ANCOM-BC bootstrapped to handle random effects
Will Macnair
Neurogenomics, Neuroscience and Rare Diseases, RocheMarch 25, 2022
Last updated: 2022-03-25
Checks: 4 3
Knit directory: MS_lesions/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
The global environment had objects present when the code in the R Markdown file was run. These objects can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment. Use wflow_publish or wflow_build to ensure that the code is always run in an empty environment.
The following objects were defined in the global environment when these results were created:
| Name | Class | Size |
|---|---|---|
| q | function | 1008 bytes |
The command set.seed(20210118) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
- calc_ancom_bootstraps
- calc_ancom_neuron_nagm
- calc_ancom_neuron_subspaces
- calc_ancom_standard
- calc_gm_layer_pcs
- calc_gm_wide_pcs
- calc_lrts_pcs
- calc_lrts_std
- calc_pcs_coefs
- calc_wide_dt
- calc_wm_oligo_grps
- combine_results
- load_ancom_bootstraps
- load_ancom_neuron_subspaces
- load_ancom_standard
- load_gpr17_ihc_data
- plot_bootstrap_vs_standard
- plot_bootstraps_all_coefs
- plot_clr_pca_neurons
- plot_effect_of_pcs_lesions
- plot_effect_of_pcs_neurons
- plot_effect_of_pcs_rest
- plot_heatmaps_clr
- plot_heatmaps_log_p
- plot_heatmaps_log_p_neurons
- plot_heatmaps_log_p_oligos
- plot_heatmaps_log_p_oligos_by_patient
- plot_layer_var_exp
- plot_no_gpr17_cells
- plot_patients_over_pcs
- plot_propns_layers
- plot_sample_splits_clrs_oligos
- plot_standard_all_coefs
- plot_standard_lesions
- plot_wm_vs_gm
- plot_wm_vs_gm_absolute
- save_pseudobulk_w_pcs
- session_info
- session-info-chunk-inserted-by-workflowr
- setup_input
- setup_outputs
- test_gpr17_ihc
To ensure reproducibility of the results, delete the cache directory ms09_ancombc_mixed_cache and re-run the analysis. To have workflowr automatically delete the cache directory prior to building the file, set delete_cache = TRUE when running wflow_build() or wflow_publish().
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 9175fdc. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .RData
Ignored: .Rhistory
Ignored: .Rprofile
Ignored: .Rproj.user/
Ignored: .log/
Ignored: MS_lesions.sublime-project
Ignored: MS_lesions.sublime-workspace
Ignored: analysis/.__site.yml
Ignored: analysis/fig_muscat_cache/
Ignored: analysis/figure/
Ignored: analysis/ms00_manuscript_figures_cache/
Ignored: analysis/ms02_doublet_id_cache/
Ignored: analysis/ms03_SampleQC_cache/
Ignored: analysis/ms03_SampleQC_summary_cache/
Ignored: analysis/ms04_conos_cache/
Ignored: analysis/ms05_splitting_cache/
Ignored: analysis/ms06_sccaf_cache/
Ignored: analysis/ms07_soup_cache/
Ignored: analysis/ms08_modules_cache/
Ignored: analysis/ms08_modules_pseudobulk_cache/
Ignored: analysis/ms09_ancombc_cache/
Ignored: analysis/ms09_ancombc_clean_1e3_cache/
Ignored: analysis/ms09_ancombc_clean_2e3_cache/
Ignored: analysis/ms09_ancombc_mixed_cache/
Ignored: analysis/ms10_muscat_run01_cache/
Ignored: analysis/ms10_muscat_run02_cache/
Ignored: analysis/ms10_muscat_template_broad_slim_cache/
Ignored: analysis/ms10_muscat_template_fine_slim_cache/
Ignored: analysis/ms11_paga_cache/
Ignored: analysis/ms11_paga_recalc_cache/
Ignored: analysis/ms11_paga_superclean_cache/
Ignored: analysis/ms12_markers_cache/
Ignored: analysis/ms13_labelling_cache/
Ignored: analysis/ms14_lesions_cache/
Ignored: analysis/ms15_mofa_gm_cache/
Ignored: analysis/ms15_mofa_gm_edger_libs_cache/
Ignored: analysis/ms15_mofa_heatmaps_cache/
Ignored: analysis/ms15_mofa_sample_gm_cache/
Ignored: analysis/ms15_mofa_sample_gm_final_meta_cache/
Ignored: analysis/ms15_mofa_sample_gm_superclean_cache/
Ignored: analysis/ms15_mofa_sample_gm_w_layers_final_meta_cache/
Ignored: analysis/ms15_mofa_sample_wm_cache/
Ignored: analysis/ms15_mofa_sample_wm_final_meta_bigger_cache/
Ignored: analysis/ms15_mofa_sample_wm_final_meta_bigger_edger_libsizes_cache/
Ignored: analysis/ms15_mofa_sample_wm_final_meta_cache/
Ignored: analysis/ms15_mofa_sample_wm_final_meta_edger_libsizes_cache/
Ignored: analysis/ms15_mofa_sample_wm_final_meta_neuro_ok_cache/
Ignored: analysis/ms15_mofa_sample_wm_new_meta_cache/
Ignored: analysis/ms15_mofa_sample_wm_superclean_cache/
Ignored: analysis/ms15_mofa_wm_bigger_cache/
Ignored: analysis/ms15_mofa_wm_cache/
Ignored: analysis/ms15_mofa_wm_coding_only_bigger_cache/
Ignored: analysis/ms15_mofa_wm_coding_only_cache/
Ignored: analysis/ms15_mofa_wm_edger_libs_cache/
Ignored: analysis/ms15_mofa_wm_neuro_ok_cache/
Ignored: analysis/ms15_mofa_wm_pearson_cache/
Ignored: analysis/ms15_patients_cache/
Ignored: analysis/ms15_patients_gm_cache/
Ignored: analysis/ms15_patients_sample_level_cache/
Ignored: analysis/ms15_patients_w_ms_cache/
Ignored: analysis/ms99_deg_figures_gm_cache/
Ignored: analysis/ms99_deg_figures_wm_cache/
Ignored: analysis/ms99_manuscript_figures_cache/
Ignored: analysis/ms99_test_cache/
Ignored: analysis/public/
Ignored: analysis/supp06_sccaf_cache/
Ignored: analysis/supp07_superclean_check_cache/
Ignored: analysis/supp09_ancombc_cache/
Ignored: analysis/supp09_ancombc_mixed_cache/
Ignored: analysis/supp09_ancombc_rowitch_cache/
Ignored: analysis/supp09_ancombc_superclean_cache/
Ignored: analysis/supp10_muscat_cache/
Ignored: analysis/supp10_muscat_ctrl_gm_vs_wm_cache/
Ignored: analysis/supp10_muscat_gm_layers_effects_cache/
