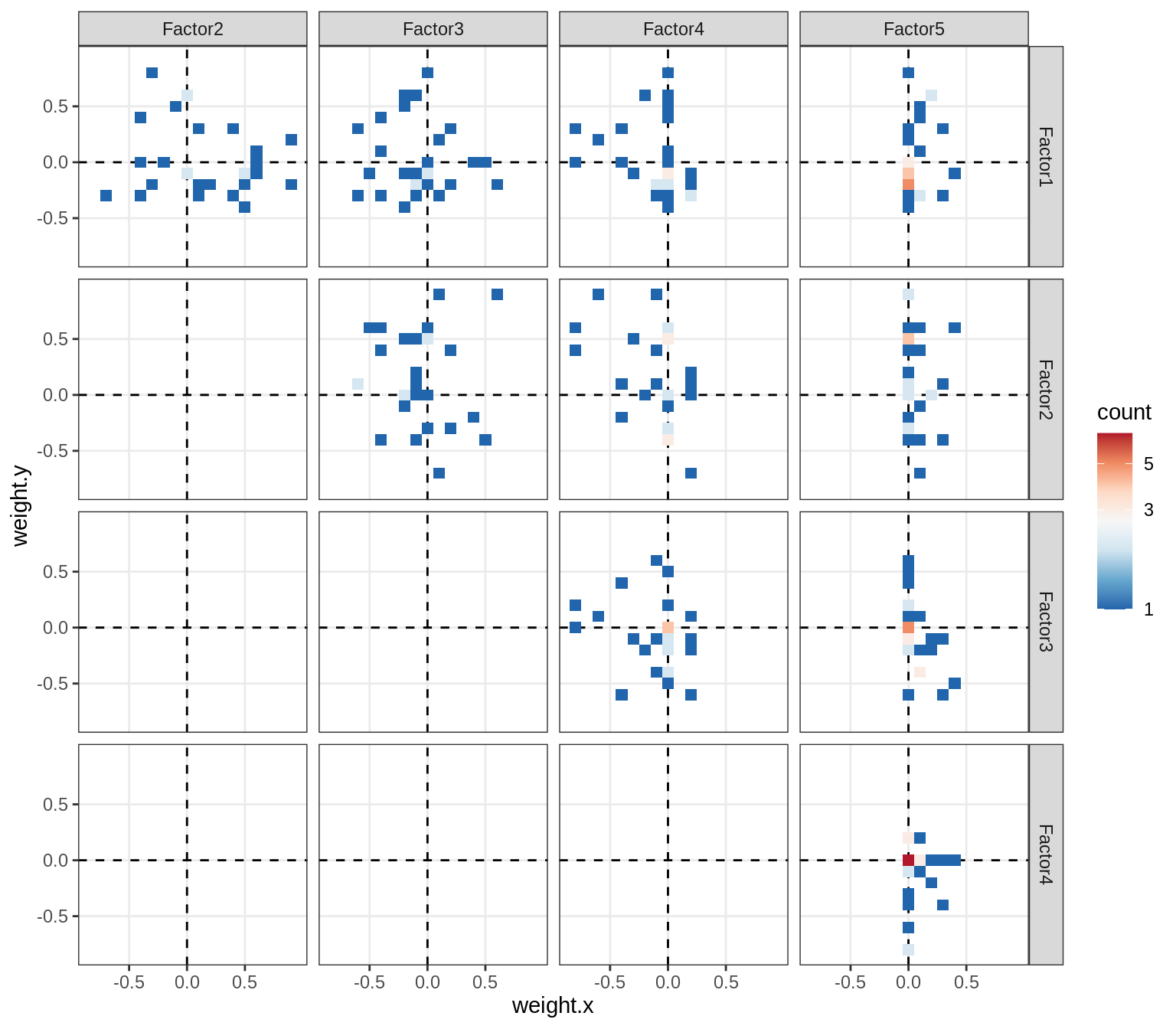
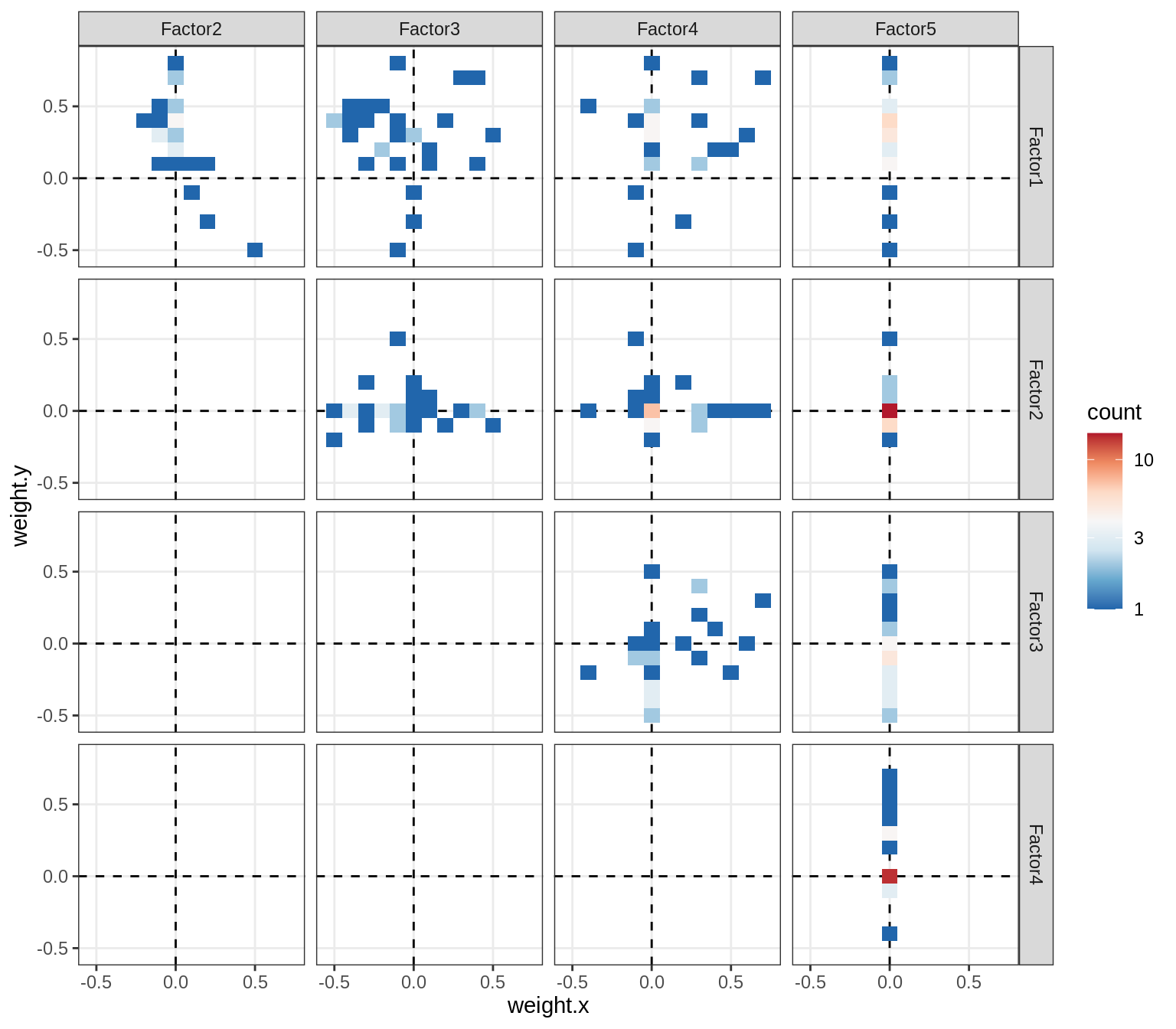
Correlations between factor weights - split by factor
for (f in factors_names(model) ) {
cat('### ', f, '\n', sep = '')
print(plot_factor_weight_corrs(model, f, by = 'factor', how = 'point'))
cat('\n\n')
}
Factor4
Variance explained
(plot_var_exp(var_exp_dt))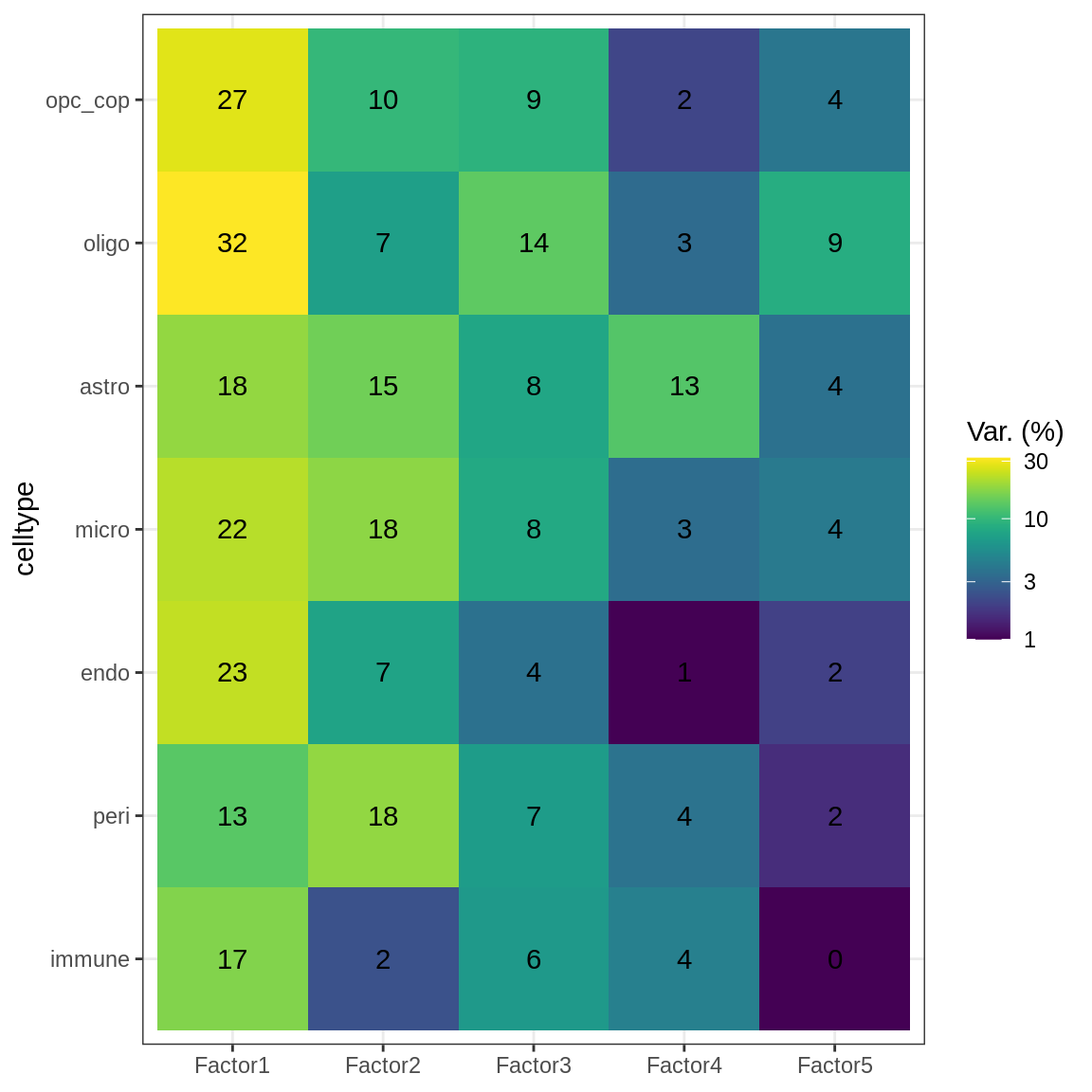
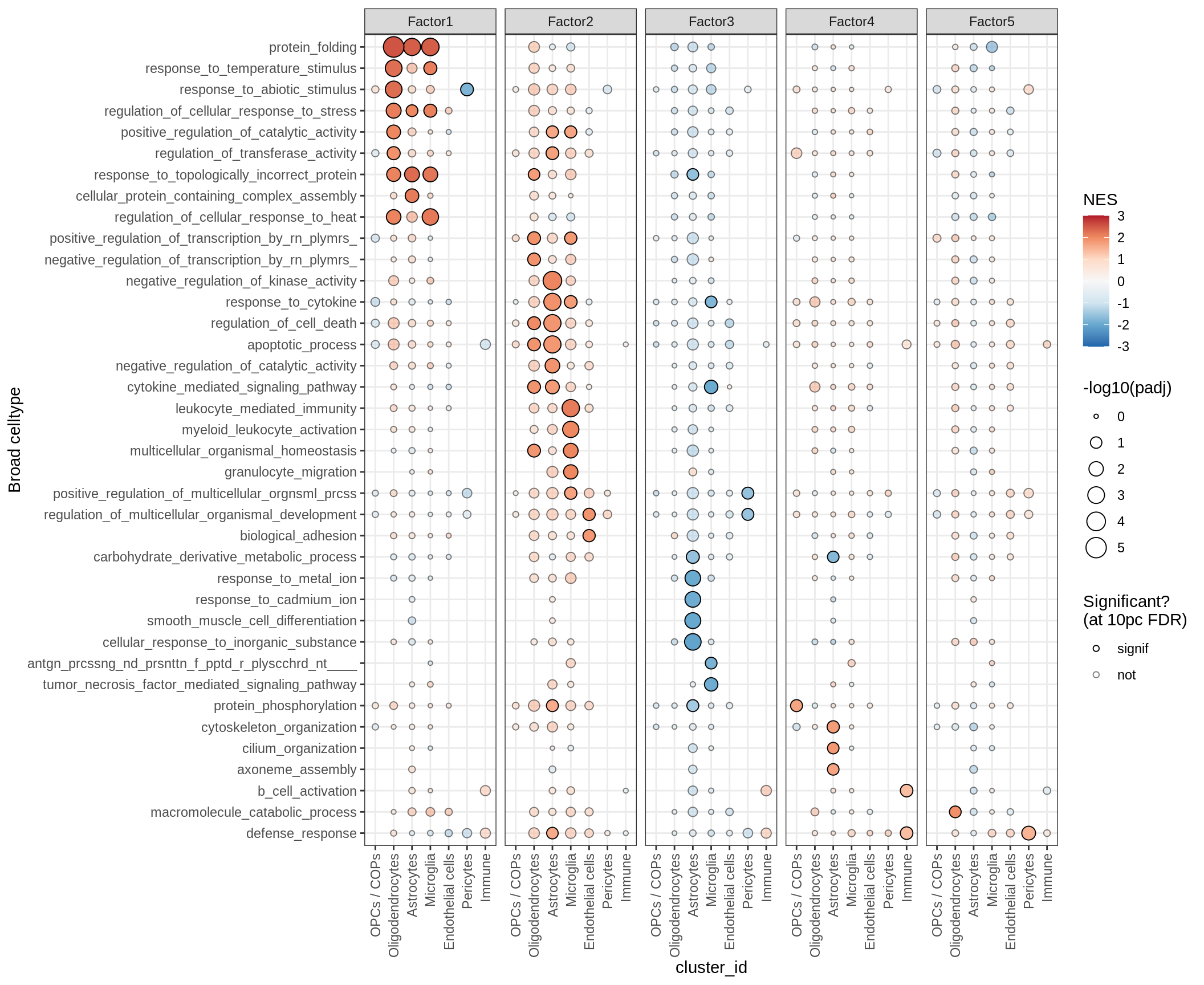
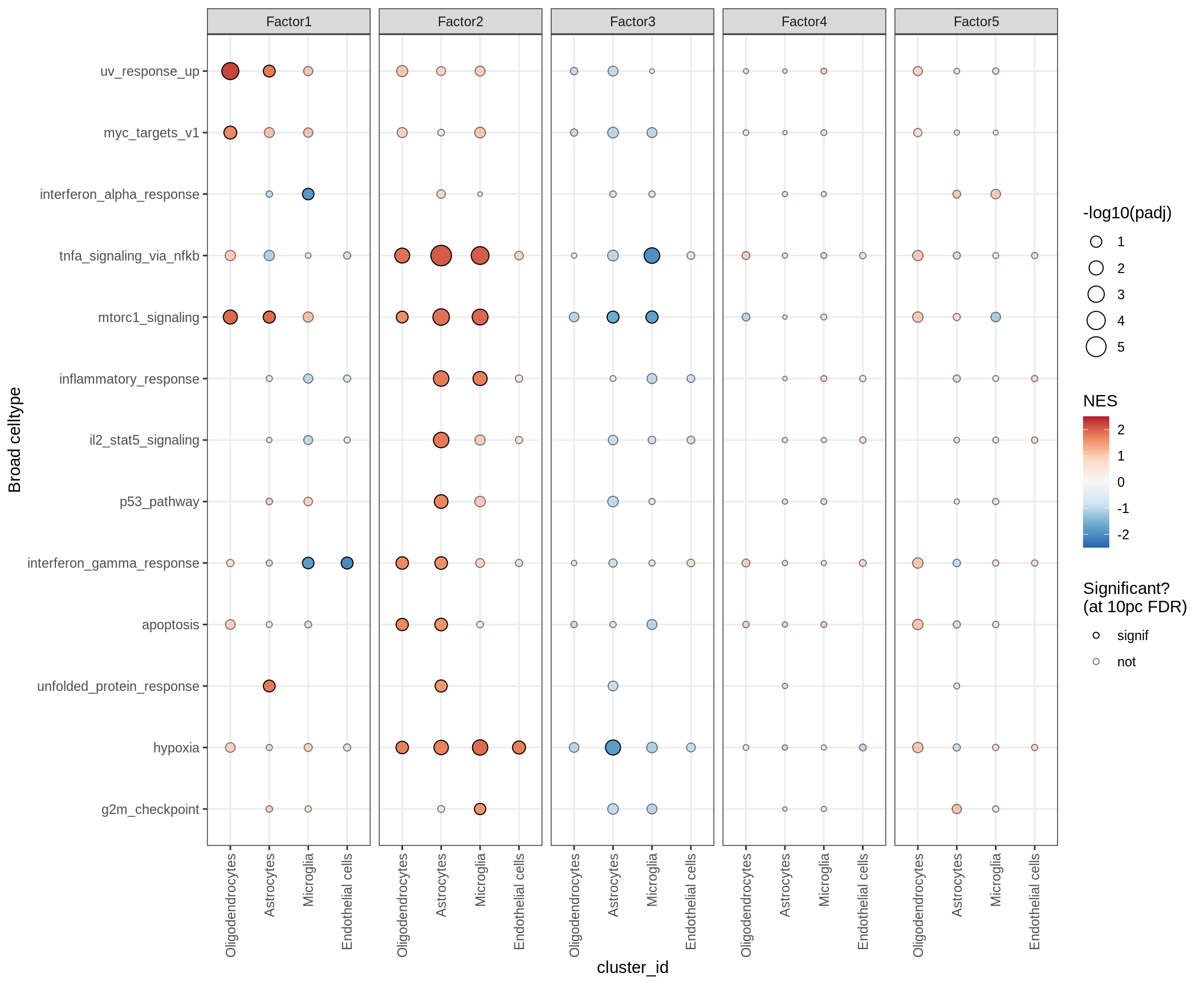
GSEA for factors
for (p in names(gsea_list)) {
# restrict to relevant GO terms
cat('### ', p, '\n', sep='')
dt = gsea_list[[p]]
if (nrow(dt[ main_path == TRUE ]) == 0)
next
# plot
print(plot_mofa_gsea_dotplot(dt, labels_dt,
fgsea_cut = fgsea_cut, n_total = 60))
cat('\n\n')
}
Outputs
Top filter genes
# merge filtered and weights
xls_dt = calc_xls_dt(model, filtered_dt)
# save outputs
write_xlsx(list(mofa_weights = xls_dt), path = interesting_f)Figures
Illustrative example
for (g in example_gs) {
cat('### ', str_extract(g, '^[^_]+'), '\n', sep = '')
suppressWarnings(print(plot_ranef_example(pb, example_cl, g)))
cat('\n\n')
}
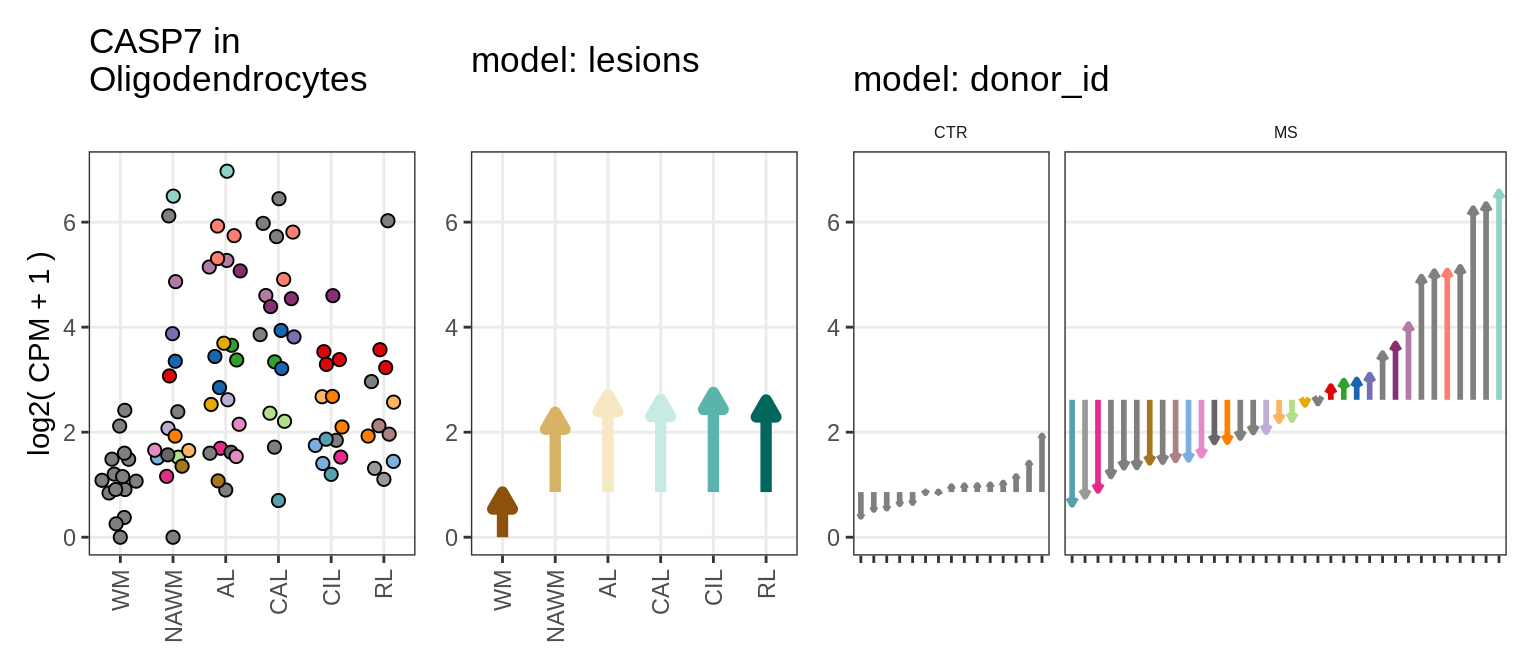
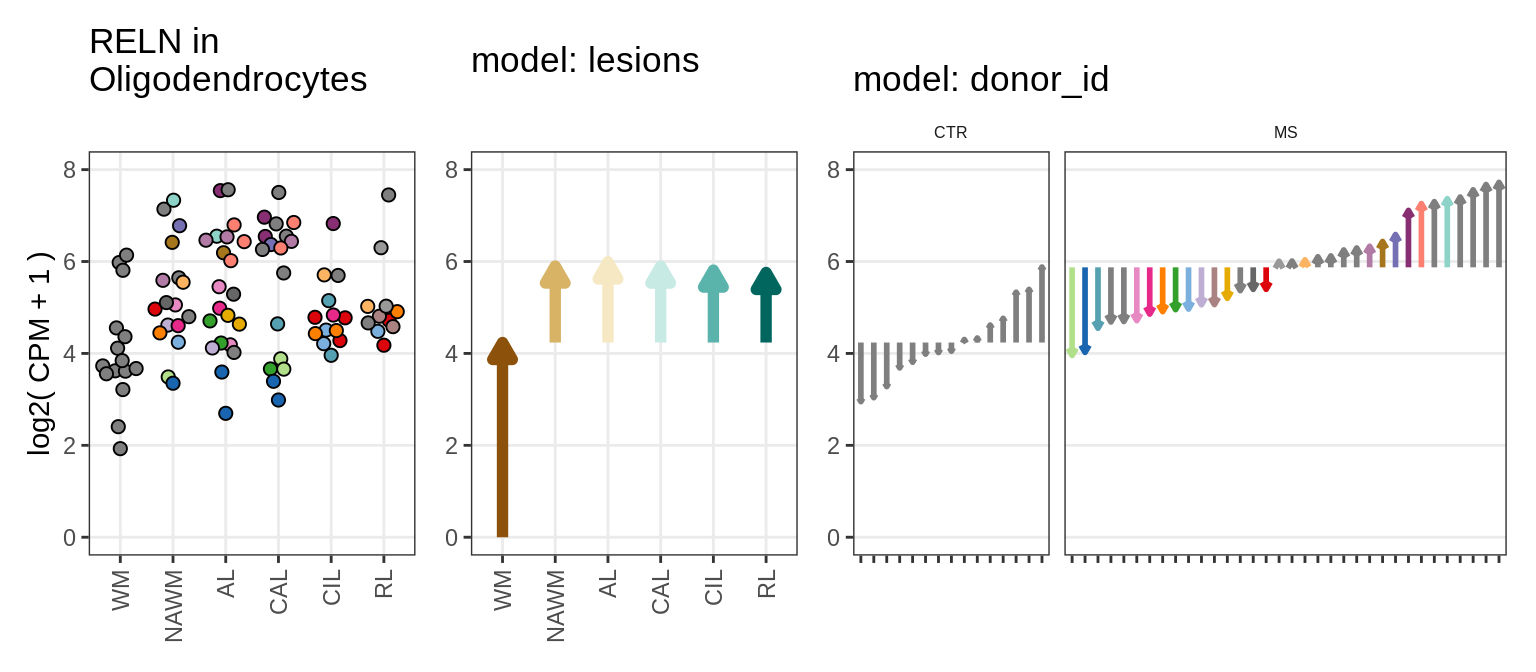
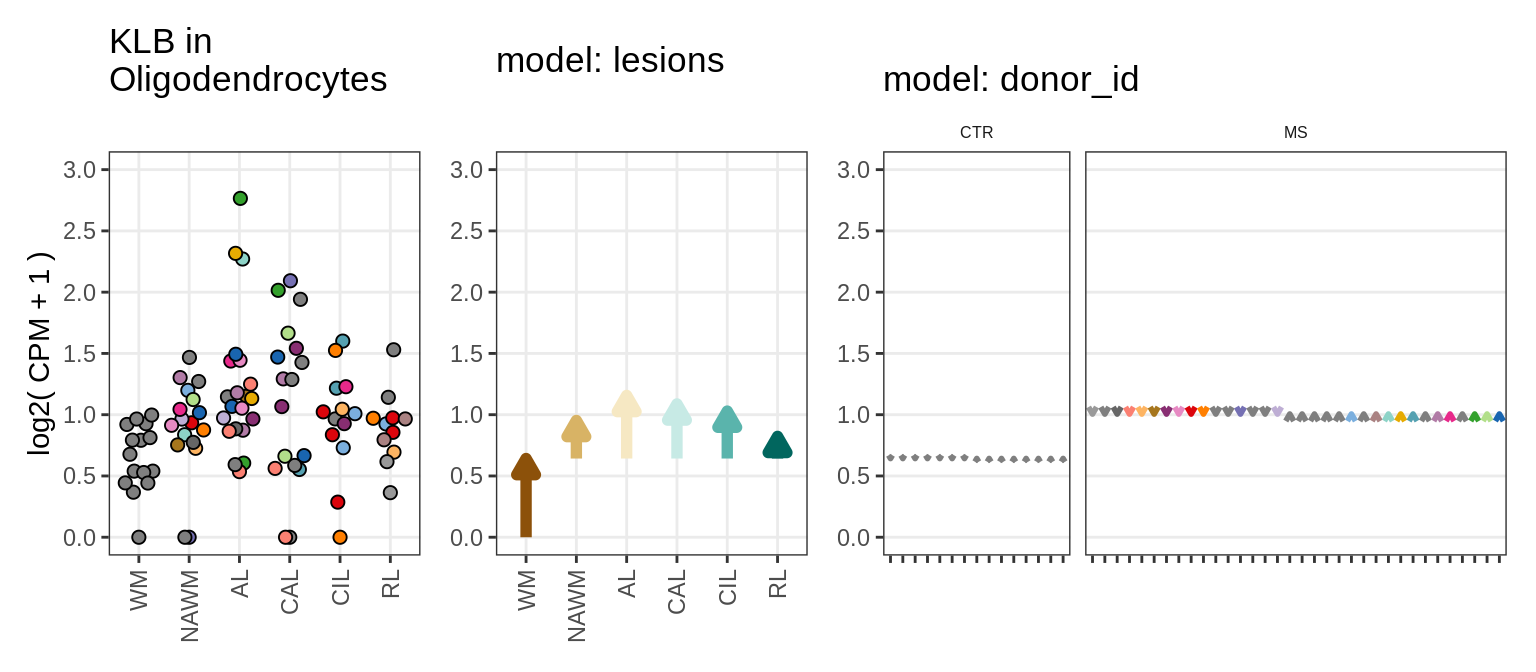
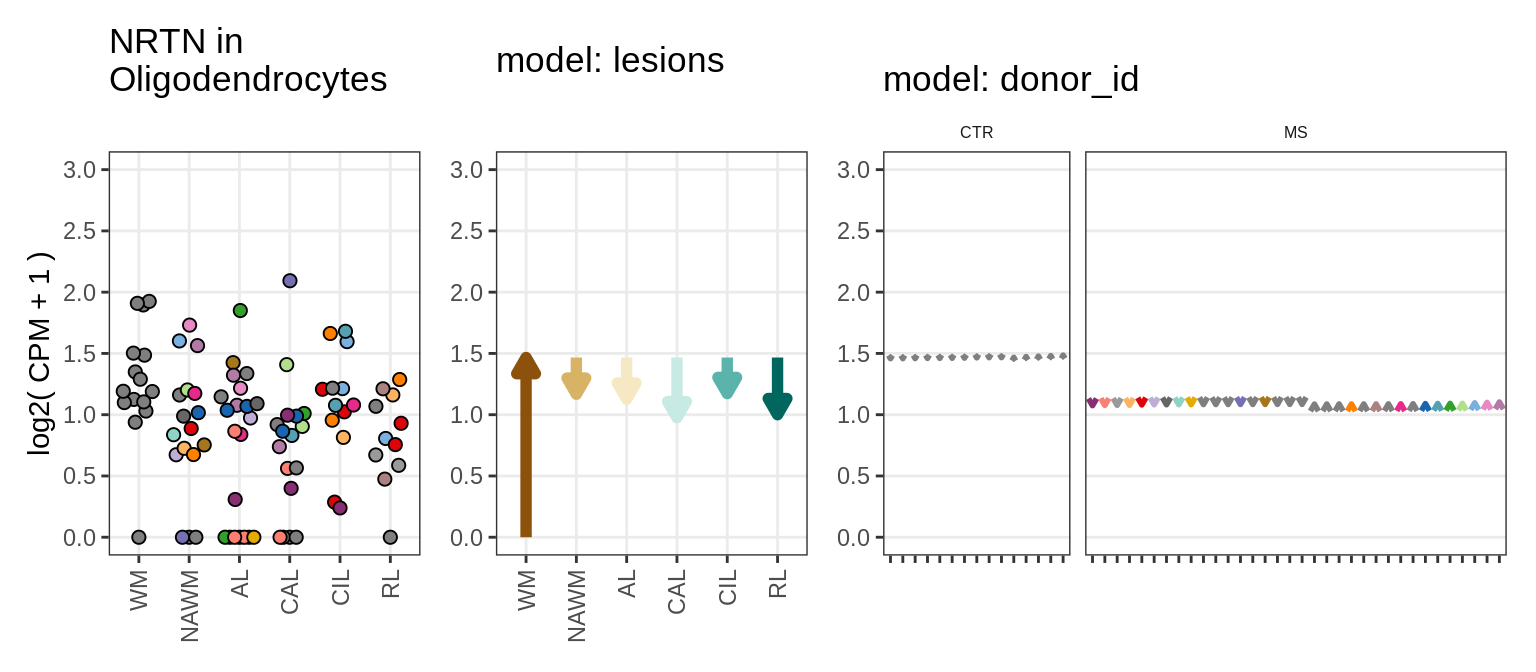
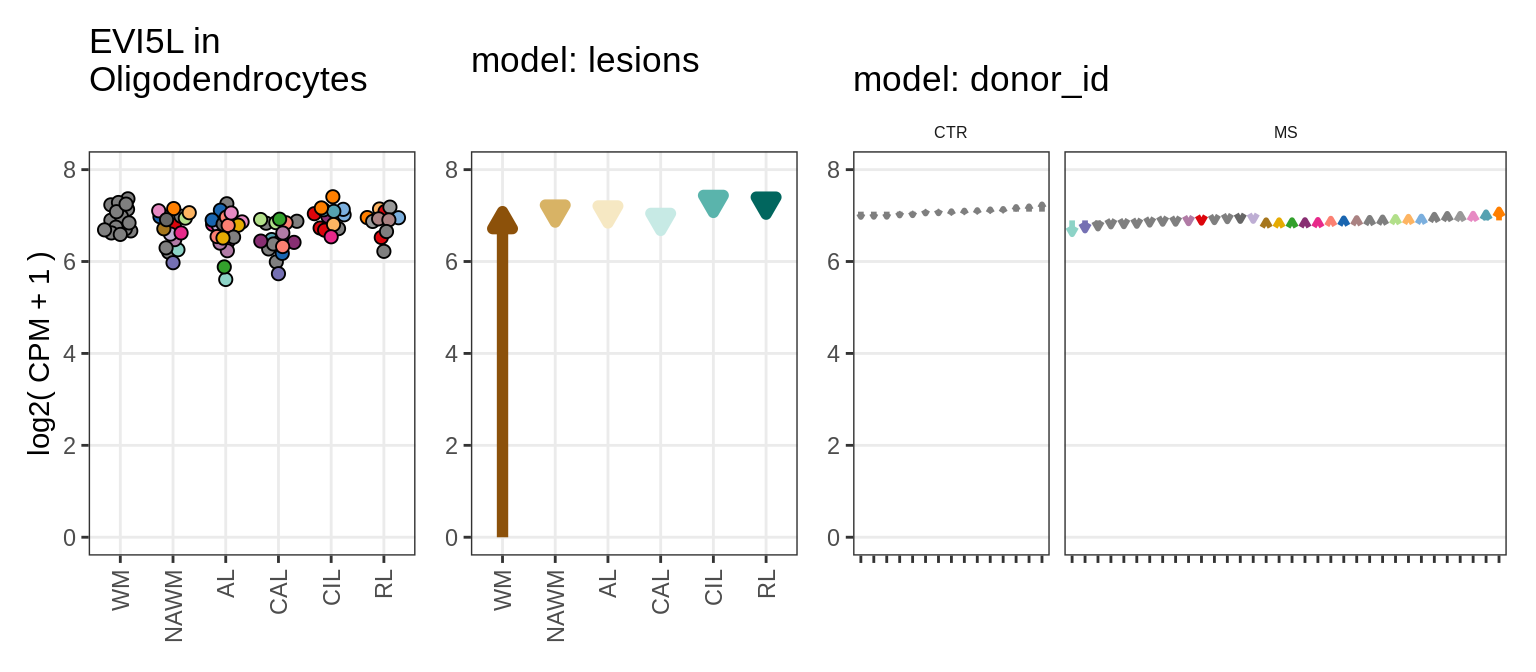
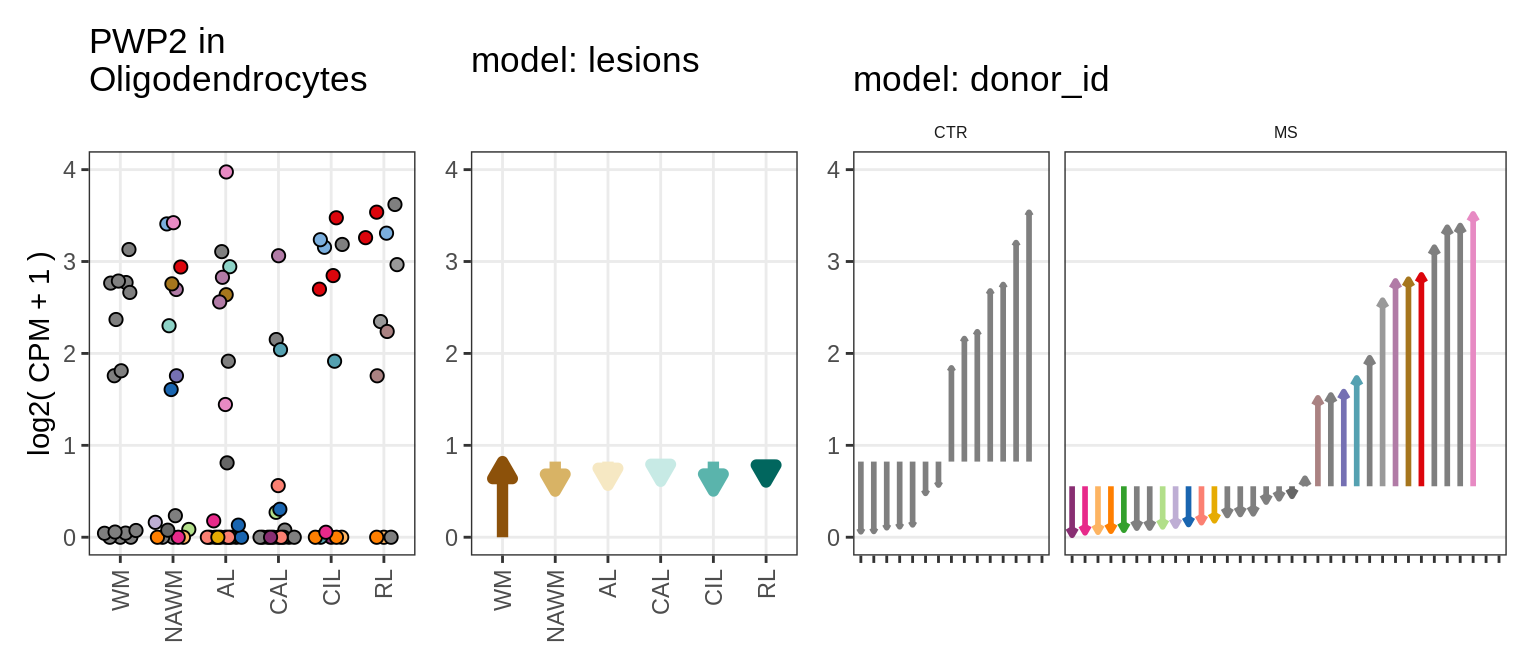
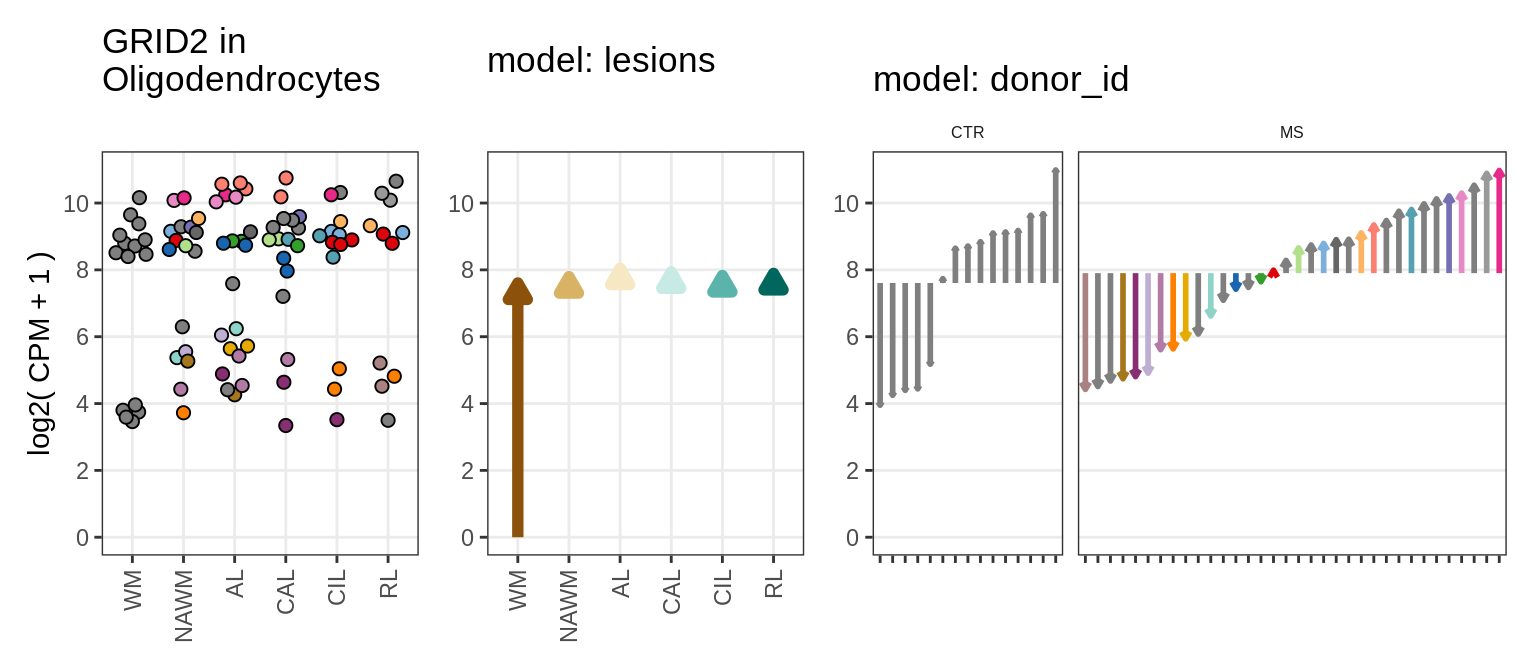
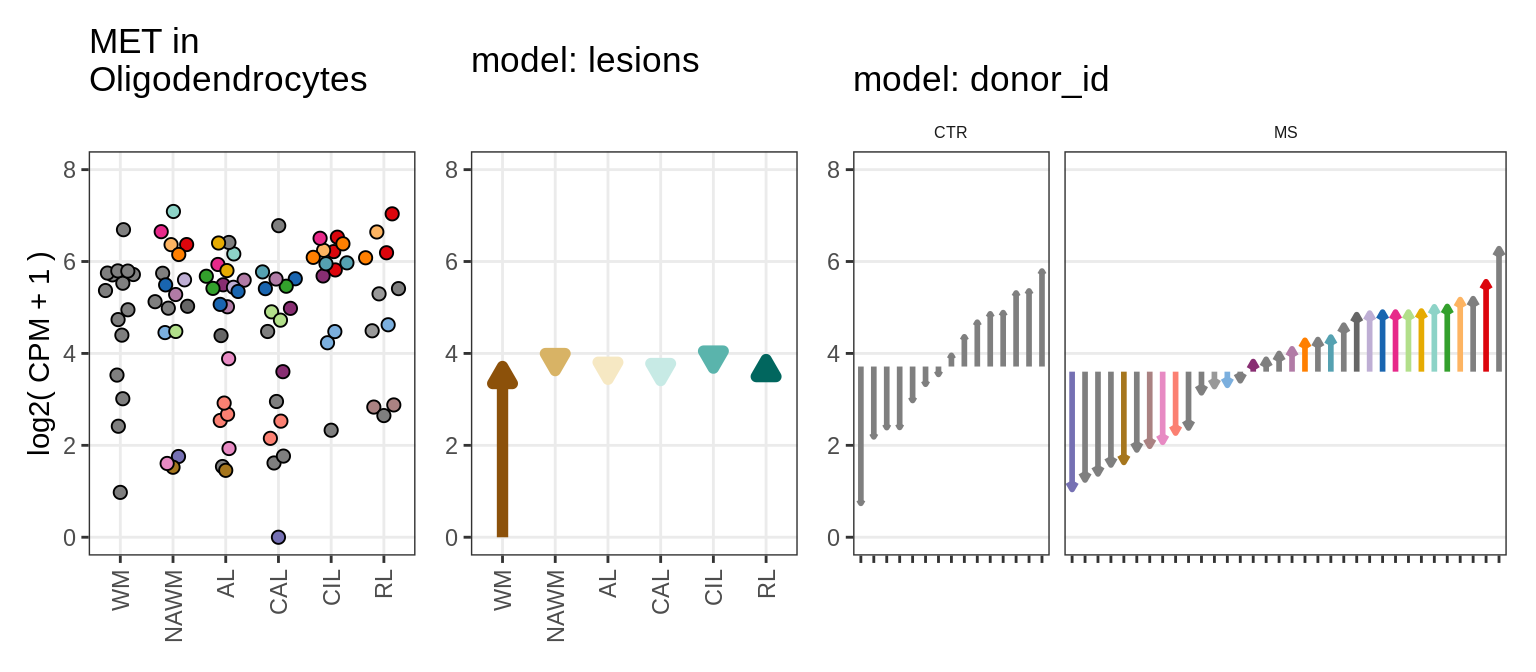
muscat results vs SD
(plot_muscat_vs_sd_min(res_dt, sd_dt, sel_cl, min_sd, max_p))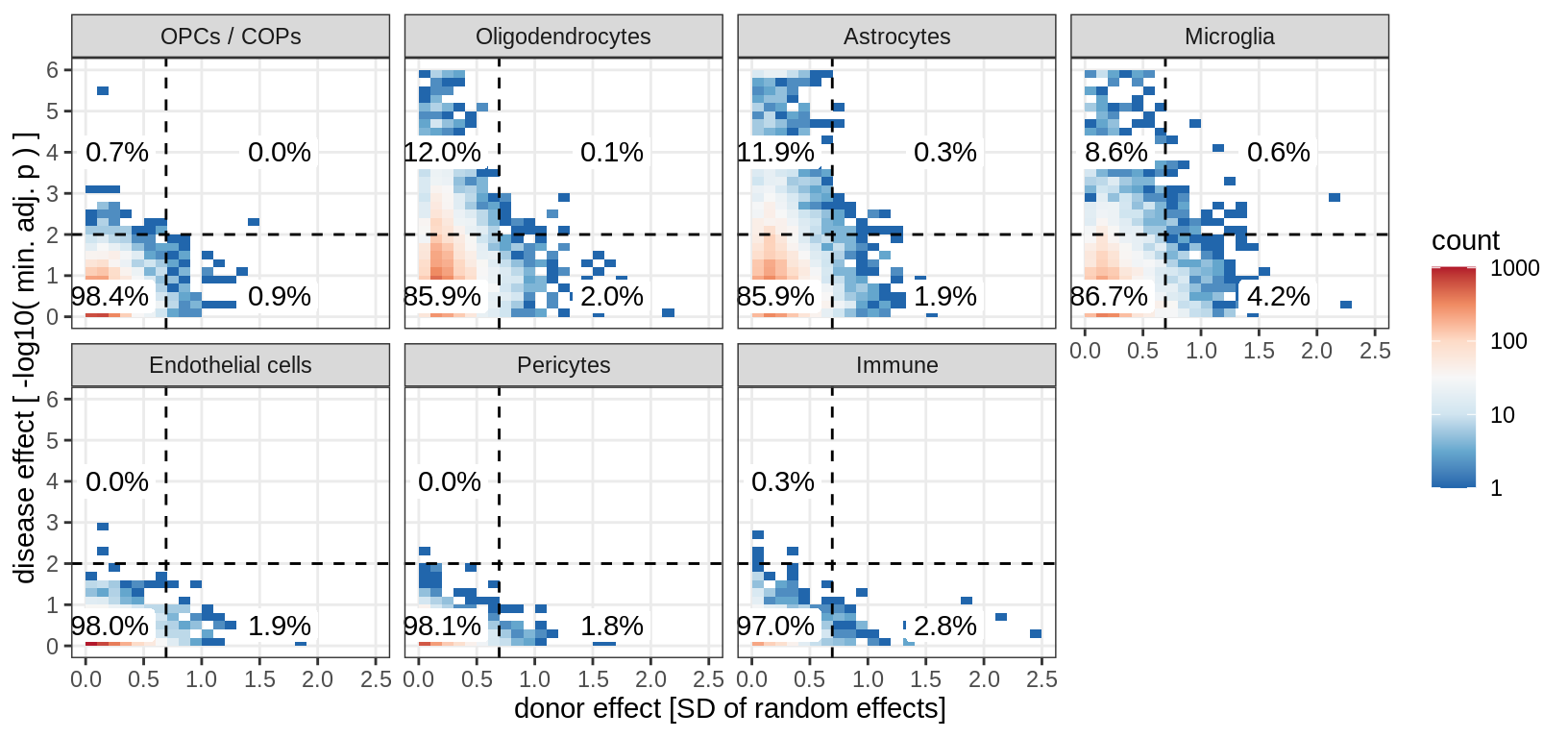
muscat results vs SD, MAGMA hits only
magma_hits = magma_dt[ p_magma_adj < 0.1 ]$gene_id
(plot_muscat_vs_sd_min(res_dt[ gene_id %in% magma_hits ], sd_dt,
sel_cl, min_sd, max_p))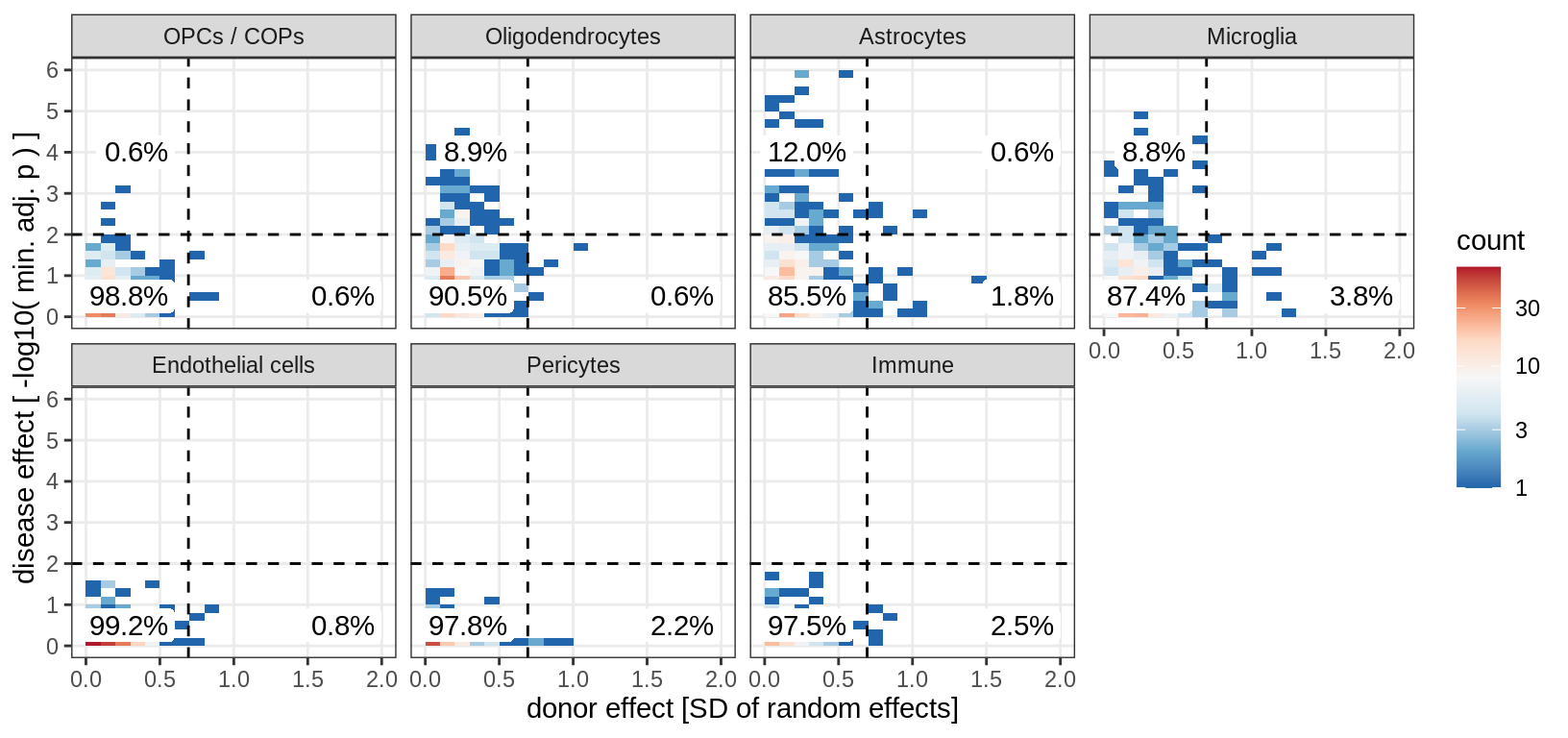
muscat results vs LoFs
(plot_muscat_and_sd_vs_lof(res_dt, sd_dt, lof_dt, sel_cl))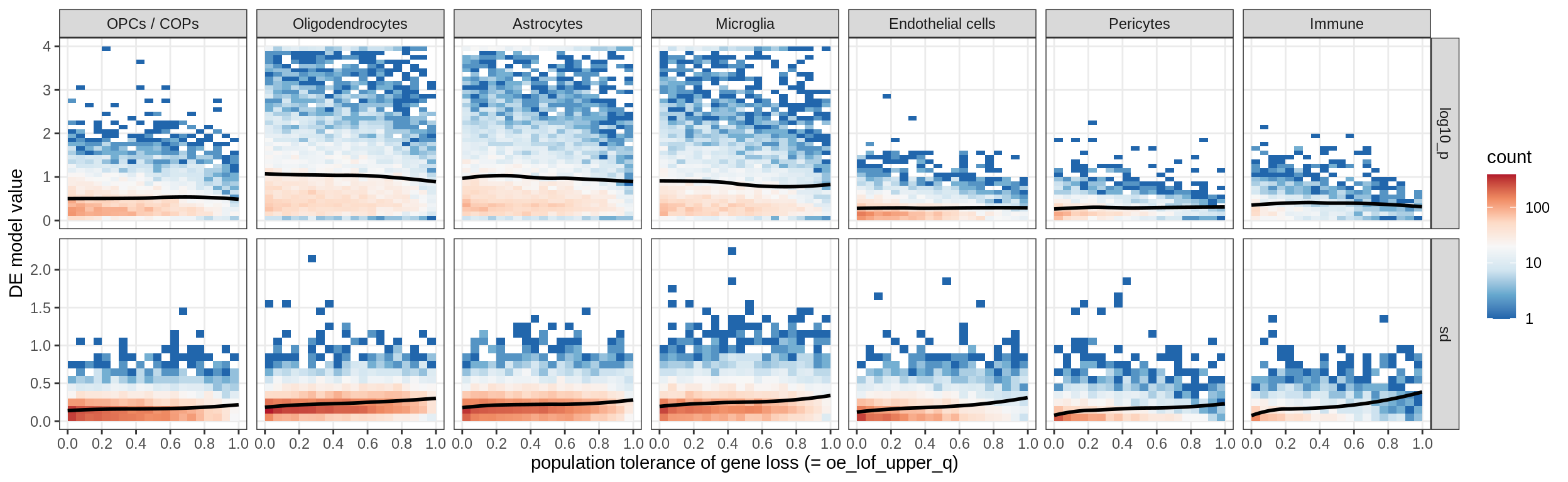
Expression heatmap
draw(plot_expression_heatmap_samples(pb, filtered_dt, annots_dt, sel_cl))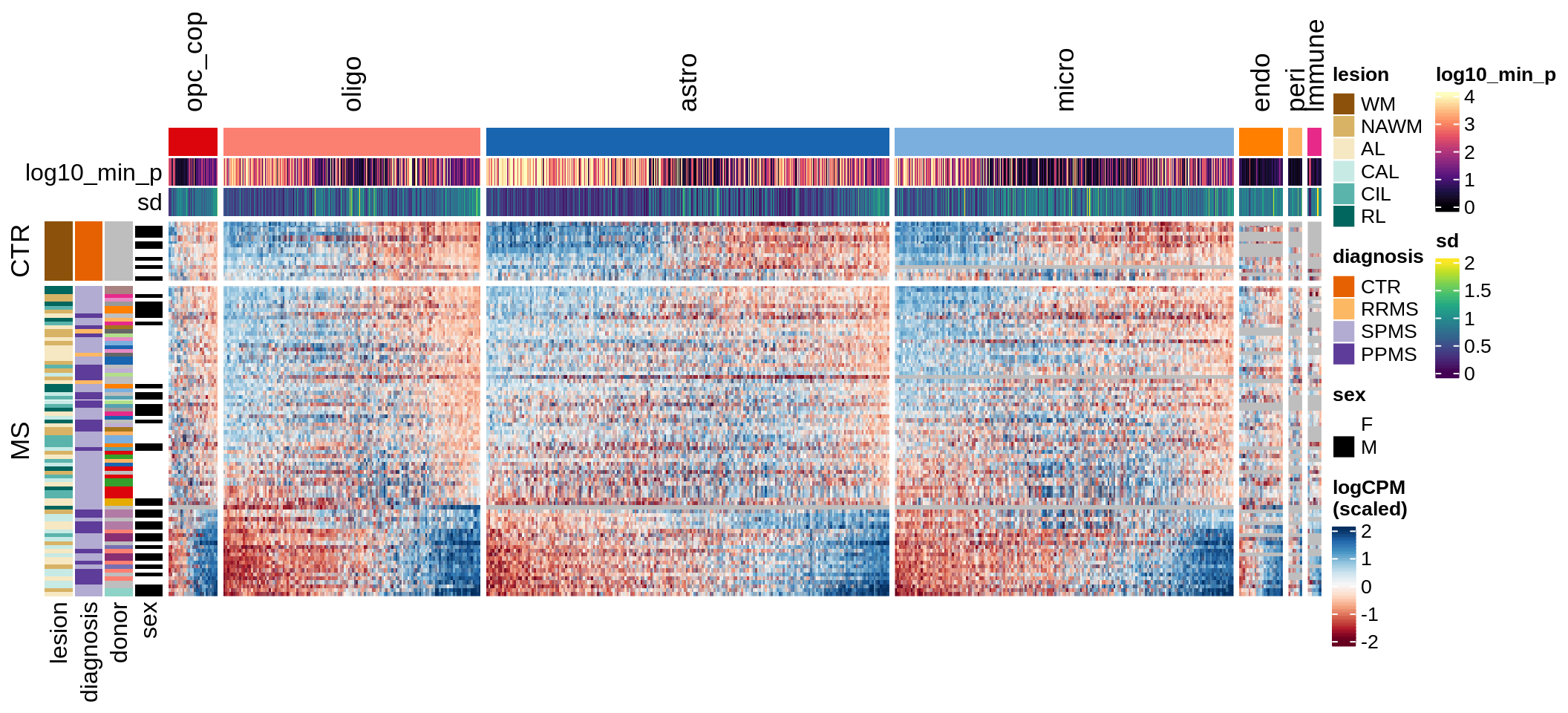
MOFA+ factors - diagnosis
(plot_factors_univariate(model, annots_dt, pb, by = 'diagnosis'))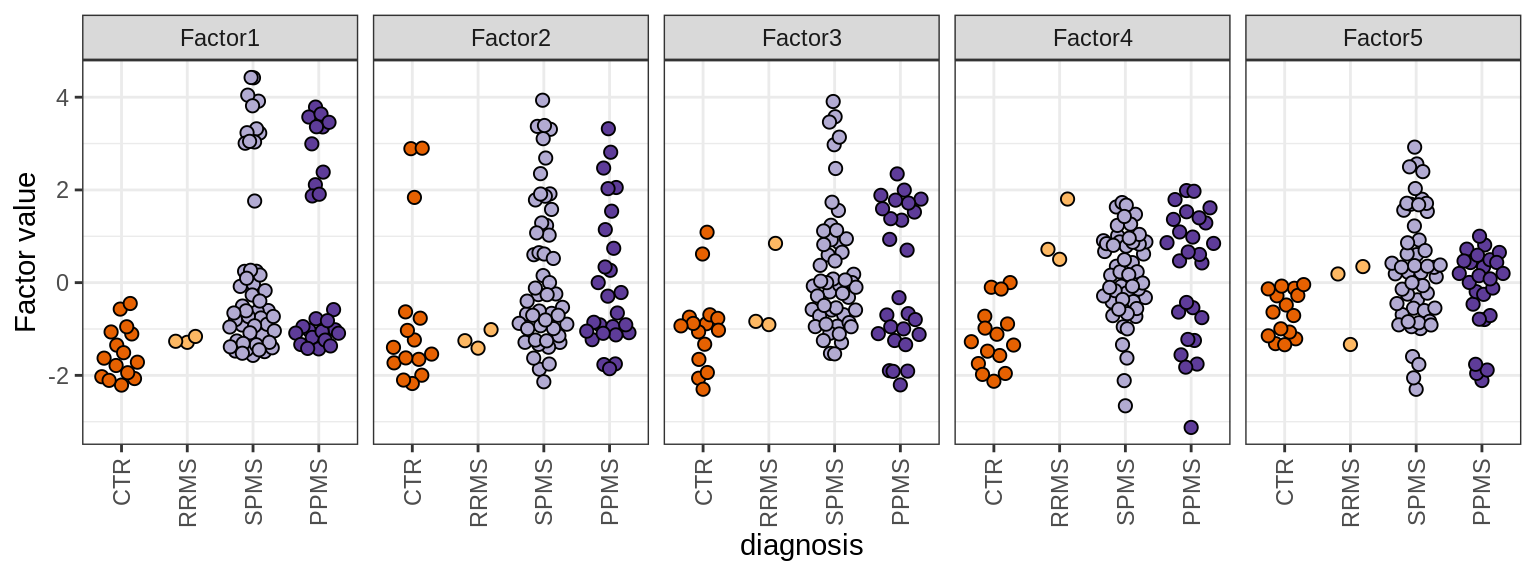
| Version | Author | Date |
|---|---|---|
| 47af3ff | Macnair | 2021-10-18 |
MOFA+ factors - lesions
(plot_factors_univariate(model, annots_dt, pb, by = 'lesion_type'))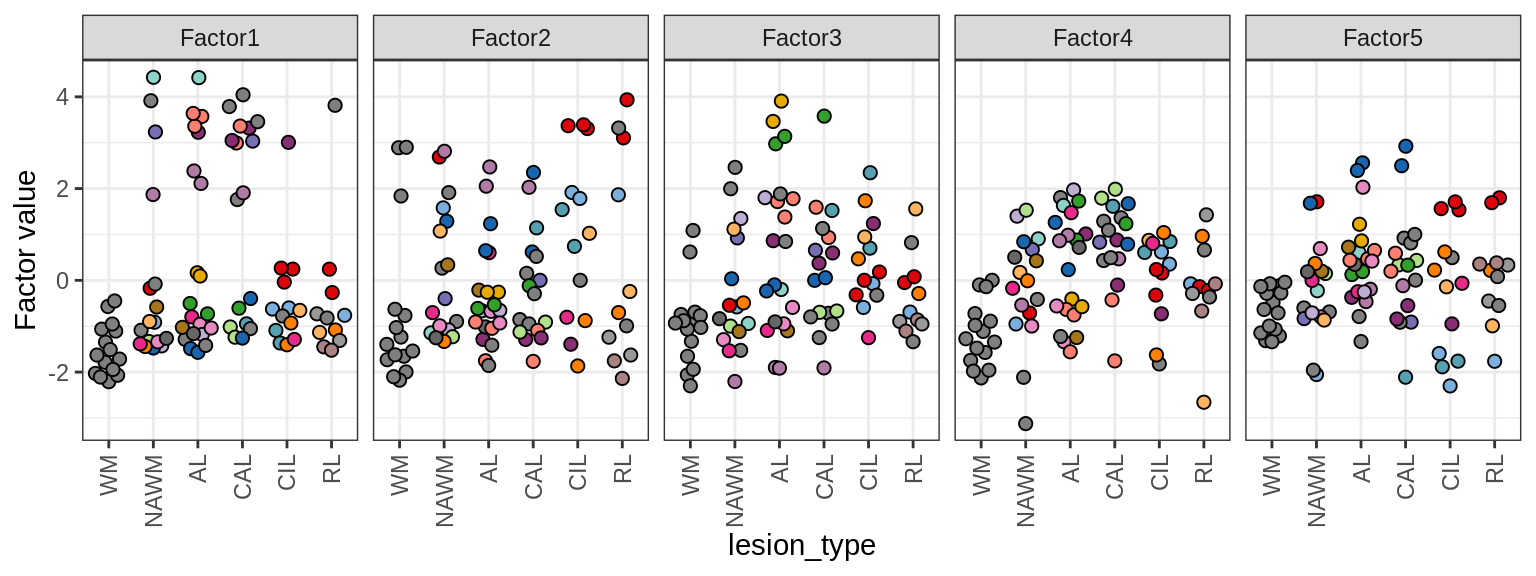
| Version | Author | Date |
|---|---|---|
| 47af3ff | Macnair | 2021-10-18 |
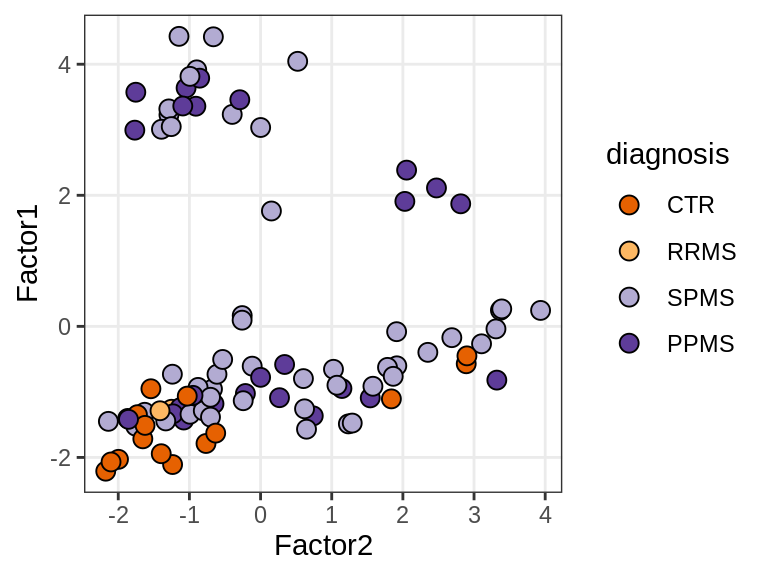
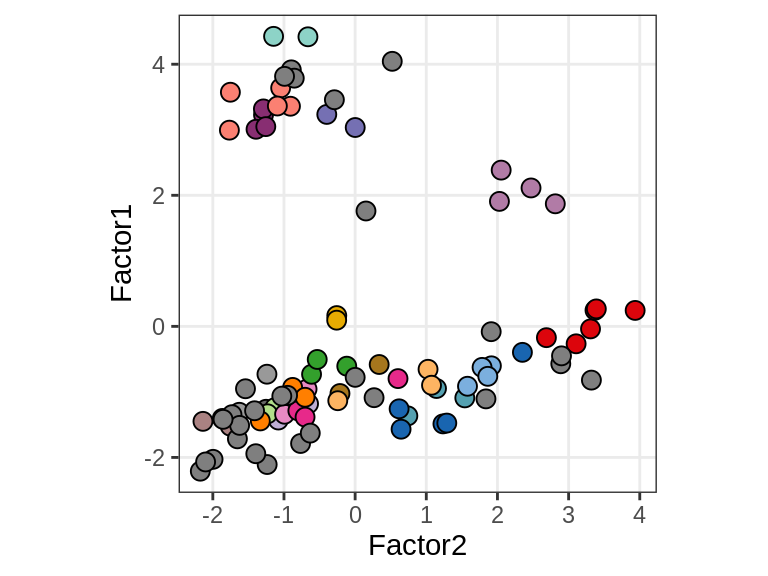
Factor 1 vs Factor 2
for (what in c("diagnosis", "lesion_type", "subject_id")) {