Ignored: analysis/supp10_muscat_gsea_cache/
Ignored: analysis/supp10_muscat_heatmaps_cache/
Ignored: analysis/supp10_muscat_olg_pc1_cache/
Ignored: analysis/supp10_muscat_olg_pc2_cache/
Ignored: analysis/supp10_muscat_olg_pc_cache/
Ignored: analysis/supp10_muscat_regression_cache/
Ignored: analysis/supp10_muscat_soup_cache/
Ignored: analysis/supp10_muscat_soup_mito_cache/
Ignored: code/._ms10_muscat_fns_recover.R
Ignored: code/.recovery/
Ignored: code/adhoc_code/
Ignored: code/jobs/._muscat_run09_2021-10-11.slurm
Ignored: data/
Ignored: figures/
Ignored: output/
Ignored: tmp/
Untracked files:
Untracked: Rplots.pdf
Untracked: analysis/_pdf.yml
Untracked: check_absolutes.png
Untracked: wm_vs_gm.png
Unstaged changes:
Modified: analysis/SampleQC_report_template_pdf.Rmd
Modified: analysis/ms03_SampleQC.Rmd
Modified: analysis/ms09_ancombc_mixed.Rmd
Modified: analysis/ms12_markers.Rmd
Modified: analysis/ms15_mofa_gm_edger_libs.Rmd
Modified: analysis/ms15_mofa_wm_edger_libs.Rmd
Modified: analysis/ms99_deg_figures_gm.Rmd
Modified: code/ms00_utils.R
Modified: code/ms03_SampleQC.R
Modified: code/ms09_ancombc_mixed.R
Modified: code/ms15_mofa.R
Modified: code/ms99_deg_figures.R
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/ms09_ancombc_mixed.Rmd) and HTML (public/ms09_ancombc_mixed.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 9175fdc | Will Macnair | 2022-03-23 | Add cell number plots to ANCOM outputs |
| html | cbfe519 | Macnair | 2022-03-18 | Add super weird git bug where all pngs appear to have changed |
| Rmd | 404cb5c | Macnair | 2022-03-18 | Make minor tweaks to plotting |
| html | 1c923eb | wmacnair | 2022-03-06 | Tweak to ancombc markdown |
| Rmd | 186f9c3 | wmacnair | 2022-02-25 | Further tweaking of CLR clustering |
| html | 186f9c3 | wmacnair | 2022-02-25 | Further tweaking of CLR clustering |
| html | de9cfc1 | wmacnair | 2022-02-25 | Improve ms09 barplots |
| Rmd | 022694d | wmacnair | 2022-02-24 | Add CLR clustered oligo groupings and update ms09 |
| html | 022694d | wmacnair | 2022-02-24 | Add CLR clustered oligo groupings and update ms09 |
| Rmd | 0d0e555 | wmacnair | 2022-02-23 | Tweak ms09 figures |
| html | 0d0e555 | wmacnair | 2022-02-23 | Tweak ms09 figures |
| Rmd | 2c2025a | wmacnair | 2022-02-18 | Add GSEA figures to manuscript figures |
| Rmd | 054f8e2 | wmacnair | 2022-02-16 | Add GPR17 IHC plot to ms09 |
| html | 054f8e2 | wmacnair | 2022-02-16 | Add GPR17 IHC plot to ms09 |
| Rmd | d8adeca | wmacnair | 2022-02-02 | Tweak various plots |
| Rmd | 5a1c0c2 | wmacnair | 2022-01-06 | Tweak oligo barplots and MOFA heatmaps |
| html | 5a1c0c2 | wmacnair | 2022-01-06 | Tweak oligo barplots and MOFA heatmaps |
| Rmd | afba18d | wmacnair | 2021-12-20 | Add more plots to ms00_manuscript_figures |
| html | afba18d | wmacnair | 2021-12-20 | Add more plots to ms00_manuscript_figures |
| Rmd | 8364a6f | wmacnair | 2021-12-13 | Regress out layer PCs in GM CLR plots |
| html | 8364a6f | wmacnair | 2021-12-13 | Regress out layer PCs in GM CLR plots |
| Rmd | 54bd2e9 | wmacnair | 2021-12-08 | Add sex to CLR heatmaps in ms09_ancombc_mixed |
| html | 54bd2e9 | wmacnair | 2021-12-08 | Add sex to CLR heatmaps in ms09_ancombc_mixed |
| Rmd | 97aa6f8 | wmacnair | 2021-12-08 | Tweak oligo barplots to include patient IDs |
| html | 97aa6f8 | wmacnair | 2021-12-08 | Tweak oligo barplots to include patient IDs |
| Rmd | 9ee5eee | wmacnair | 2021-12-08 | Add oligo groupings to ms09_ancombc_mixed CLR plots |
| html | 9ee5eee | wmacnair | 2021-12-08 | Add oligo groupings to ms09_ancombc_mixed CLR plots |
| Rmd | 315cf3b | wmacnair | 2021-12-08 | Tidy up ms09_ancombc_mixed.Rmd |
| html | 315cf3b | wmacnair | 2021-12-08 | Tidy up ms09_ancombc_mixed.Rmd |
| Rmd | 47a3ec8 | wmacnair | 2021-12-08 | Add oligo barplots to ms09_ancombc_mixed.Rmd |
| html | 47a3ec8 | wmacnair | 2021-12-08 | Add oligo barplots to ms09_ancombc_mixed.Rmd |
| Rmd | 1f4bea5 | wmacnair | 2021-11-29 | Fix plot bug in ms09_ancombc_mixed |
| html | 1f4bea5 | wmacnair | 2021-11-29 | Fix plot bug in ms09_ancombc_mixed |
| Rmd | 270e5fc | wmacnair | 2021-11-26 | Add signif_only option to ANCOM-BC plots |
| html | 270e5fc | wmacnair | 2021-11-26 | Add signif_only option to ANCOM-BC plots |
| html | 7fb1b95 | wmacnair | 2021-11-25 | Host with GitLab. |
| Rmd | 8b7ce6e | Macnair | 2021-11-24 | Final update to ANCOM-BC analysis, including layer PCs |
| Rmd | afe32c6 | Macnair | 2021-11-16 | Final version of ANCOM-BC analysis |
| Rmd | 8d1ece6 | Macnair | 2021-10-28 | Update ms09_ancombc_mixed with paired analysis |
| Rmd | a73a2fa | Macnair | 2021-10-23 | Update ANCOM bootstrapping analysis |
Setup / definitions
Libraries
Helper functions
source('code/ms00_utils.R')
source('code/ms04_conos.R')
source('code/ms07_soup.R')
source('code/ms09_ancombc_mixed.R')
setDTthreads(8)Inputs
# define run
labels_f = 'data/byhand_markers/validation_markers_2021-05-31.csv'
labelled_f = 'output/ms13_labelling/conos_labelled_2021-05-31.txt.gz'
meta_f = "data/metadata/metadata_checked_assumptions_2022-03-22.xlsx"
# define pseudobulk data
soup_dir = 'output/ms07_soup'
pb_broad_f = file.path(soup_dir, 'pb_sum_broad_2021-10-11.rds')
pb_fine_f = file.path(soup_dir, 'pb_sum_fine_2021-10-11.rds')
pb_f_ls = c(broad = pb_broad_f, fine = pb_fine_f)
# celltype proportions data
prop_fine_f = file.path(soup_dir, 'pb_prop_fine_2021-10-11.rds')