cat('### ', what, '\n', sep = '')
print(plot_factors_pair(model, annots_dt, pb,
f_pair = c("Factor2", "Factor1"), by = what))
cat('\n\n')
}
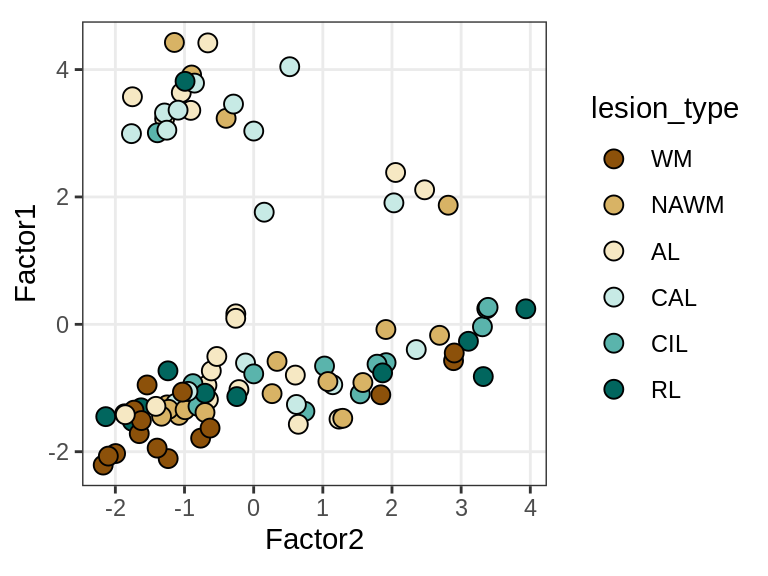
Interactions between factors and model components
(plot_factor_r2s(r2_dt, var_exp_dt))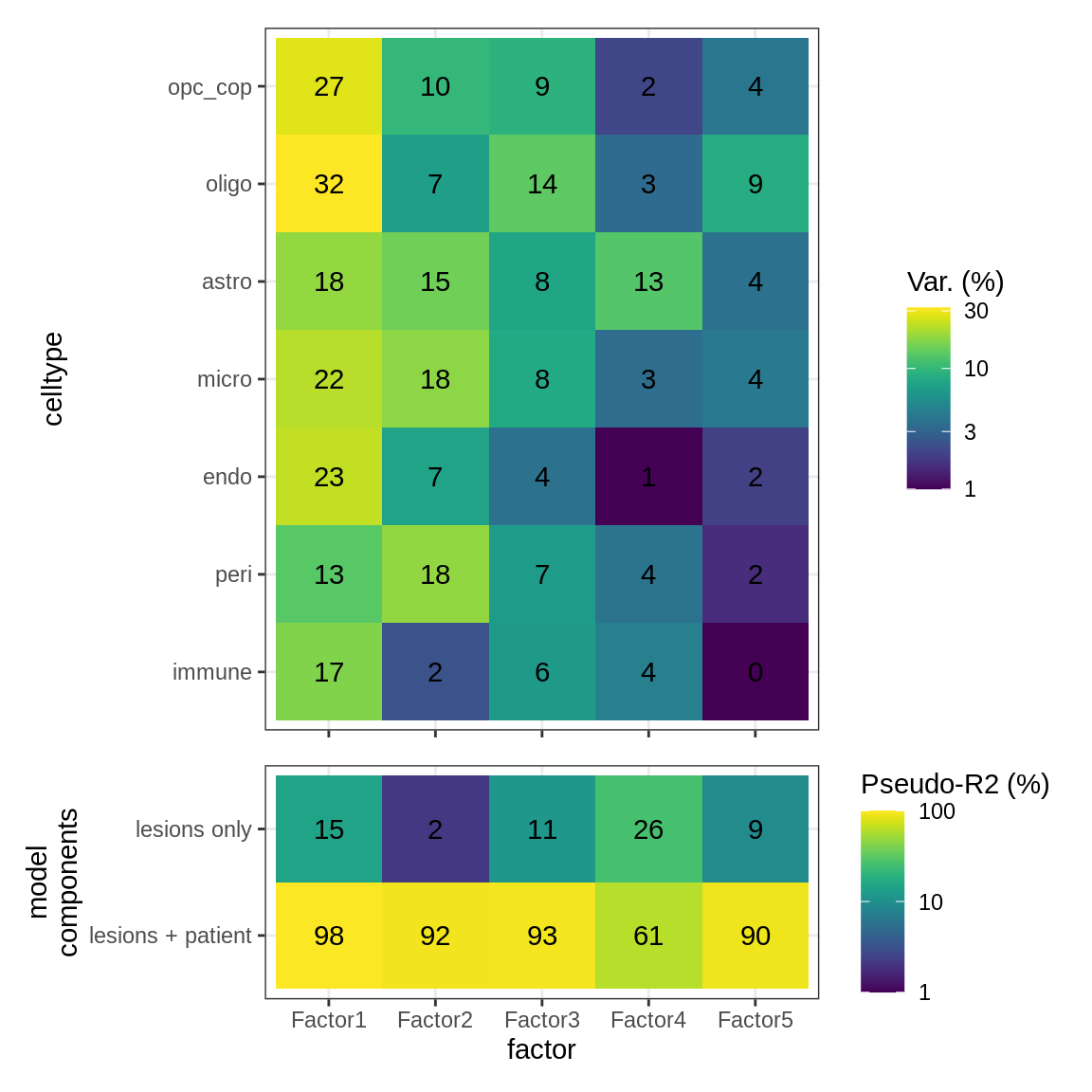
GO terms for factors
print(plot_mofa_gsea_dotplot(gsea_list[['go_bp']], labels_dt,
fgsea_cut = fgsea_cut, n_total = 50))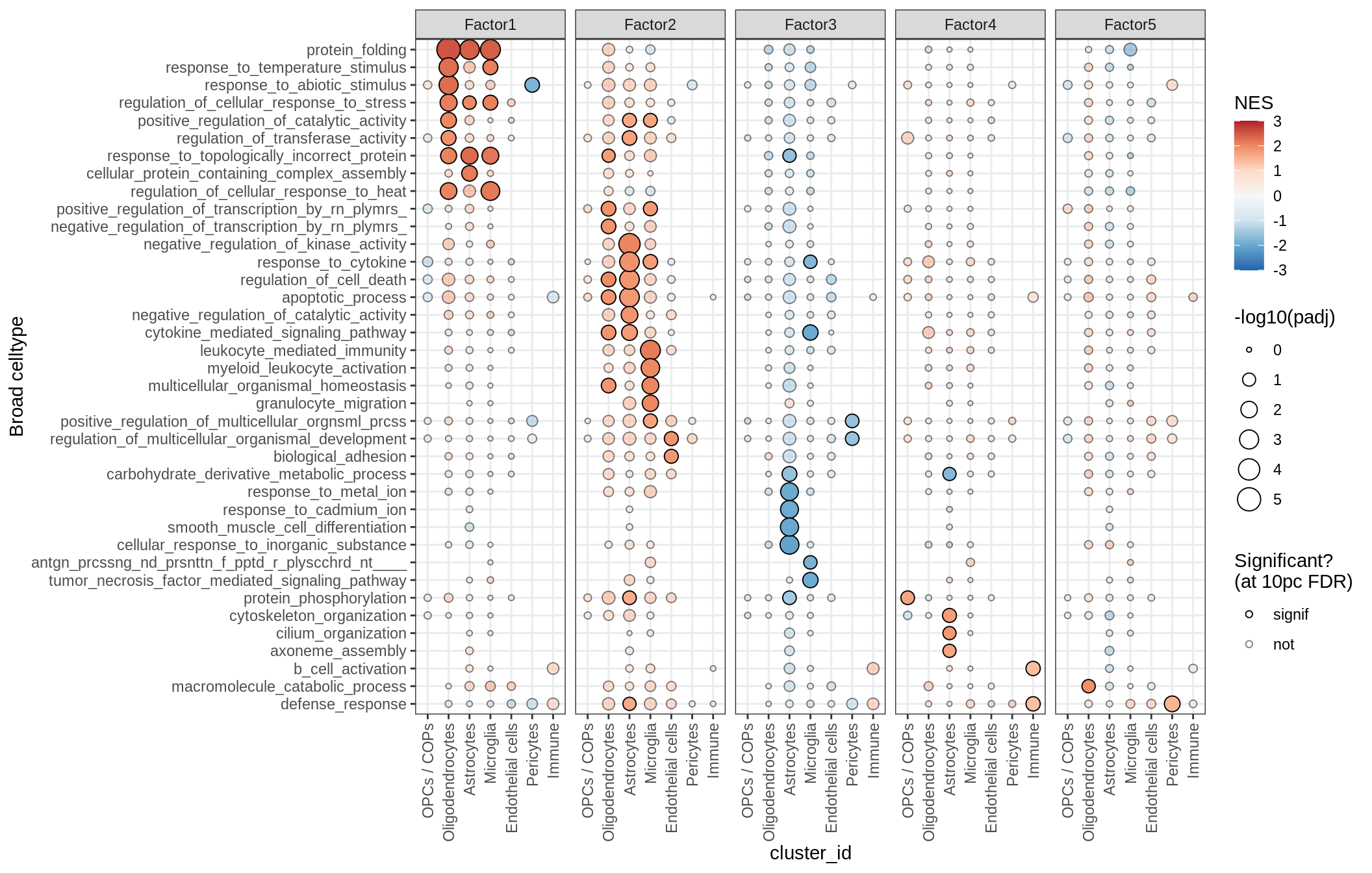
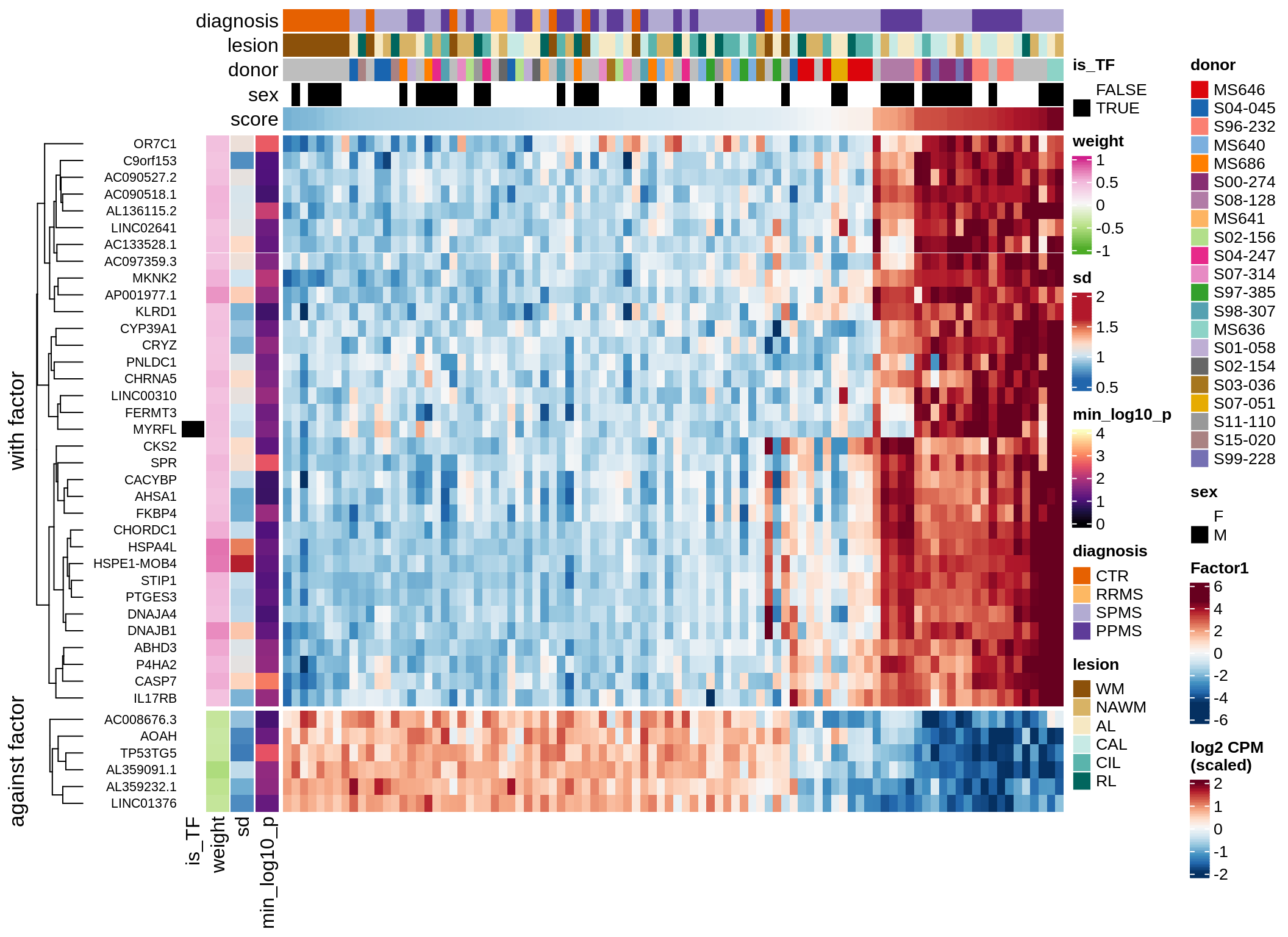
Top genes for Factor 1
draw( plot_top_genes_expression(model, pb, annots_dt,
filter_dt, tfs_dt, var_exp_dt,
sel_f = 'Factor1', min_var = 10, min_w = 0.2, n_top = 10) )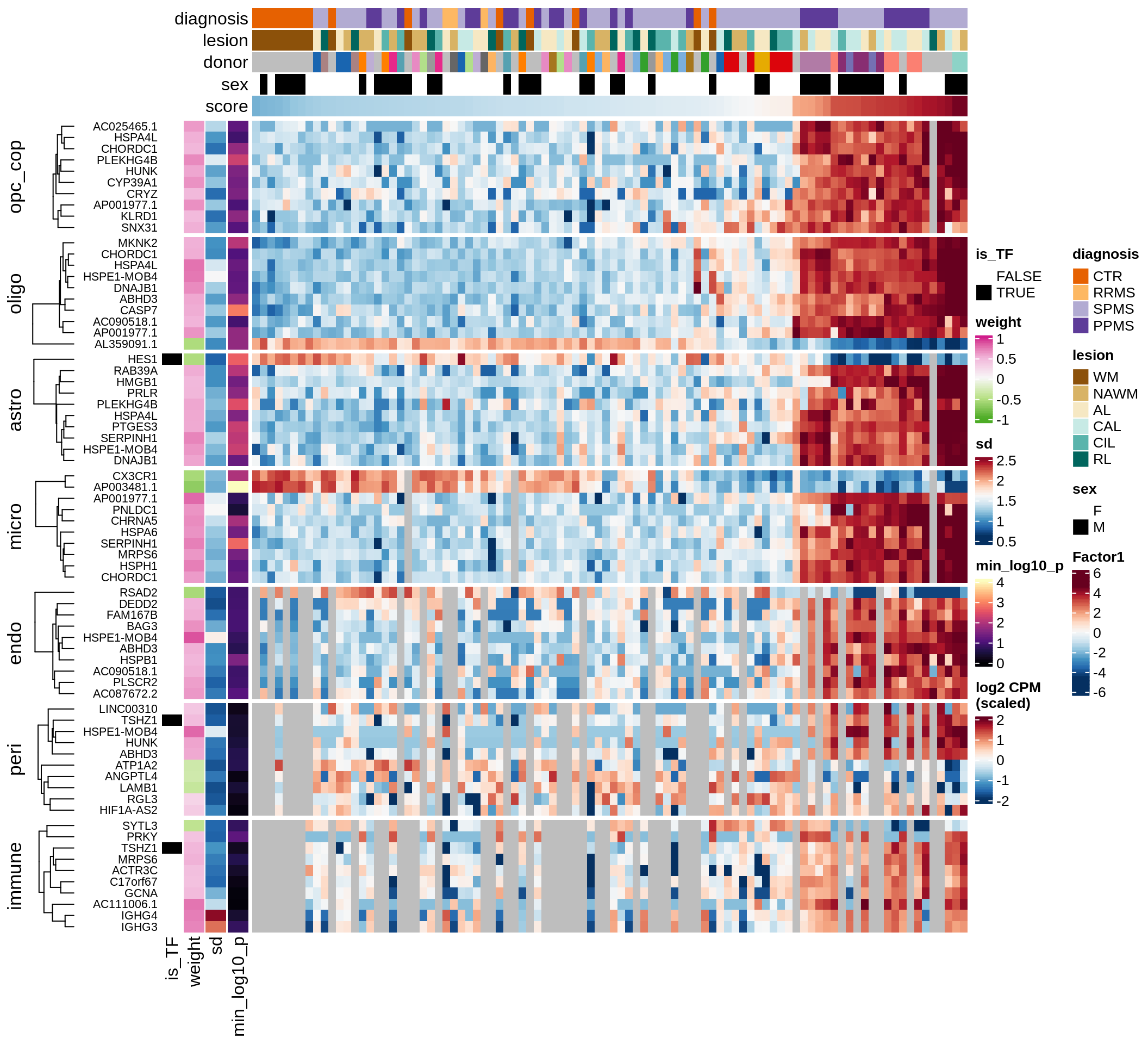
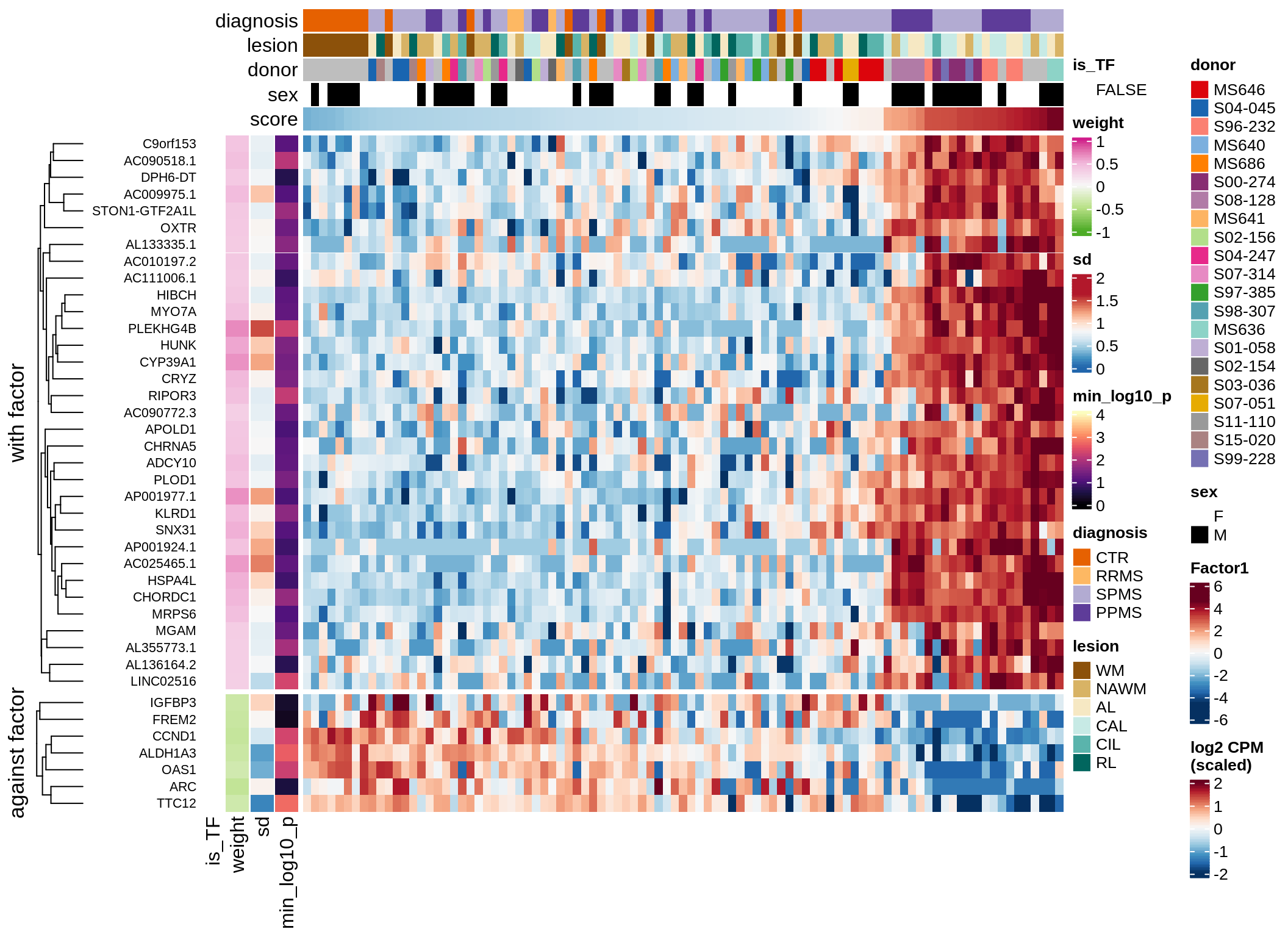
Top genes for Factor 2
draw( plot_top_genes_expression(model, pb, annots_dt,
filter_dt, tfs_dt, var_exp_dt,
sel_f = 'Factor2', min_var = 10, min_w = 0.2, n_top = 10) )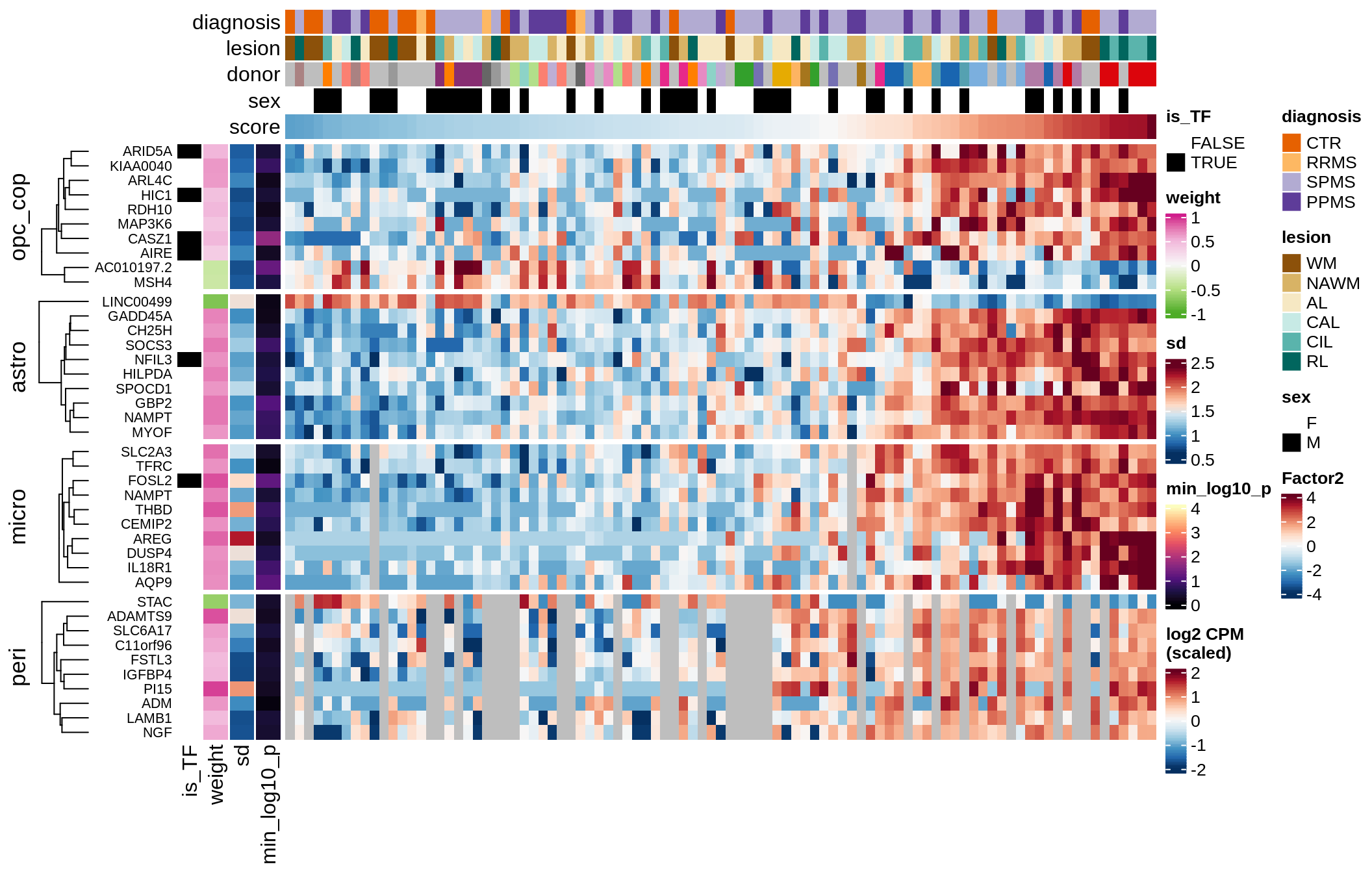
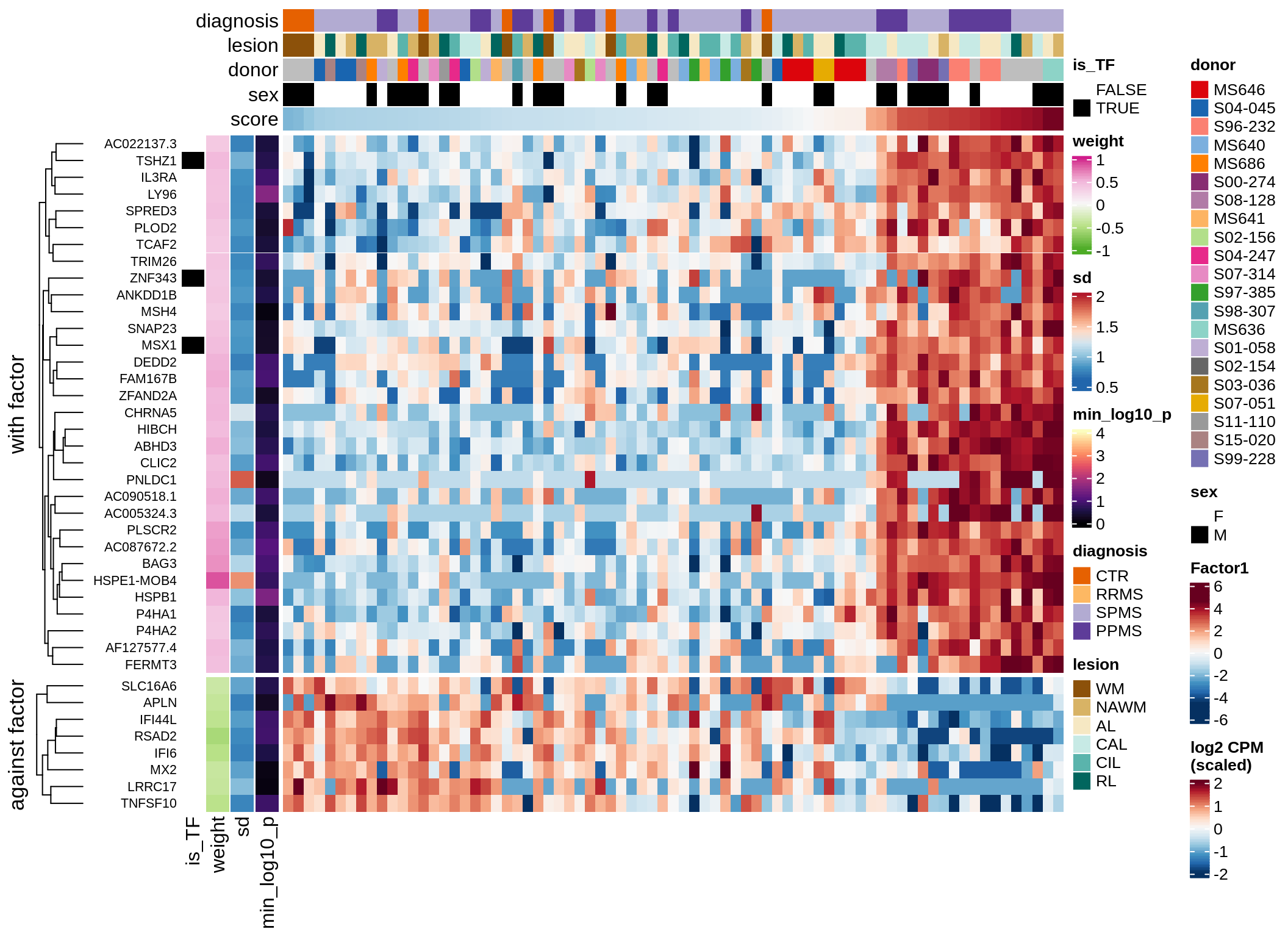
Top genes for Factor 3
draw( plot_top_genes_expression(model, pb, annots_dt,
filter_dt, tfs_dt, var_exp_dt,
sel_f = 'Factor3', min_var = 5, min_w = 0.2, n_top = 10) )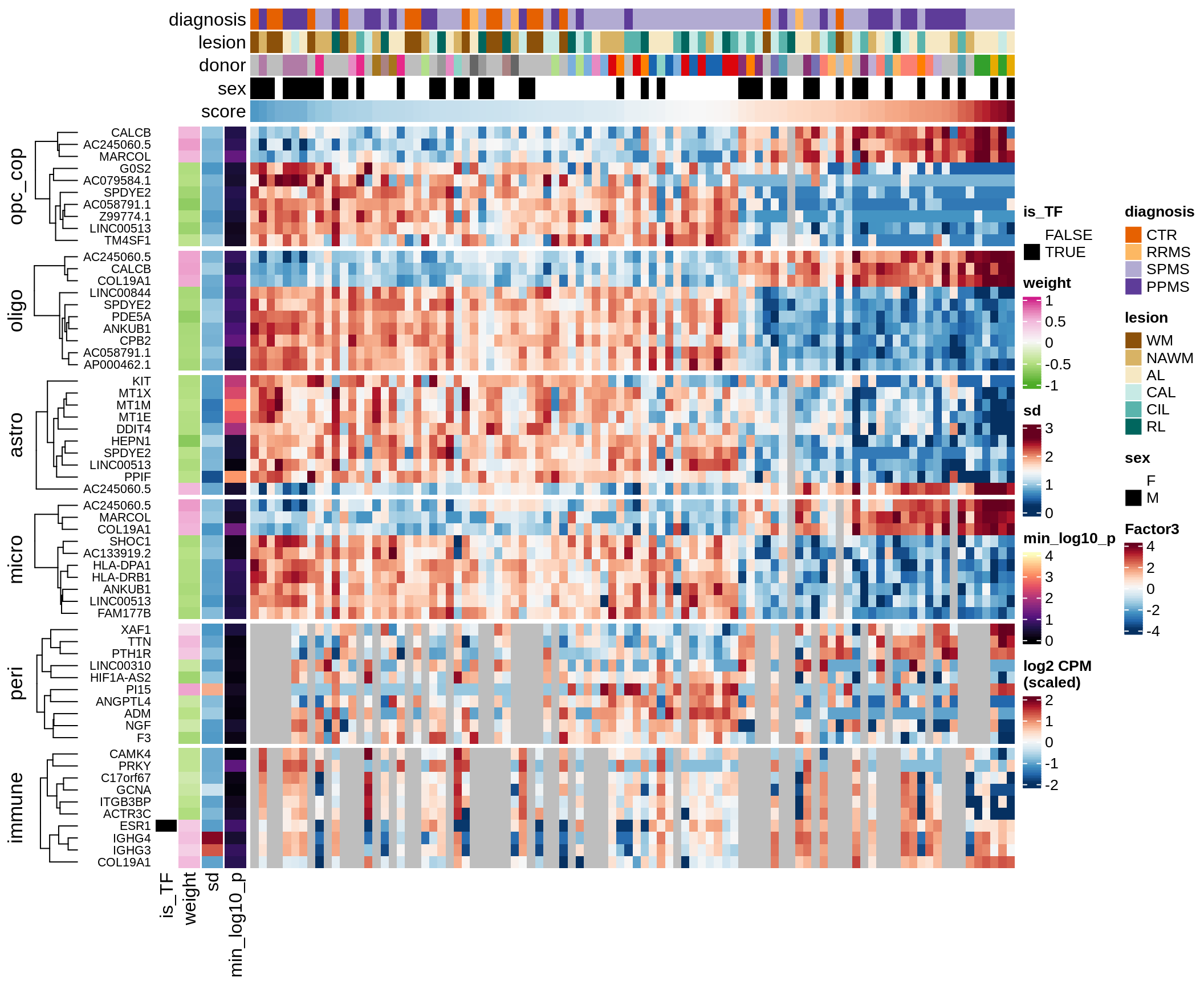
Top genes for Factor 4
draw( plot_top_genes_expression(model, pb, annots_dt,
filter_dt, tfs_dt, var_exp_dt,
sel_f = 'Factor4', min_var = 5, min_w = 0.2, n_top = 20) )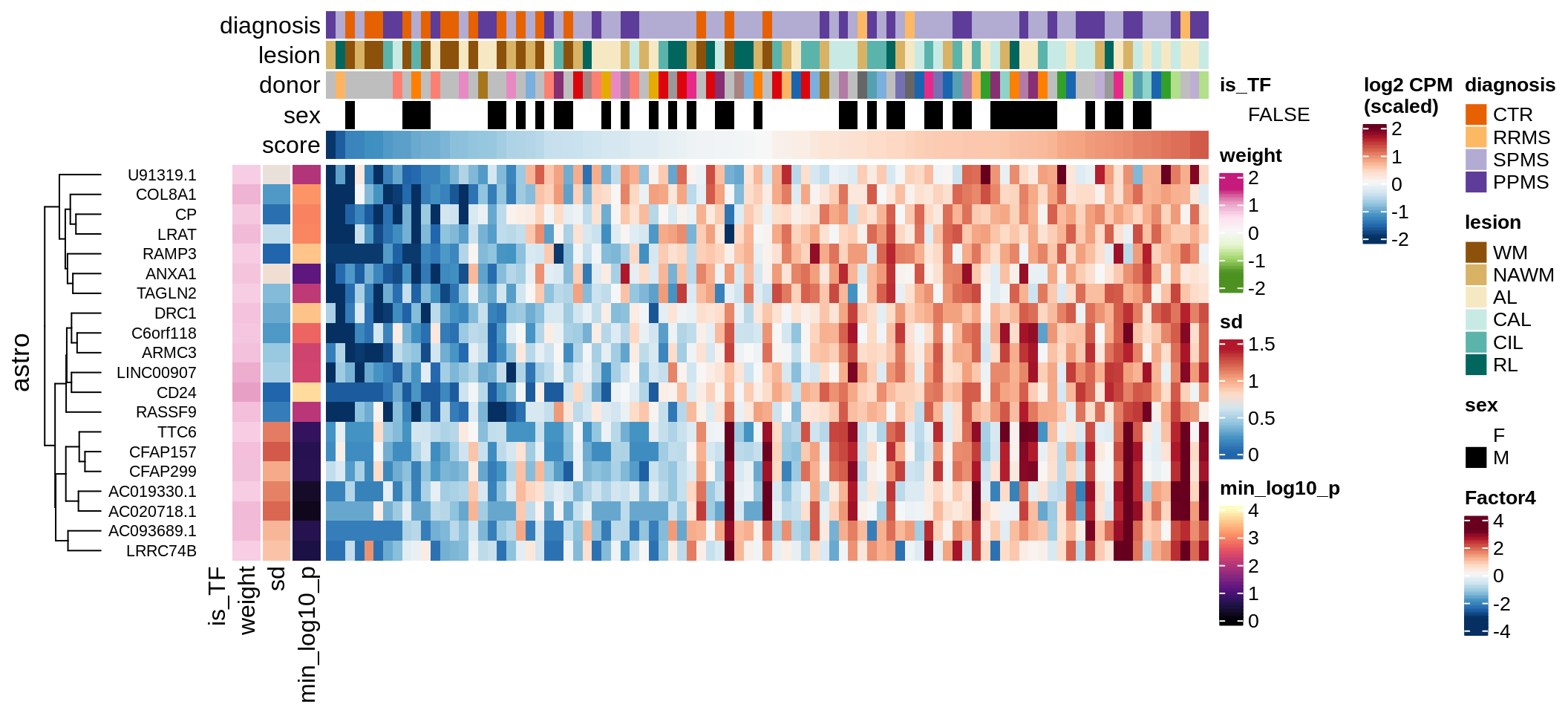
Top genes for Factor 5
draw( plot_top_genes_expression(model, pb, annots_dt,
filter_dt, tfs_dt, var_exp_dt,
sel_f = 'Factor5', min_var = 5, min_w = 0.2, n_top = 20) )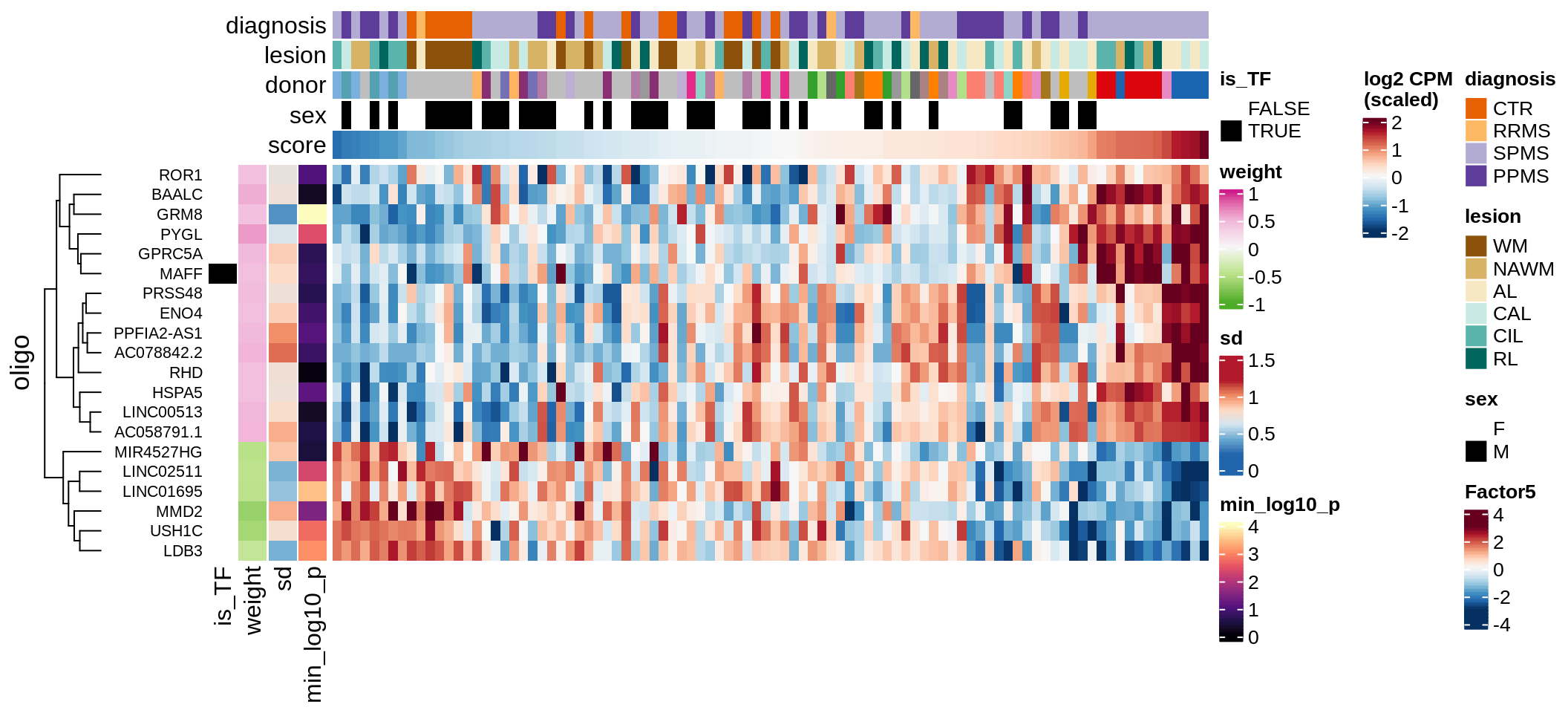
3D plots under duress
combn1
plot_factors_3d(model, annots_dt, sel_fs = c('Factor1', 'Factor2', 'Factor3'),
annot_v = 'oligo_grp')combn2
plot_factors_3d(model, annots_dt, sel_fs = c('Factor1', 'Factor5', 'Factor3'),
annot_v = 'oligo_grp')combn3
plot_factors_3d(model, annots_dt, sel_fs = c('Factor2', 'Factor5', 'Factor3'),
annot_v = 'oligo_grp')combn4
plot_factors_3d(model, annots_dt, sel_fs = c('Factor1', 'Factor2', 'Factor5'),
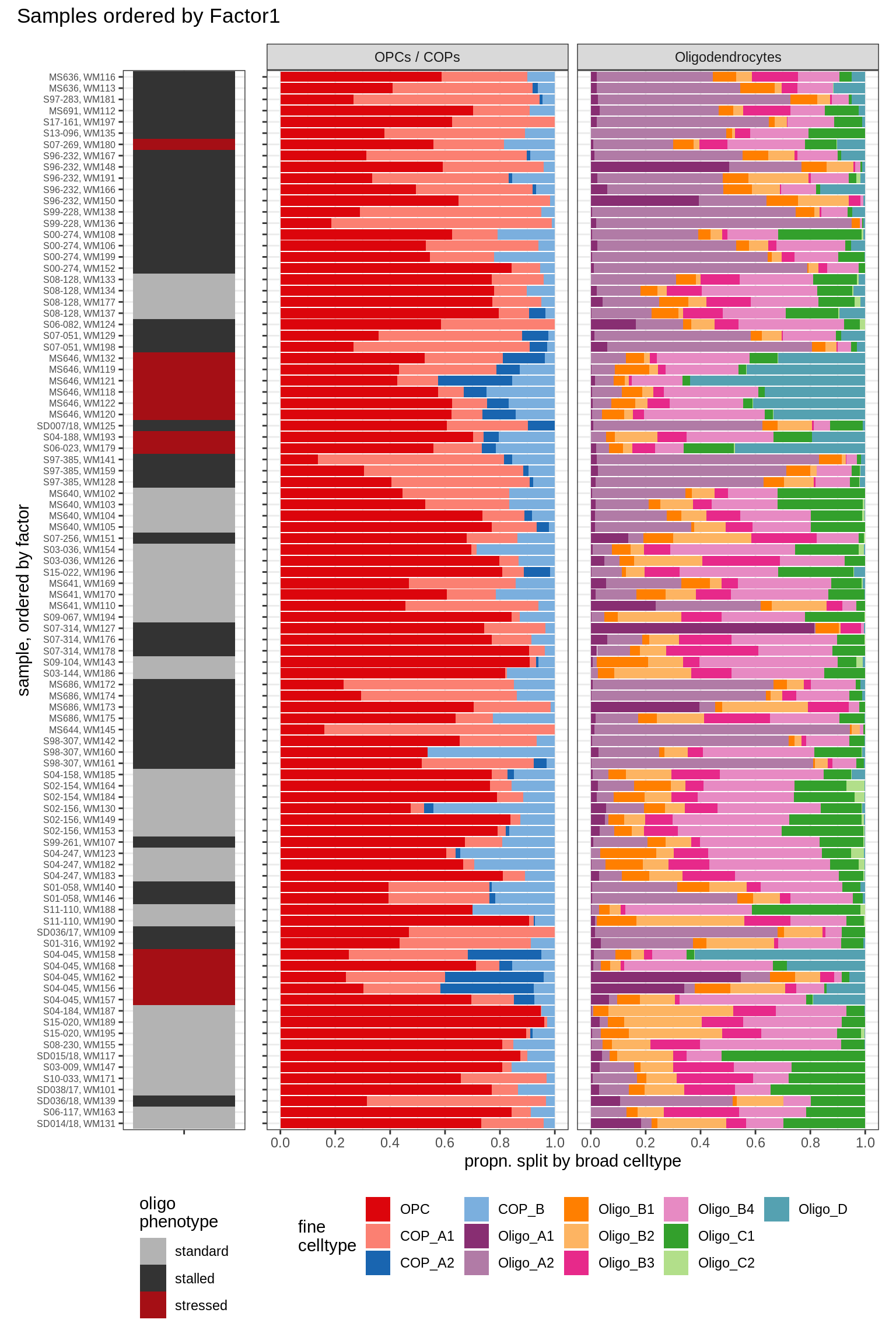
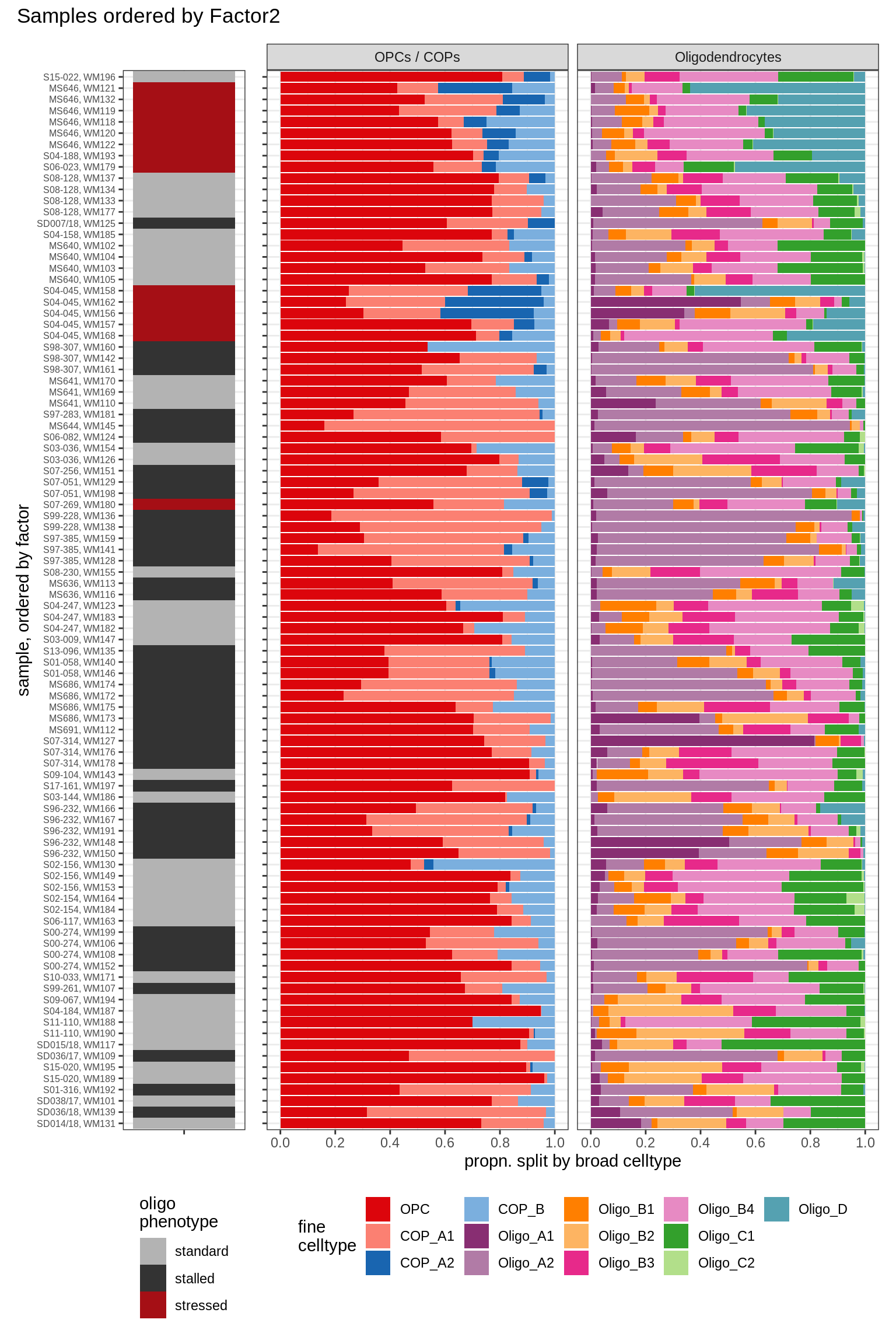
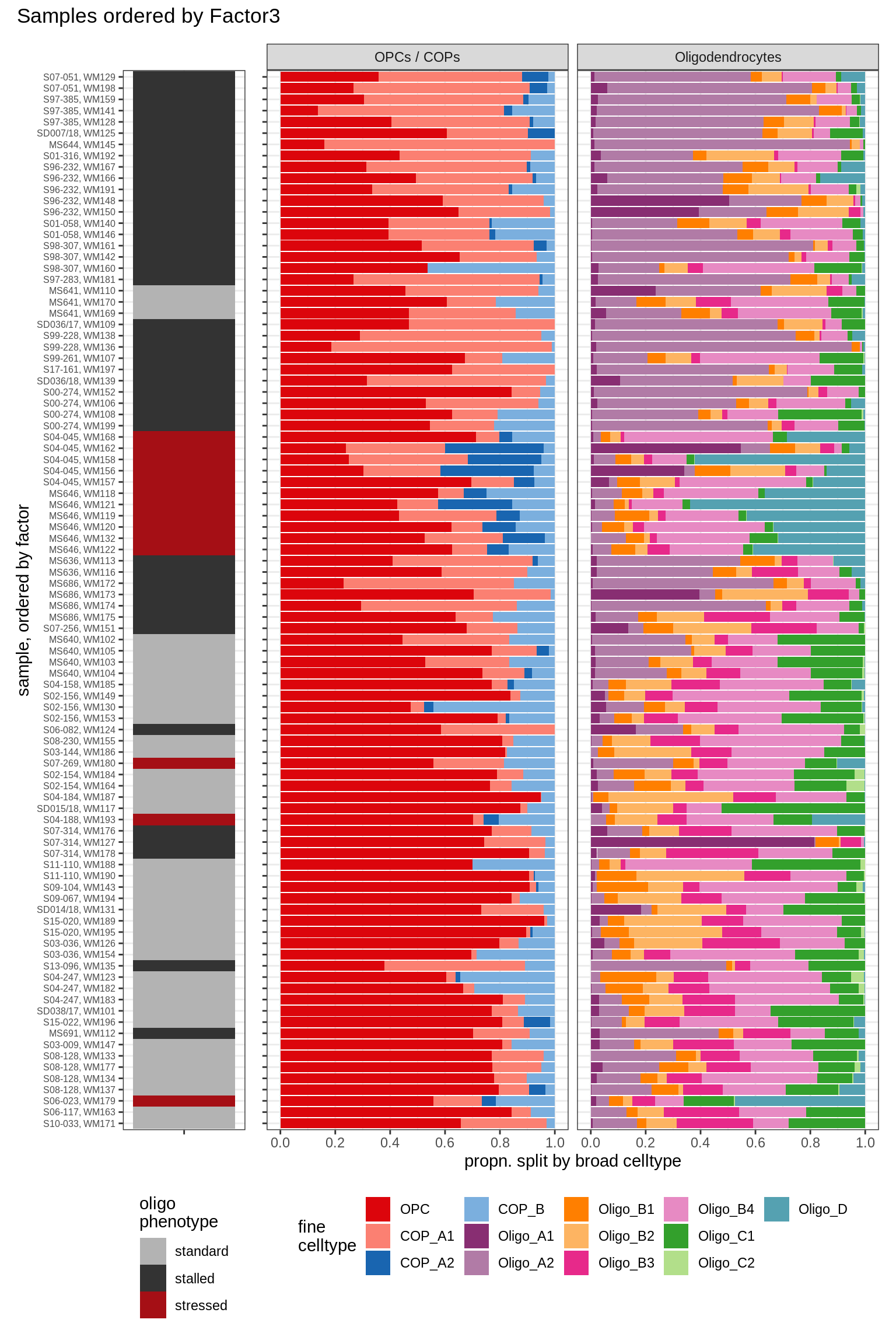
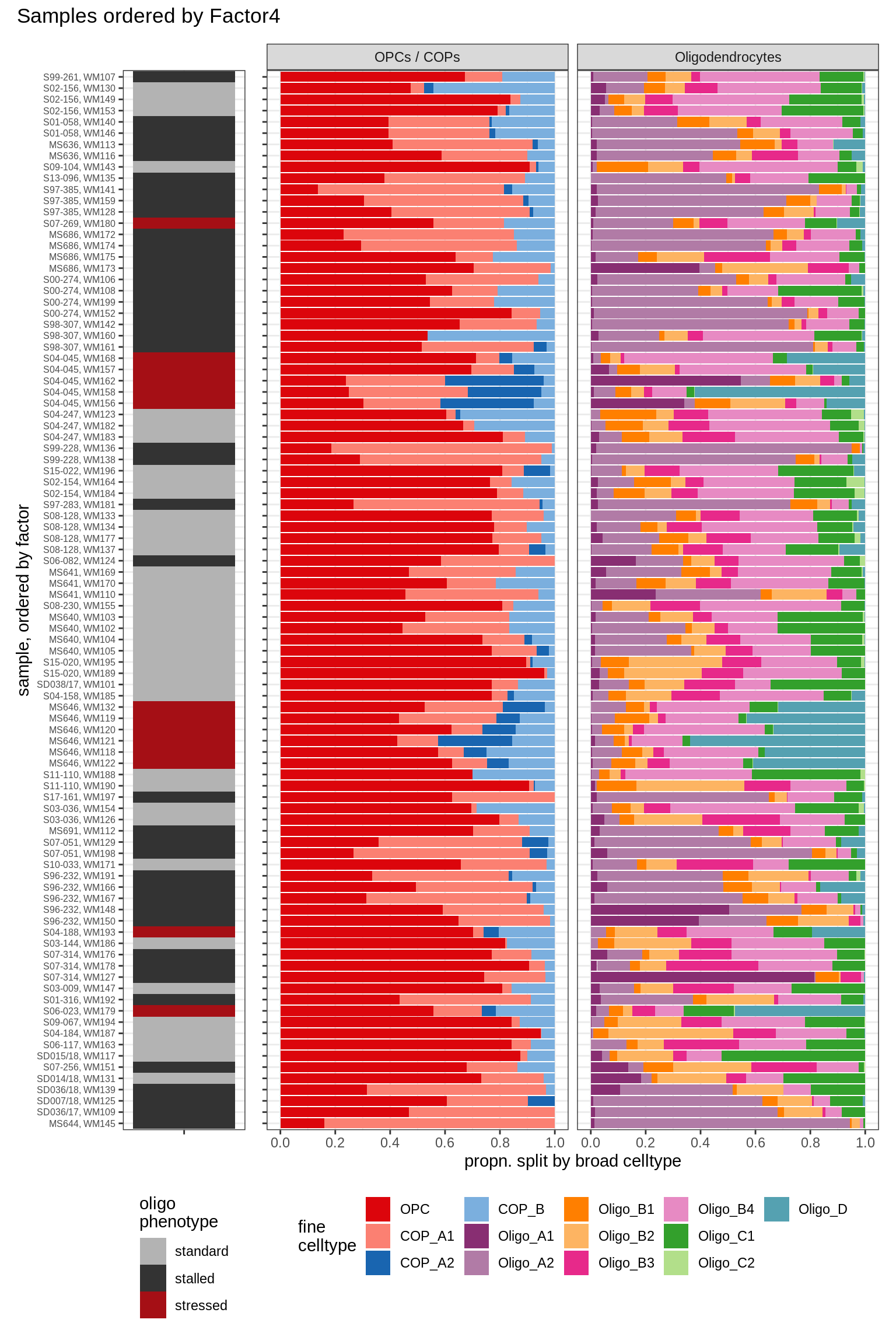
annot_v = 'oligo_grp')Barplots of celltype proportions ordered by factors
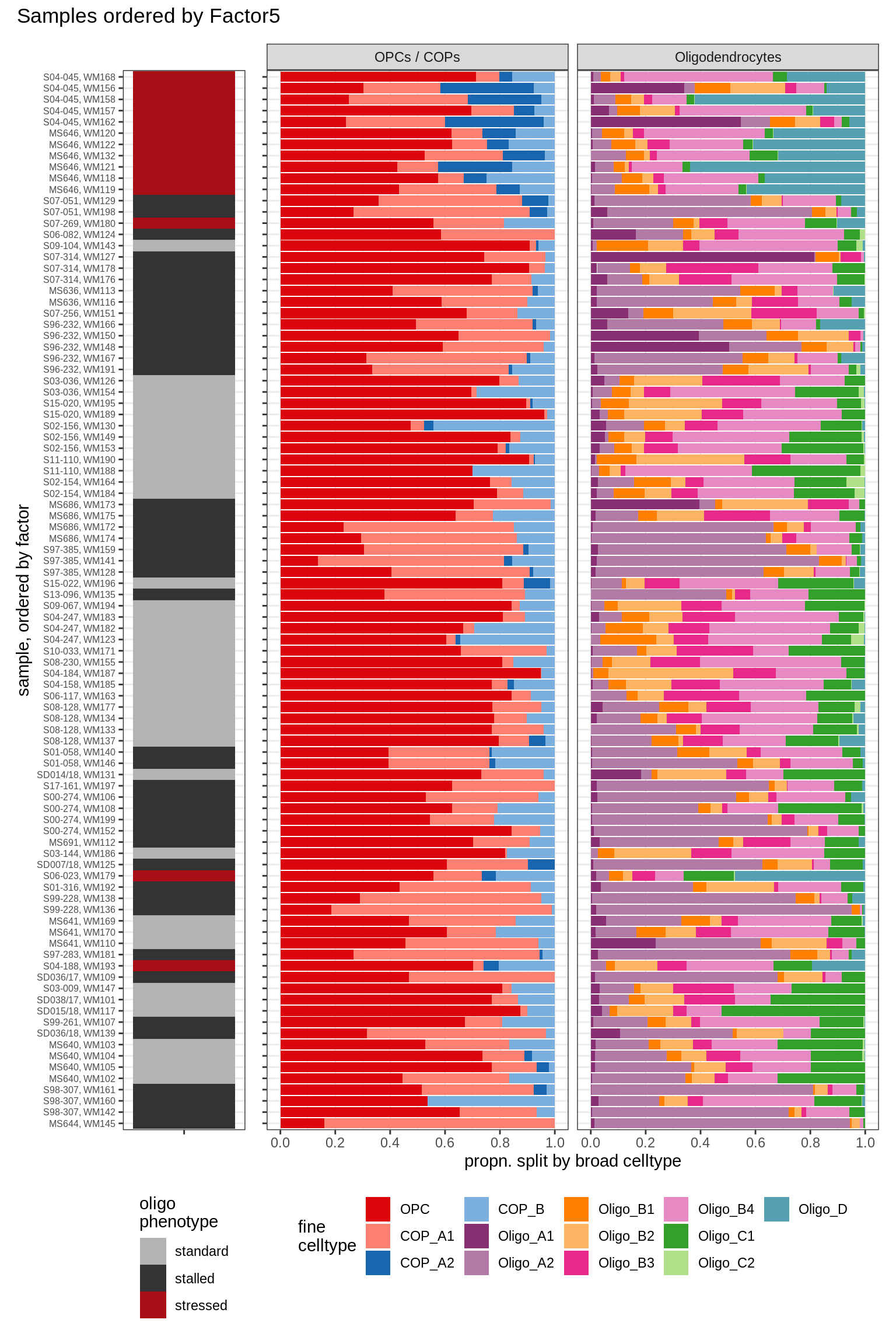
conos_dt = load_labelled_dt(labelled_f, labels_f)
types = c('OPCs / COPs', 'Oligodendrocytes')
m = "WM"
oligos_dt = conos_dt[ type_broad %in% types & str_detect(sample_id, "WM") ] %>%
.[, N_sample := .N, by = sample_id] %>%
.[, .N, by = .(sample_id, N_sample, type_broad, type_fine)] %>%