# GPR17 IHC staining data
gpr17_f = 'data/gpr17_ihc/gpr17_ihc_data_2022-02-16.csv'Outputs
# where to save?
save_dir = 'output/ms09_ancombc'
date_tag = '2021-11-12'
if (!dir.exists(save_dir))
dir.create(save_dir)
# sample variables
sample_vars = c('sample_id', 'matter', 'lesion_type',
'neuro_ok', 'neuro_prop', 'sample_source', 'subject_id',
'sex', 'age_scale', 'pmi_cat', 'pmi_cat2')
# identifying strange samples
neuro_mad_cut = 2
log_n_mad_cut = 3
# define how to select PCs
cut_var_exp = 0.01
cut_layer_cor = 0.2
# define WM data
wm_spec = list(
name = 'lesions_WM',
subset = list(matter = 'WM', neuro_ok = TRUE),
size = list(min_count = 10, min_prop = 0.1),
exc_regex = '^(Ex_|Inh_|Neuro_oligo)',
formula = '~ lesion_type + sex + age_scale + pmi_cat',
fixef_test = 'lesion_type',
fixef_covar = c('sex', 'age_scale', 'pmi_cat'),
ranef_var = 'subject_id'
)
gm_spec = list(
name = 'lesions_GM',
subset = list(matter = 'GM', neuro_ok = TRUE),
size = list(min_count = 10, min_prop = 0.1),
exc_regex = NULL,
formula = '~ lesion_type + sex + age_scale + pmi_cat2',
fixef_test = 'lesion_type',
fixef_covar = c('sex', 'age_scale', 'pmi_cat2'),
ranef_var = 'subject_id'
)
# define multiple different ways to do subspaces
gm_pc_spec = list(
name_str = 'lesions_GM_',
subset = list(matter = 'GM', neuro_ok = TRUE),
size = list(min_count = 10, min_prop = 0.1),
exc_regex = NULL,
formula_pat = '~ lesion_type + %s + sex + age_scale + pmi_cat2',
fixef_test = 'lesion_type',
fixef_covar = c('sex', 'age_scale', 'pmi_cat2'),
ranef_var = 'subject_id',
broad_sel = c("Excitatory neurons", "Inhibitory neurons"),
lesion_ctrl = "GM",
n_pcs = NA
)
# define multiple different ways to do subspaces
nagm_pc_spec = list(
name_str = 'lesions_NAGM_',
subset = list(matter = 'GM', neuro_ok = TRUE),
size = list(min_count = 10, min_prop = 0.1),
exc_regex = NULL,
formula_pat = '~ lesion_type + %s + sex + age_scale + pmi_cat2',
fixef_test = 'lesion_type',
fixef_covar = c('sex', 'age_scale', 'pmi_cat2'),
ranef_var = 'subject_id',
broad_sel = c("Excitatory neurons", "Inhibitory neurons"),
lesion_ctrl = "GM",
n_pcs = NA
)
# gather things
spec_list = list(wm_spec, gm_spec)
names(spec_list) = sapply(spec_list, function(l) l$name)
# bootstrapping parameters
n_boots = 2e4
n_cores = 4
# define files for saving outputs
lrt_pat = file.path(save_dir, 'abundance_nb_lrt_model_%s_%s.rds')
clr_pat = file.path(save_dir, 'clr_clustering_%s_%s.txt')
ancom_pat = file.path(save_dir, 'ancombc_standard_%s_%s.rds')
boots_pat = file.path(save_dir, 'ancombc_bootstrap_%s_%s.txt.gz')
# define files for saving outputs
pb_pcs_ls = c(
broad = sprintf('%s/pb_gm_w_pcs_sum_broad_%s.rds', soup_dir, date_tag),
fine = sprintf('%s/pb_gm_w_pcs_sum_fine_%s.rds', soup_dir, date_tag)
)
# define various groups of celltypes
olg_types = c('OPCs / COPs', 'Oligodendrocytes')
neu_types = c('Excitatory neurons', 'Inhibitory neurons')
wm_types = setdiff(broad_ord, c('Excitatory neurons', 'Inhibitory neurons'))
gm_types = setdiff(broad_ord, c('Immune'))
# define parameters for CLR plots
min_cells = 50
olg_grps_f = 'data/metadata/oligo_groupings.txt'
olg_clust_f = sprintf("%s/oligo_clr_clusters_%s.txt", save_dir, date_tag)
# params for plotting GPR17 cell abundances
sel_g = "GPR17"Load inputs
meta_dt = load_meta_dt_from_xls(meta_f)
labels_dt = load_names_dt(labels_f) %>%
.[, cluster_id := type_fine]Warning in FUN(X[[i]], ...): unable to translate '<U+00C4>' to native encodingWarning in FUN(X[[i]], ...): unable to translate '<U+00D6>' to native encodingWarning in FUN(X[[i]], ...): unable to translate '<U+00DC>' to native encodingWarning in FUN(X[[i]], ...): unable to translate '<U+00E4>' to native encodingWarning in FUN(X[[i]], ...): unable to translate '<U+00F6>' to native encodingWarning in FUN(X[[i]], ...): unable to translate '<U+00FC>' to native encodingWarning in FUN(X[[i]], ...): unable to translate '<U+00DF>' to native encodingWarning in FUN(X[[i]], ...): unable to translate '<U+00C6>' to native encodingWarning in FUN(X[[i]], ...): unable to translate '<U+00E6>' to native encodingWarning in FUN(X[[i]], ...): unable to translate '<U+00D8>' to native encodingWarning in FUN(X[[i]], ...): unable to translate '<U+00F8>' to native encodingWarning in FUN(X[[i]], ...): unable to translate '<U+00C5>' to native encodingWarning in FUN(X[[i]], ...): unable to translate '<U+00E5>' to native encodingconos_all = load_labelled_dt(labelled_f, labels_f) %>%