.[, prop := N / sum(N), by = .(sample_id, type_broad)] %>%
.[, type_fine := fct_relevel(type_fine, 'OPC')]
for (sel_f in factors_names(model)) {
cat('### ', sel_f, '\n', sep = '')
print(plot_barplots_ordered_by_factors(oligos_dt, model, sel_f))
cat('\n\n')
}Factor1
Factor2
Factor3
Factor4
Factor5
devtools::session_info()- Session info ---------------------------------------------------------------
setting value
version R version 4.0.5 (2021-03-31)
os CentOS Linux 7 (Core)
system x86_64, linux-gnu
ui X11
language (EN)
collate en_US.UTF-8
ctype C
tz Europe/Zurich
date 2021-11-25
- Packages -------------------------------------------------------------------
package * version date lib
ade4 1.7-18 2021-09-16 [1]
ANCOMBC * 1.0.5 2021-03-09 [1]
annotate 1.68.0 2020-10-27 [1]
AnnotationDbi 1.52.0 2020-10-27 [1]
ape 5.5 2021-04-25 [1]
assertthat * 0.2.1 2019-03-21 [2]
backports 1.2.1 2020-12-09 [2]
basilisk 1.2.1 2020-12-16 [1]
basilisk.utils 1.2.2 2021-01-27 [1]
beachmat 2.6.4 2020-12-20 [1]
beeswarm 0.4.0 2021-06-01 [1]
Biobase * 2.50.0 2020-10-27 [1]
BiocGenerics * 0.36.1 2021-04-16 [1]
BiocManager 1.30.16 2021-06-15 [1]
BiocNeighbors 1.8.2 2020-12-07 [1]
BiocParallel * 1.24.1 2020-11-06 [1]
BiocSingular 1.6.0 2020-10-27 [1]
BiocStyle * 2.18.1 2020-11-24 [1]
biomformat 1.18.0 2020-10-27 [1]
Biostrings 2.58.0 2020-10-27 [1]
bit 4.0.4 2020-08-04 [2]
bit64 4.0.5 2020-08-30 [2]
bitops 1.0-7 2021-04-24 [2]
blme 1.0-5 2021-01-05 [1]
blob 1.2.2 2021-07-23 [2]
boot 1.3-28 2021-05-03 [2]
broom 0.7.9 2021-07-27 [2]
bslib 0.3.1 2021-10-06 [2]
cachem 1.0.6 2021-08-19 [1]
Cairo 1.5-12.2 2020-07-07 [2]
callr 3.7.0 2021-04-20 [2]
caTools 1.18.2 2021-03-28 [2]
cellranger 1.1.0 2016-07-27 [2]
circlize * 0.4.13 2021-06-09 [1]
cli 3.0.1 2021-07-17 [1]
clue 0.3-60 2021-10-11 [1]
cluster 2.1.2 2021-04-17 [2]
codetools 0.2-18 2020-11-04 [2]
colorout * 1.2-2 2021-04-15 [1]
colorRamps 2.3 2012-10-29 [1]
colorspace 2.0-2 2021-06-24 [1]
ComplexHeatmap * 2.6.2 2020-11-12 [1]
corrplot 0.90 2021-06-30 [1]
cowplot 1.1.1 2020-12-30 [2]
crayon 1.4.1 2021-02-08 [2]
data.table * 1.14.2 2021-09-27 [2]
DBI 1.1.1 2021-01-15 [2]
dbplyr 2.1.1 2021-04-06 [2]
DelayedArray 0.16.3 2021-03-24 [1]
DelayedMatrixStats 1.12.3 2021-02-03 [1]
desc 1.4.0 2021-09-28 [1]
DESeq2 1.30.1 2021-02-19 [1]
devtools 2.4.2 2021-06-07 [1]
digest 0.6.28 2021-09-23 [2]
doParallel 1.0.16 2020-10-16 [1]
dplyr * 1.0.7 2021-06-18 [2]
edgeR * 3.32.1 2021-01-14 [1]
ellipsis 0.3.2 2021-04-29 [2]
evaluate 0.14 2019-05-28 [2]
fansi 0.5.0 2021-05-25 [2]
farver 2.1.0 2021-02-28 [2]
fastmap 1.1.0 2021-01-25 [2]
fastmatch 1.1-3 2021-07-23 [1]