merge(meta_dt, by = 'sample_id') %>%
add_neuro_props(mad_cut = neuro_mad_cut)# check for any outliers
size_chks = calc_size_outliers(conos_all, mad_cut = log_n_mad_cut)
message("these samples excluded to outlier sample sizes:")these samples excluded to outlier sample sizes:print(size_chks[ size_ok == FALSE ]) matter sample_id N med_log_N mad_log_N size_ok
1: GM EU034 911 8.519391 0.423111 FALSE
2: GM EU043 687 8.519391 0.423111 FALSE# exclude them from conos
ok_samples = size_chks[ size_ok == TRUE ]$sample_id
conos_all = conos_all[ sample_id %in% ok_samples ]
conos_dt = conos_all[ (sample_id %in% ok_samples) & (neuro_ok == TRUE) ]props_dt = calc_props_dt(conos_dt, sample_vars)
wide_dt = calc_counts_wide(props_dt, sample_vars)# get neuronal proportions for all samples
props_neu = conos_dt %>%
.[ (type_broad %in% gm_pc_spec$broad_sel) ] %>%
calc_props_dt(sample_vars)
# calc PCAs
ctrl_pcs_dt = props_neu %>%
.[ lesion_type == gm_pc_spec$lesion_ctrl ] %>%
calc_ctrl_pcs_dt(layers_dt)# apply pcs
all_pcs_dt = apply_ctrl_pcs(props_neu[ matter == "GM" ], ctrl_pcs_dt,
cut_var_exp, cut_layer_cor)
wide_neu = merge(all_pcs_dt, wide_dt, 'sample_id')
pc_vars = str_subset(names(wide_neu), "ctrl_PC")gpr17_dt = load_gpr17_dt(gpr17_f)Processing / calculations
# negative-binomial model on unadjusted counts, WM
nb_wm_f = sprintf(lrt_pat, wm_spec$name, date_tag)
if (file.exists(nb_wm_f)) {
nb_wm_ls = readRDS(nb_wm_f)
} else {
nb_wm_ls = calc_celltype_mixed_models(wide_dt, sample_vars,
wm_spec$subset, wm_spec$size, wm_spec$exc_regex, wm_spec$inc_regex,
wm_spec$fixef_test, wm_spec$fixef_covar, wm_spec$ranef_var,
n_cores = n_cores, offset_var = NULL)
saveRDS(nb_wm_ls, file = nb_wm_f)
}
# negative-binomial model on unadjusted counts, GM
nb_gm_f = sprintf(lrt_pat, gm_spec$name, date_tag)
if (file.exists(nb_gm_f)) {
nb_gm_ls = readRDS(nb_gm_f)
} else {
nb_gm_ls = calc_celltype_mixed_models(wide_dt, sample_vars,
gm_spec$subset, gm_spec$size, gm_spec$exc_regex, gm_spec$inc_regex,
gm_spec$fixef_test, gm_spec$fixef_covar, gm_spec$ranef_var,
n_cores = n_cores, offset_var = NULL)
saveRDS(nb_gm_ls, file = nb_gm_f)
}# make spec
for (n_pcs in seq_along(pc_vars)) {
# which PCs?
layer_spec = make_layer_pc_spec(gm_pc_spec, pc_vars, n_pcs = n_pcs)
# negative-binomial model on unadjusted counts, GM, w layers
nb_layers_f = sprintf(lrt_pat, layer_spec$name, date_tag)
if (!file.exists(nb_layers_f)) {
nb_layers_ls = calc_celltype_mixed_models(wide_neu, c(sample_vars, pc_vars),
layer_spec$subset, layer_spec$size, layer_spec$exc_regex, layer_spec$inc_regex,
layer_spec$fixef_test, layer_spec$fixef_covar, layer_spec$ranef_var,
n_cores = n_cores, offset_var = NULL)
saveRDS(nb_layers_ls, file = nb_layers_f)
}
}
# load them
nb_pcs_ls = lapply(seq_along(pc_vars), function(n_pcs) {
# make file
layer_spec = make_layer_pc_spec(gm_pc_spec, pc_vars, n_pcs = n_pcs)
nb_gm_ls = sprintf(lrt_pat, layer_spec$name, date_tag) %>%
readRDS
return(nb_gm_ls)
}) %>% setNames(paste0(gm_pc_spec$name_str, seq_along(pc_vars), 'pcs'))
# make list of models
lrt_ls = list(WM = nb_wm_ls, GM = nb_gm_ls) %>%
c(nb_pcs_ls)# loop through specified models
for (nn in names(spec_list)) {
# make file
ancom_f = sprintf(ancom_pat, spec_list[[nn]]$name, date_tag)
# if necessary, run thing
if (!file.exists(ancom_f)) {
message('running standard ANCOM-BC for ', nn)
# define things we need
spec = spec_list[[nn]]
# do standard ANCOM, save results
ancom_obj = calc_ancom_standard(wide_dt, sample_vars,
spec$subset, spec$size, spec$exc_regex, spec$inc_regex, spec$ref_type,
spec$fixef_test, spec$fixef_covar)
saveRDS(ancom_obj, file = ancom_f)
}
}# loop through specified models
for (nn in names(spec_list)) {
# make file
boots_f = sprintf(boots_pat, spec_list[[nn]]$name, date_tag)
# if necessary, run thing
if (!file.exists(boots_f)) {
message('running bootstrapped ANCOM-BC for ', nn)
# define things we need
spec = spec_list[[nn]]
# do bootstrapping, save resulst
boots_dt = calc_ancom_bootstrap(wide_dt, sample_vars,
spec$subset, spec$size, spec$exc_regex, spec$inc_regex, spec$ref_type,
spec$fixef_test, spec$fixef_covar, spec$ranef_var,
seed = 1, n_boots, n_cores)
fwrite(boots_dt, file = boots_f)