fgsea * 1.16.0 2020-10-27 [1]
filelock 1.0.2 2018-10-05 [1]
forcats * 0.5.1 2021-01-27 [2]
foreach 1.5.1 2020-10-15 [2]
fs 1.5.0 2020-07-31 [2]
future 1.22.1 2021-08-25 [2]
future.apply 1.8.1 2021-08-10 [2]
genefilter 1.72.1 2021-01-21 [1]
geneplotter 1.68.0 2020-10-27 [1]
generics 0.1.1 2021-10-25 [2]
GenomeInfoDb * 1.26.7 2021-04-08 [1]
GenomeInfoDbData 1.2.4 2021-04-15 [1]
GenomicAlignments 1.26.0 2020-10-27 [1]
GenomicRanges * 1.42.0 2020-10-27 [1]
GetoptLong 1.0.5 2020-12-15 [1]
ggbeeswarm * 0.6.0 2017-08-07 [1]
ggplot2 * 3.3.5 2021-06-25 [1]
ggrepel * 0.9.1 2021-01-15 [2]
git2r 0.28.0 2021-01-10 [1]
glmmTMB 1.1.2.3 2021-09-20 [1]
GlobalOptions 0.1.2 2020-06-10 [1]
globals 0.14.0 2020-11-22 [2]
glue 1.4.2 2020-08-27 [2]
gplots 3.1.1 2020-11-28 [2]
gridExtra 2.3 2017-09-09 [2]
grr 0.9.5 2016-08-26 [1]
gtable 0.3.0 2019-03-25 [2]
gtools 3.9.2 2021-06-06 [2]
haven 2.4.3 2021-08-04 [2]
HDF5Array 1.18.1 2021-02-04 [1]
here 1.0.1 2020-12-13 [2]
highr 0.9 2021-04-16 [2]
hms 1.1.1 2021-09-26 [1]
htmltools 0.5.2 2021-08-25 [2]
htmlwidgets 1.5.4 2021-09-08 [2]
httpuv 1.6.3 2021-09-09 [2]
httr 1.4.2 2020-07-20 [2]
igraph 1.2.7 2021-10-15 [2]
insight 0.14.5 2021-10-16 [1]
IRanges * 2.24.1 2020-12-12 [1]
irlba 2.3.3 2019-02-05 [2]
iterators 1.0.13 2020-10-15 [2]
janitor 2.1.0 2021-01-05 [1]
jquerylib 0.1.4 2021-04-26 [2]
jsonlite 1.7.2 2020-12-09 [2]
KernSmooth 2.23-20 2021-05-03 [2]
knitr 1.36 2021-09-29 [1]
labeling 0.4.2 2020-10-20 [2]
later 1.3.0 2021-08-18 [2]
lattice 0.20-45 2021-09-22 [2]
lifecycle 1.0.1 2021-09-24 [2]
limma * 3.46.0 2020-10-27 [1]
listenv 0.8.0 2019-12-05 [2]
lme4 1.1-27.1 2021-06-22 [1]
lmerTest 3.1-3 2020-10-23 [1]
locfit 1.5-9.4 2020-03-25 [1]
lubridate 1.8.0 2021-10-07 [2]
magick 2.7.3 2021-08-18 [2]
magrittr * 2.0.1 2020-11-17 [1]
MASS * 7.3-54 2021-05-03 [2]
Matrix * 1.3-4 2021-06-01 [2]
Matrix.utils * 0.9.8 2020-02-26 [1]
MatrixGenerics * 1.2.1 2021-01-30 [1]
matrixStats * 0.61.0 2021-09-17 [1]
memoise 2.0.0 2021-01-26 [1]
mgcv 1.8-38 2021-10-06 [1]
microbiome 1.12.0 2020-10-27 [1]
minqa 1.2.4 2014-10-09 [1]
modelr 0.1.8 2020-05-19 [2]
MOFA2 * 1.0.1 2020-11-03 [1]
multtest 2.46.0 2020-10-27 [1]
munsell 0.5.0 2018-06-12 [2]
muscat * 1.5.1 2021-04-15 [1]
nlme 3.1-153 2021-09-07 [2]
nloptr 1.2.2.2 2020-07-02 [1]
numDeriv 2016.8-1.1 2019-06-06 [2]
parallelly 1.28.1 2021-09-09 [2]
patchwork * 1.1.1 2020-12-17 [2]
pbkrtest 0.5.1 2021-03-09 [1]
performance * 0.8.0 2021-10-01 [1]
permute 0.9-5 2019-03-12 [1]
pheatmap 1.0.12 2019-01-04 [1]
phyloseq * 1.34.0 2020-10-27 [1]
pillar 1.6.4 2021-10-18 [1]
pkgbuild 1.2.0 2020-12-15 [1]
pkgconfig 2.0.3 2019-09-22 [2]