}
}# set up this run
for (n_pcs in seq_along(pc_vars)) {
# which PCs?
layer_spec = make_layer_pc_spec(gm_pc_spec, pc_vars, n_pcs = n_pcs)
# make file
ancom_f = sprintf(ancom_pat, layer_spec$name, date_tag)
# if necessary, run thing
if (file.exists(ancom_f)) {
message('standard ANCOM-BC for ', layer_spec$name, ' already done')
} else {
# do bootstrapping, save results
message('running standard ANCOM-BC for ', layer_spec$name)
ancom_neu = calc_ancom_standard(wide_neu, c(sample_vars, pc_vars),
layer_spec$subset, layer_spec$size, layer_spec$exc_regex,
layer_spec$inc_regex, layer_spec$ref_type,
layer_spec$fixef_test, layer_spec$fixef_covar)
saveRDS(ancom_neu, file = ancom_f)
}
# do bootstrapping
boots_f = sprintf(boots_pat, layer_spec$name, date_tag)
# if necessary, run thing
if (file.exists(boots_f)) {
message('bootstrapped ANCOM-BC for ', layer_spec$name, ' already done')
} else {
# do bootstrapping, save resulst
message('running bootstrapped ANCOM-BC for ', layer_spec$name)
t_start = Sys.time()
boots_neu = calc_ancom_bootstrap(wide_neu, c(sample_vars, pc_vars),
layer_spec$subset, layer_spec$size, layer_spec$exc_regex,
layer_spec$inc_regex, layer_spec$ref_type,
layer_spec$fixef_test, layer_spec$fixef_covar, layer_spec$ranef_var,
seed = 1, n_boots, n_cores)
t_stop = Sys.time()
fwrite(boots_neu, file = boots_f)
# report how long it took
t_elapsed = difftime(t_stop, t_start, units = 'mins') %>% unclass
message(sprintf(
paste0(' (bootstrapping %d boots with %d cores took %.1f minutes;',
' %.1f boots / min / core)'),
n_boots, n_cores, t_elapsed, n_boots / t_elapsed / n_cores))
}