pkgload 1.2.3 2021-10-13 [2]
plyr 1.8.6 2020-03-03 [2]
png 0.1-7 2013-12-03 [2]
prettyunits 1.1.1 2020-01-24 [2]
processx 3.5.2 2021-04-30 [2]
progress 1.2.2 2019-05-16 [2]
promises 1.2.0.1 2021-02-11 [2]
ps 1.6.0 2021-02-28 [2]
purrr * 0.3.4 2020-04-17 [2]
R.methodsS3 1.8.1 2020-08-26 [1]
R.oo 1.24.0 2020-08-26 [1]
R.utils 2.11.0 2021-09-26 [1]
R6 2.5.1 2021-08-19 [2]
rappdirs 0.3.3 2021-01-31 [2]
rbibutils 2.2.4 2021-10-11 [1]
RColorBrewer * 1.1-2 2014-12-07 [2]
Rcpp 1.0.7 2021-07-07 [1]
RCurl 1.98-1.5 2021-09-17 [1]
Rdpack 2.1.2 2021-06-01 [1]
readr * 2.0.2 2021-09-27 [2]
readxl * 1.3.1 2019-03-13 [2]
registry 0.5-1 2019-03-05 [1]
remotes 2.4.1 2021-09-29 [1]
reprex 2.0.1 2021-08-05 [2]
reshape2 * 1.4.4 2020-04-09 [2]
reticulate * 1.22 2021-09-17 [2]
rgl * 0.107.14 2021-08-21 [1]
rhdf5 2.34.0 2020-10-27 [1]
rhdf5filters 1.2.1 2021-05-03 [1]
Rhdf5lib 1.12.1 2021-01-26 [1]
rjson 0.2.20 2018-06-08 [1]
rlang 0.4.12 2021-10-18 [2]
rmarkdown * 2.11 2021-09-14 [1]
rprojroot 2.0.2 2020-11-15 [2]
Rsamtools 2.6.0 2020-10-27 [1]
RSQLite 2.2.8 2021-08-21 [1]
rstudioapi 0.13 2020-11-12 [2]
rsvd 1.0.5 2021-04-16 [1]
rtracklayer * 1.50.0 2020-10-27 [1]
Rtsne 0.15 2018-11-10 [2]
rvest 1.0.2 2021-10-16 [2]
S4Vectors * 0.28.1 2020-12-09 [1]
sass 0.4.0 2021-05-12 [2]
scales * 1.1.1 2020-05-11 [2]
scater * 1.18.6 2021-02-26 [1]
sctransform 0.3.2 2020-12-16 [2]
scuttle 1.0.4 2020-12-17 [1]
seriation * 1.3.1 2021-10-16 [1]
sessioninfo 1.1.1 2018-11-05 [1]
shape 1.4.6 2021-05-19 [1]
SingleCellExperiment * 1.12.0 2020-10-27 [1]
snakecase 0.11.0 2019-05-25 [1]
sparseMatrixStats 1.2.1 2021-02-02 [1]
stringi 1.7.4 2021-08-25 [1]
stringr * 1.4.0 2019-02-10 [2]
SummarizedExperiment * 1.20.0 2020-10-27 [1]
survival 3.2-13 2021-08-24 [2]
testthat 3.1.0 2021-10-04 [2]
tibble * 3.1.5 2021-09-30 [1]
tictoc * 1.0.1 2021-04-19 [1]
tidyr * 1.1.4 2021-09-27 [2]
tidyselect 1.1.1 2021-04-30 [2]
tidyverse * 1.3.1 2021-04-15 [2]
TMB 1.7.22 2021-09-28 [1]
TSP 1.1-11 2021-10-06 [1]
tzdb 0.1.2 2021-07-20 [2]
UpSetR * 1.4.0 2019-05-22 [1]
usethis 2.1.2 2021-10-25 [1]
utf8 1.2.2 2021-07-24 [1]
uwot 0.1.10 2020-12-15 [2]
variancePartition 1.20.0 2020-10-27 [1]
vctrs 0.3.8 2021-04-29 [2]
vegan 2.5-7 2020-11-28 [1]
vipor 0.4.5 2017-03-22 [1]
viridis * 0.6.2 2021-10-13 [1]
viridisLite * 0.4.0 2021-04-13 [1]
whisker 0.4 2019-08-28 [1]
withr 2.4.2 2021-04-18 [2]
workflowr * 1.6.2 2020-04-30 [1]
writexl * 1.4.0 2021-04-20 [1]
xfun 0.27 2021-10-18 [1]
XML 3.99-0.8 2021-09-17 [1]
xml2 1.3.2 2020-04-23 [2]
xtable 1.8-4 2019-04-21 [2]
XVector 0.30.0 2020-10-27 [1]
yaml 2.2.1 2020-02-01 [2]
zlibbioc 1.36.0 2020-10-27 [1]
source
CRAN (R 4.0.5)
Bioconductor
Bioconductor
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
Bioconductor
Bioconductor
Bioconductor
CRAN (R 4.0.3)
Bioconductor
Bioconductor
CRAN (R 4.0.3)
Bioconductor
Bioconductor
Bioconductor
Bioconductor
Bioconductor
Bioconductor
CRAN (R 4.0.2)
CRAN (R 4.0.2)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.5)
CRAN (R 4.0.3)
CRAN (R 4.0.5)
CRAN (R 4.0.5)
CRAN (R 4.0.5)
CRAN (R 4.0.2)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.5)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Github (jalvesaq/colorout@79931fd)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.5)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.5)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Bioconductor
Bioconductor
CRAN (R 4.0.5)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.5)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.5)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.2)
CRAN (R 4.0.5)
CRAN (R 4.0.5)
Bioconductor
Bioconductor
CRAN (R 4.0.5)
Bioconductor
Bioconductor
Bioconductor
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.5)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.5)
Bioconductor
CRAN (R 4.0.5)
CRAN (R 4.0.3)
CRAN (R 4.0.5)
CRAN (R 4.0.5)
CRAN (R 4.0.5)
CRAN (R 4.0.5)
CRAN (R 4.0.2)
CRAN (R 4.0.5)
CRAN (R 4.0.5)
Bioconductor
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.5)
CRAN (R 4.0.3)
CRAN (R 4.0.5)
CRAN (R 4.0.5)
CRAN (R 4.0.5)
Bioconductor
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.5)
CRAN (R 4.0.5)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.5)
CRAN (R 4.0.3)
CRAN (R 4.0.5)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.0)
Bioconductor
Bioconductor
CRAN (R 4.0.0)
Github (HelenaLC/muscat@c939663)
CRAN (R 4.0.5)
CRAN (R 4.0.3)
CRAN (R 4.0.2)
CRAN (R 4.0.5)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.5)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.5)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.5)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.5)
CRAN (R 4.0.5)
CRAN (R 4.0.3)
CRAN (R 4.0.5)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.5)
CRAN (R 4.0.3)
CRAN (R 4.0.5)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.5)
CRAN (R 4.0.5)
CRAN (R 4.0.0)
CRAN (R 4.0.5)
CRAN (R 4.0.5)
Bioconductor
Bioconductor
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.5)
CRAN (R 4.0.5)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.5)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.0)
CRAN (R 4.0.5)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.5)
CRAN (R 4.0.3)
CRAN (R 4.0.1)