}standard ANCOM-BC for lesions_GM_1pcs already donebootstrapped ANCOM-BC for lesions_GM_1pcs already donestandard ANCOM-BC for lesions_GM_2pcs already donebootstrapped ANCOM-BC for lesions_GM_2pcs already donestandard ANCOM-BC for lesions_GM_3pcs already donebootstrapped ANCOM-BC for lesions_GM_3pcs already donestandard ANCOM-BC for lesions_GM_4pcs already donebootstrapped ANCOM-BC for lesions_GM_4pcs already donestandard ANCOM-BC for lesions_GM_5pcs already donebootstrapped ANCOM-BC for lesions_GM_5pcs already donestandard ANCOM-BC for lesions_GM_6pcs already donebootstrapped ANCOM-BC for lesions_GM_6pcs already donestandard ANCOM-BC for lesions_GM_7pcs already donebootstrapped ANCOM-BC for lesions_GM_7pcs already done# set up this run
nagm_pcs = 4
layer_spec = make_layer_pc_spec(nagm_pc_spec, pc_vars, n_pcs = nagm_pcs)
wide_nagm = wide_neu %>% copy %>%
.[, lesion_type := lesion_type %>% fct_relevel("NAGM") %>% fct_drop ]
# make file
ancom_f = sprintf(ancom_pat, layer_spec$name, date_tag)
# if necessary, run thing
if (file.exists(ancom_f)) {
message('standard ANCOM-BC for ', layer_spec$name, ' already done')
ancom_nagm = ancom_f %>% readRDS
} else {
# do bootstrapping, save results
message('running standard ANCOM-BC for ', layer_spec$name)
ancom_nagm = calc_ancom_standard(wide_nagm, c(sample_vars, pc_vars),
layer_spec$subset, layer_spec$size, layer_spec$exc_regex,
layer_spec$inc_regex, layer_spec$ref_type,
layer_spec$fixef_test, layer_spec$fixef_covar)
saveRDS(ancom_nagm, file = ancom_f)
}standard ANCOM-BC for lesions_NAGM_4pcs already done# do bootstrapping
boots_f = sprintf(boots_pat, layer_spec$name, date_tag)
# if necessary, run thing
if (file.exists(boots_f)) {
message('bootstrapped ANCOM-BC for ', layer_spec$name, ' already done')
boots_nagm = boots_f %>% fread
} else {
# do bootstrapping, save resulst
message('running bootstrapped ANCOM-BC for ', layer_spec$name)
t_start = Sys.time()
boots_nagm = calc_ancom_bootstrap(wide_nagm, c(sample_vars, pc_vars),
layer_spec$subset, layer_spec$size, layer_spec$exc_regex,
layer_spec$inc_regex, layer_spec$ref_type,
layer_spec$fixef_test, layer_spec$fixef_covar, layer_spec$ranef_var,
seed = 1, n_boots, n_cores)
t_stop = Sys.time()
fwrite(boots_nagm, file = boots_f)
# report how long it took
t_elapsed = difftime(t_stop, t_start, units = 'mins') %>% unclass
message(sprintf(
paste0(' (bootstrapping %d boots with %d cores took %.1f minutes;',
' %.1f boots / min / core)'),
n_boots, n_cores, t_elapsed, n_boots / t_elapsed / n_cores))
}bootstrapped ANCOM-BC for lesions_NAGM_4pcs already doneancom_ls = lapply(names(spec_list), function(nn) {
# make file
ancom_obj = sprintf(ancom_pat, spec_list[[nn]]$name, date_tag) %>%
readRDS
return(ancom_obj)
}) %>% setNames(names(spec_list))boots_ls = lapply(names(spec_list), function(nn) {
# make file
boots_dt = sprintf(boots_pat, spec_list[[nn]]$name, date_tag) %>% fread
return(boots_dt)
}) %>% setNames(names(spec_list))# load std
ancom_pcs_ls = lapply(seq_along(pc_vars), function(n_pcs) {
# make file
layer_spec = make_layer_pc_spec(gm_pc_spec, pc_vars, n_pcs = n_pcs)
ancom_obj = sprintf(ancom_pat, layer_spec$name, date_tag) %>%
readRDS
return(ancom_obj)
}) %>% setNames(paste0(gm_pc_spec$name_str, seq_along(pc_vars), 'pcs'))