Bioconductor
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.5)
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.5)
CRAN (R 4.0.5)
CRAN (R 4.0.5)
CRAN (R 4.0.3)
CRAN (R 4.0.5)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.5)
CRAN (R 4.0.5)
CRAN (R 4.0.5)
CRAN (R 4.0.3)
CRAN (R 4.0.5)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.5)
CRAN (R 4.0.1)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.5)
CRAN (R 4.0.5)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.3)
Bioconductor
[1] /pstore/home/macnairw/lib/conda_r3.12
[2] /pstore/home/macnairw/.conda/envs/r_4.0.3/lib/R/library
sessionInfo()R version 4.0.5 (2021-03-31)
Platform: x86_64-conda-linux-gnu (64-bit)
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS/LAPACK: /pstore/home/macnairw/.conda/envs/r_4.0.3/lib/libopenblasp-r0.3.12.so
locale:
[1] LC_CTYPE=C LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] grid parallel stats4 stats graphics grDevices utils
[8] datasets methods base
other attached packages:
[1] rgl_0.107.14 MOFA2_1.0.1
[3] rmarkdown_2.11 writexl_1.4.0
[5] ComplexHeatmap_2.6.2 fgsea_1.16.0
[7] tictoc_1.0.1 performance_0.8.0
[9] edgeR_3.32.1 limma_3.46.0
[11] reshape2_1.4.4 scater_1.18.6
[13] Matrix.utils_0.9.8 Matrix_1.3-4
[15] SingleCellExperiment_1.12.0 SummarizedExperiment_1.20.0
[17] Biobase_2.50.0 MatrixGenerics_1.2.1
[19] matrixStats_0.61.0 seriation_1.3.1
[21] UpSetR_1.4.0 BiocParallel_1.24.1
[23] muscat_1.5.1 dplyr_1.0.7
[25] readr_2.0.2 tidyr_1.1.4
[27] tibble_3.1.5 tidyverse_1.3.1
[29] rtracklayer_1.50.0 GenomicRanges_1.42.0
[31] GenomeInfoDb_1.26.7 IRanges_2.24.1
[33] S4Vectors_0.28.1 BiocGenerics_0.36.1
[35] ggbeeswarm_0.6.0 ggrepel_0.9.1
[37] reticulate_1.22 MASS_7.3-54
[39] phyloseq_1.34.0 ANCOMBC_1.0.5
[41] purrr_0.3.4 patchwork_1.1.1
[43] readxl_1.3.1 forcats_0.5.1
[45] ggplot2_3.3.5 scales_1.1.1
[47] viridis_0.6.2 viridisLite_0.4.0
[49] assertthat_0.2.1 stringr_1.4.0
[51] data.table_1.14.2 magrittr_2.0.1
[53] circlize_0.4.13 RColorBrewer_1.1-2
[55] BiocStyle_2.18.1 colorout_1.2-2
[57] workflowr_1.6.2
loaded via a namespace (and not attached):
[1] rappdirs_0.3.3 R.methodsS3_1.8.1
[3] bit64_4.0.5 knitr_1.36
[5] R.utils_2.11.0 irlba_2.3.3
[7] DelayedArray_0.16.3 RCurl_1.98-1.5
[9] doParallel_1.0.16 generics_0.1.1
[11] callr_3.7.0 cowplot_1.1.1
[13] microbiome_1.12.0 usethis_2.1.2
[15] RSQLite_2.2.8 future_1.22.1
[17] bit_4.0.4 tzdb_0.1.2
[19] xml2_1.3.2 lubridate_1.8.0
[21] httpuv_1.6.3 xfun_0.27
[23] hms_1.1.1 jquerylib_0.1.4
[25] TSP_1.1-11 evaluate_0.14
[27] promises_1.2.0.1 fansi_0.5.0
[29] progress_1.2.2 caTools_1.18.2
[31] dbplyr_2.1.1 htmlwidgets_1.5.4
[33] igraph_1.2.7 DBI_1.1.1
[35] geneplotter_1.68.0 ellipsis_0.3.2
[37] corrplot_0.90 backports_1.2.1
[39] insight_0.14.5 permute_0.9-5
[41] annotate_1.68.0 sparseMatrixStats_1.2.1
[43] vctrs_0.3.8 remotes_2.4.1
[45] here_1.0.1 Cairo_1.5-12.2
[47] cachem_1.0.6 withr_2.4.2
[49] grr_0.9.5 sctransform_0.3.2
[51] vegan_2.5-7 GenomicAlignments_1.26.0
[53] prettyunits_1.1.1 cluster_2.1.2
[55] ape_5.5 crayon_1.4.1
[57] basilisk.utils_1.2.2 genefilter_1.72.1
[59] labeling_0.4.2 pkgconfig_2.0.3
[61] pkgload_1.2.3 nlme_3.1-153
[63] vipor_0.4.5 devtools_2.4.2
[65] blme_1.0-5 rlang_0.4.12
[67] globals_0.14.0 lifecycle_1.0.1
[69] filelock_1.0.2 registry_0.5-1
[71] modelr_0.1.8 rsvd_1.0.5
[73] cellranger_1.1.0 rprojroot_2.0.2
[75] Rhdf5lib_1.12.1 boot_1.3-28
[77] reprex_2.0.1 beeswarm_0.4.0
[79] processx_3.5.2 pheatmap_1.0.12
[81] whisker_0.4 GlobalOptions_0.1.2
[83] png_0.1-7 rjson_0.2.20
[85] bitops_1.0-7 R.oo_1.24.0
[87] KernSmooth_2.23-20 rhdf5filters_1.2.1
[89] Biostrings_2.58.0 blob_1.2.2
[91] DelayedMatrixStats_1.12.3 shape_1.4.6
[93] parallelly_1.28.1 beachmat_2.6.4
[95] memoise_2.0.0 plyr_1.8.6
[97] gplots_3.1.1 zlibbioc_1.36.0
[99] compiler_4.0.5 clue_0.3-60
[101] lme4_1.1-27.1 DESeq2_1.30.1
[103] snakecase_0.11.0 Rsamtools_2.6.0
[105] cli_3.0.1 ade4_1.7-18
[107] XVector_0.30.0 lmerTest_3.1-3
[109] listenv_0.8.0 ps_1.6.0
[111] TMB_1.7.22 mgcv_1.8-38
[113] tidyselect_1.1.1 stringi_1.7.4
[115] highr_0.9 yaml_2.2.1
[117] BiocSingular_1.6.0 locfit_1.5-9.4
[119] sass_0.4.0 fastmatch_1.1-3
[121] tools_4.0.5 future.apply_1.8.1
[123] rstudioapi_0.13 foreach_1.5.1
[125] git2r_0.28.0 janitor_2.1.0
[127] gridExtra_2.3 farver_2.1.0
[129] Rtsne_0.15 digest_0.6.28
[131] BiocManager_1.30.16 Rcpp_1.0.7
[133] broom_0.7.9 scuttle_1.0.4
[135] later_1.3.0 httr_1.4.2
[137] AnnotationDbi_1.52.0 Rdpack_2.1.2
[139] colorspace_2.0-2 rvest_1.0.2
[141] XML_3.99-0.8 fs_1.5.0
[143] splines_4.0.5 uwot_0.1.10
[145] basilisk_1.2.1 multtest_2.46.0
[147] sessioninfo_1.1.1 xtable_1.8-4
[149] jsonlite_1.7.2 nloptr_1.2.2.2
[151] testthat_3.1.0 R6_2.5.1
[153] pillar_1.6.4 htmltools_0.5.2
[155] glue_1.4.2 fastmap_1.1.0
[157] minqa_1.2.4 BiocNeighbors_1.8.2
[159] codetools_0.2-18 pkgbuild_1.2.0
[161] utf8_1.2.2 lattice_0.20-45
[163] bslib_0.3.1 numDeriv_2016.8-1.1
[165] pbkrtest_0.5.1 colorRamps_2.3
[167] gtools_3.9.2 magick_2.7.3
[169] survival_3.2-13 glmmTMB_1.1.2.3
[171] desc_1.4.0 biomformat_1.18.0
[173] munsell_0.5.0 GetoptLong_1.0.5
[175] rhdf5_2.34.0 GenomeInfoDbData_1.2.4
[177] iterators_1.0.13 HDF5Array_1.18.1
[179] variancePartition_1.20.0 haven_2.4.3
[181] gtable_0.3.0 rbibutils_2.2.4