# load boots
boots_pcs_ls = lapply(seq_along(pc_vars), function(n_pcs) {
# make file
layer_spec = make_layer_pc_spec(gm_pc_spec, pc_vars, n_pcs = n_pcs)
boots_dt = sprintf(boots_pat, layer_spec$name, date_tag) %>% fread
return(boots_dt)
}) %>% setNames(paste0(gm_pc_spec$name_str, seq_along(pc_vars), 'pcs'))ancom_ls = c(ancom_ls, ancom_pcs_ls, list(lesions_NAGM_4pcs = ancom_nagm))
boots_ls = c(boots_ls, boots_pcs_ls, list(lesions_NAGM_4pcs = boots_nagm))pcs_coefs_dt = calc_pcs_coefs_dt(pc_vars, boots_ls, labels_dt) # load hand-picked oligo groupings
olg_grps_dt = fread(olg_grps_f)
# restrict to WM oligos, also cell types with min. no. of cells
input_dt = conos_all %>%
.[(matter == "WM") & (type_broad %in% olg_types)] %>%
.[, n_type := .N, by = type_fine ] %>%
.[ n_type >= min_cells ] %>% .[ order(sample_id, type_fine) ]
# calc clusters
set.seed(20211112)
olg_clust_dt = calc_olg_grps_from_clrs(input_dt, k = 3, prior_size = 1e3)
fwrite(olg_clust_dt, file = olg_clust_f)Analysis
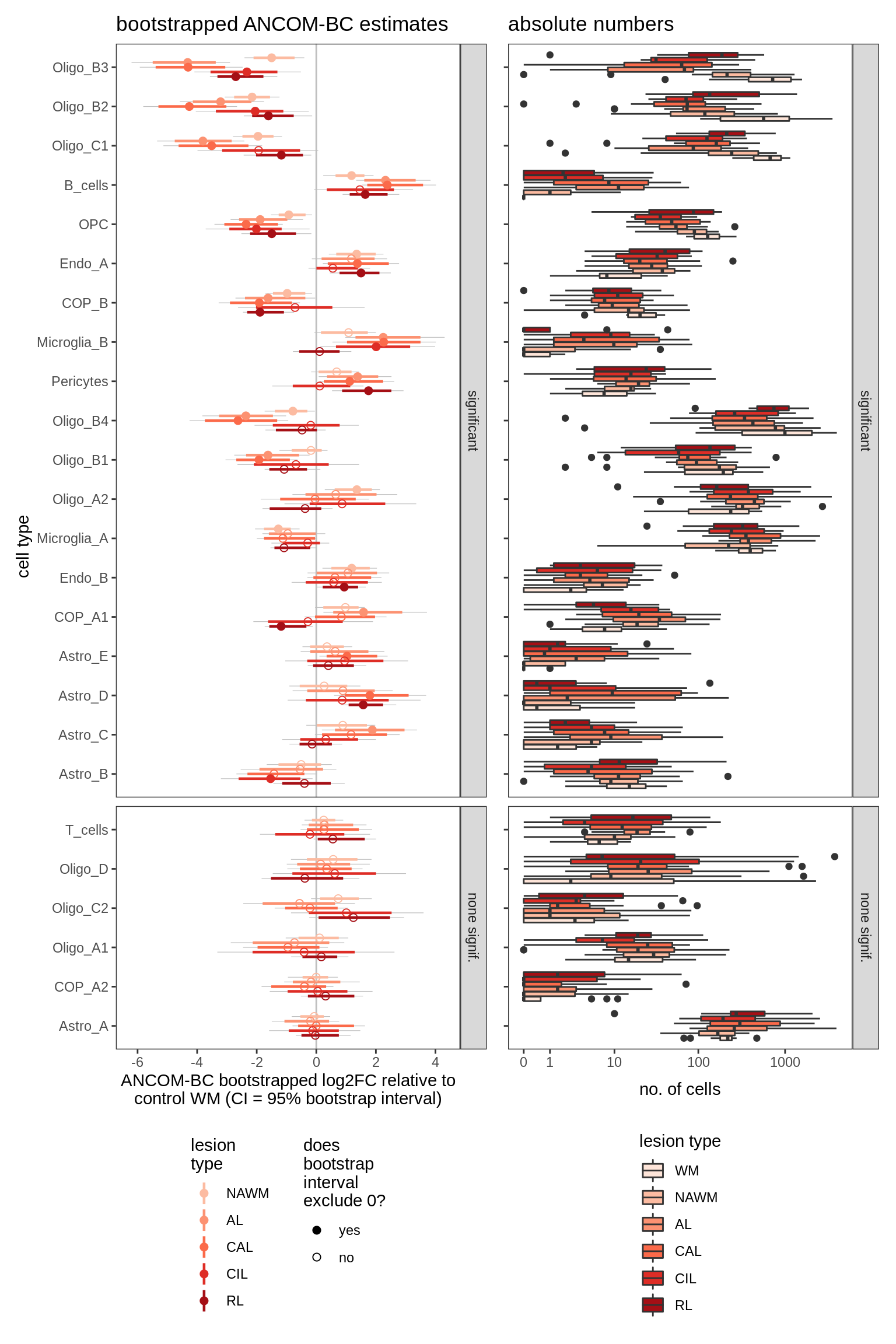
Ctrl GM vs WM
for (m in c('nb', 'edger', 'poisson', 'beta')) {
cat('### ', m, '\n')
print(plot_wm_vs_gm(conos_dt, model = m))
cat('\n\n')
}nb
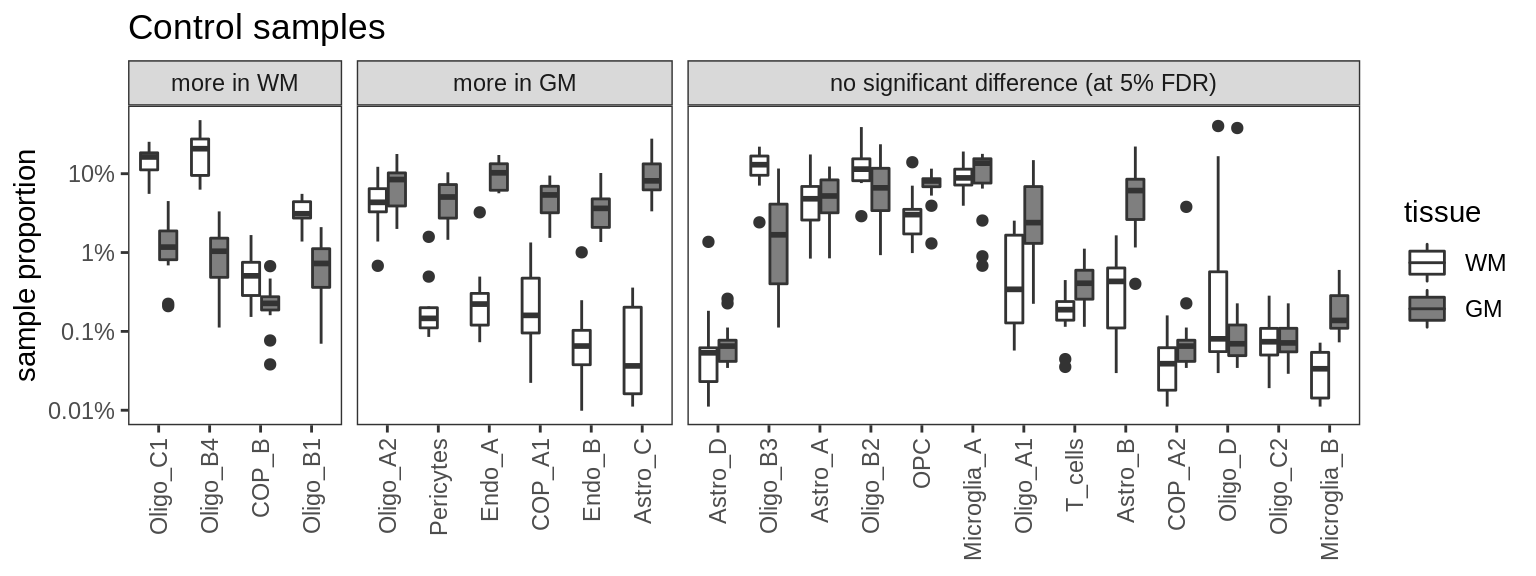
edger
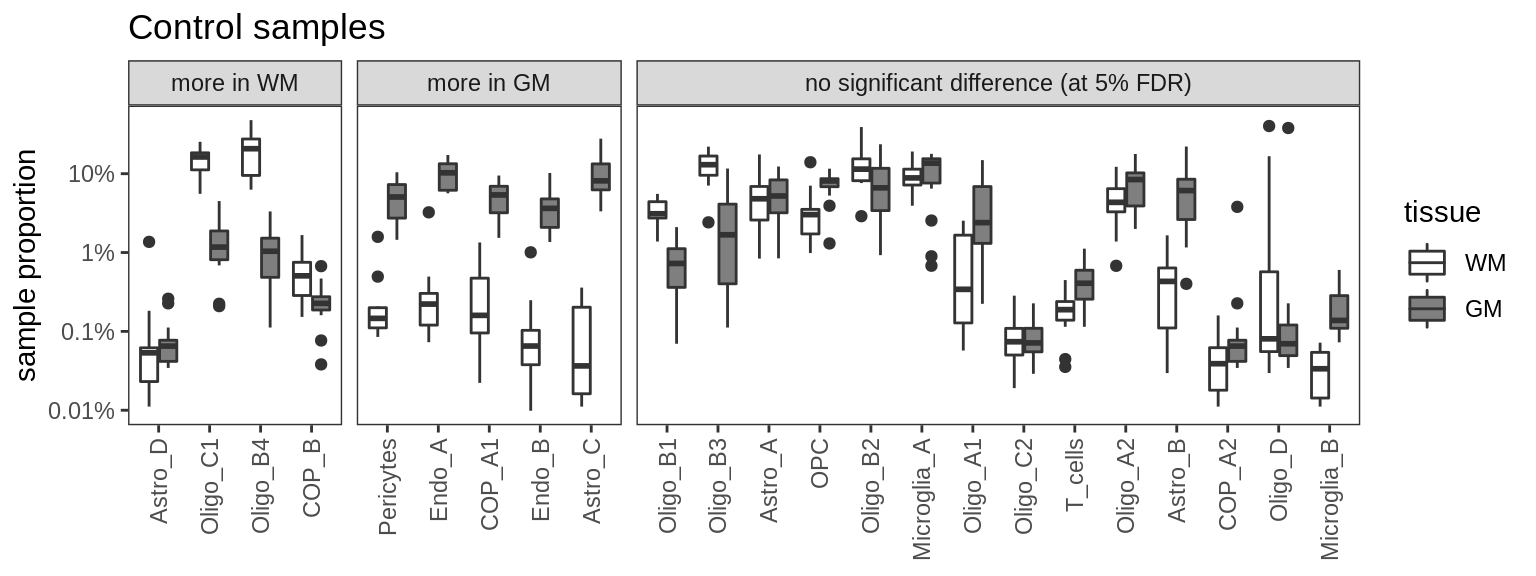
poisson

beta

m = 'nb_w_absolute'
cat('### ', m, '\n')nb_w_absolute
print(plot_wm_vs_gm(conos_dt, model = m))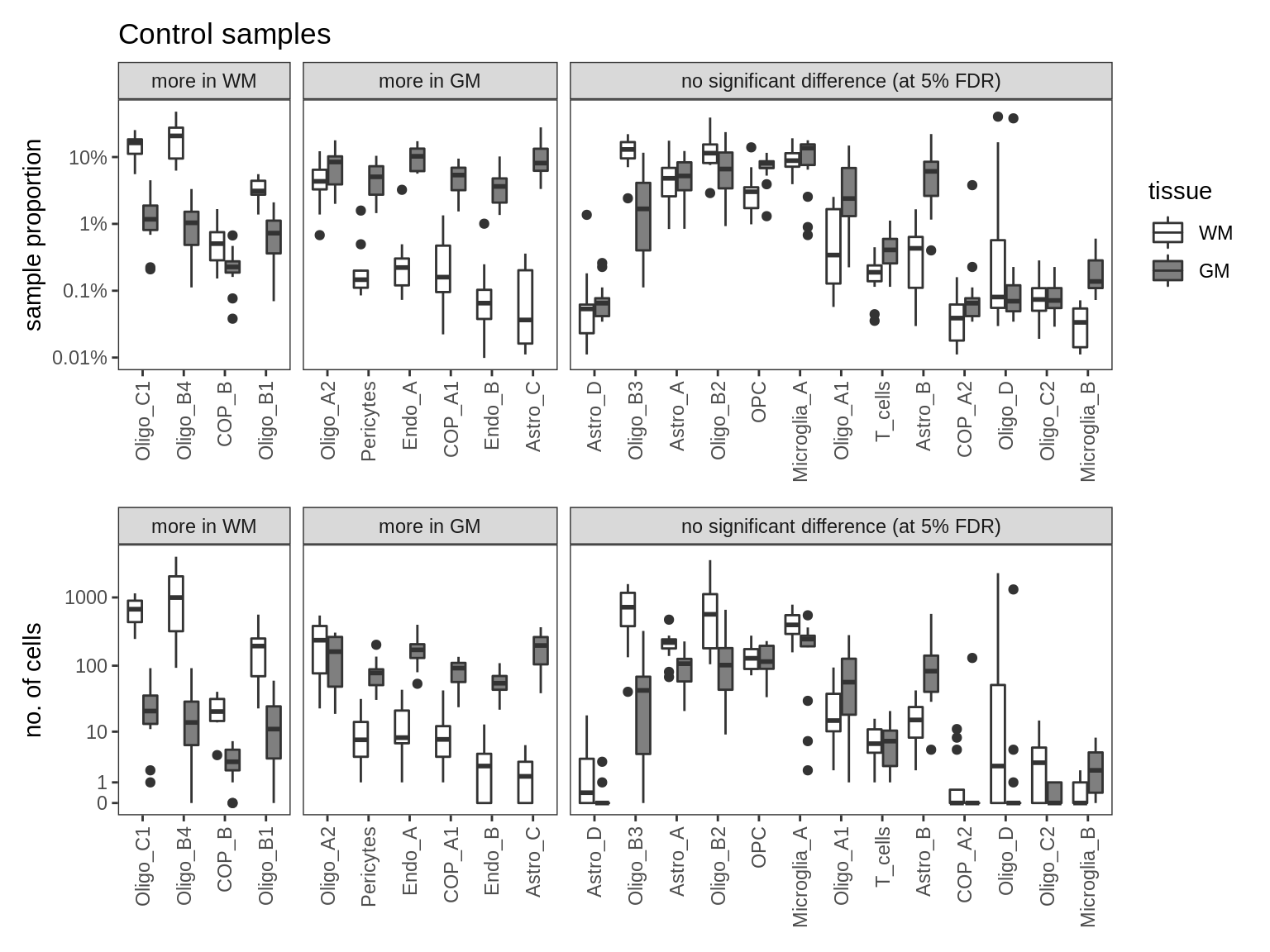
cat('\n\n') Contribution of lesion type and donor to variability in celltype counts
for (nn in names(lrt_ls)) {
cat('### ', nn, '\n')
print(plot_lrt_results(nn, lrt_ls, labels_dt))
cat('\n\n')
}WM
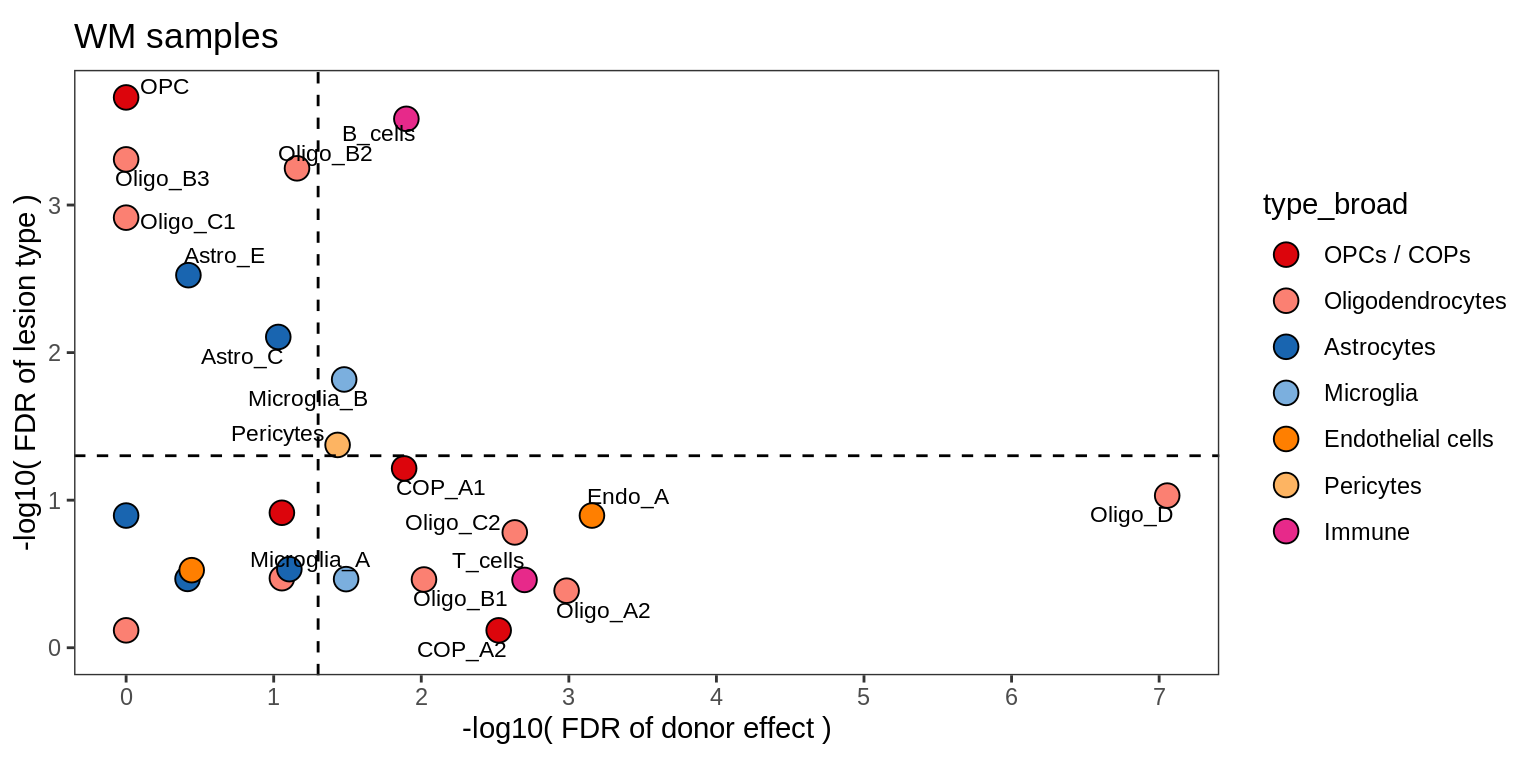
GM
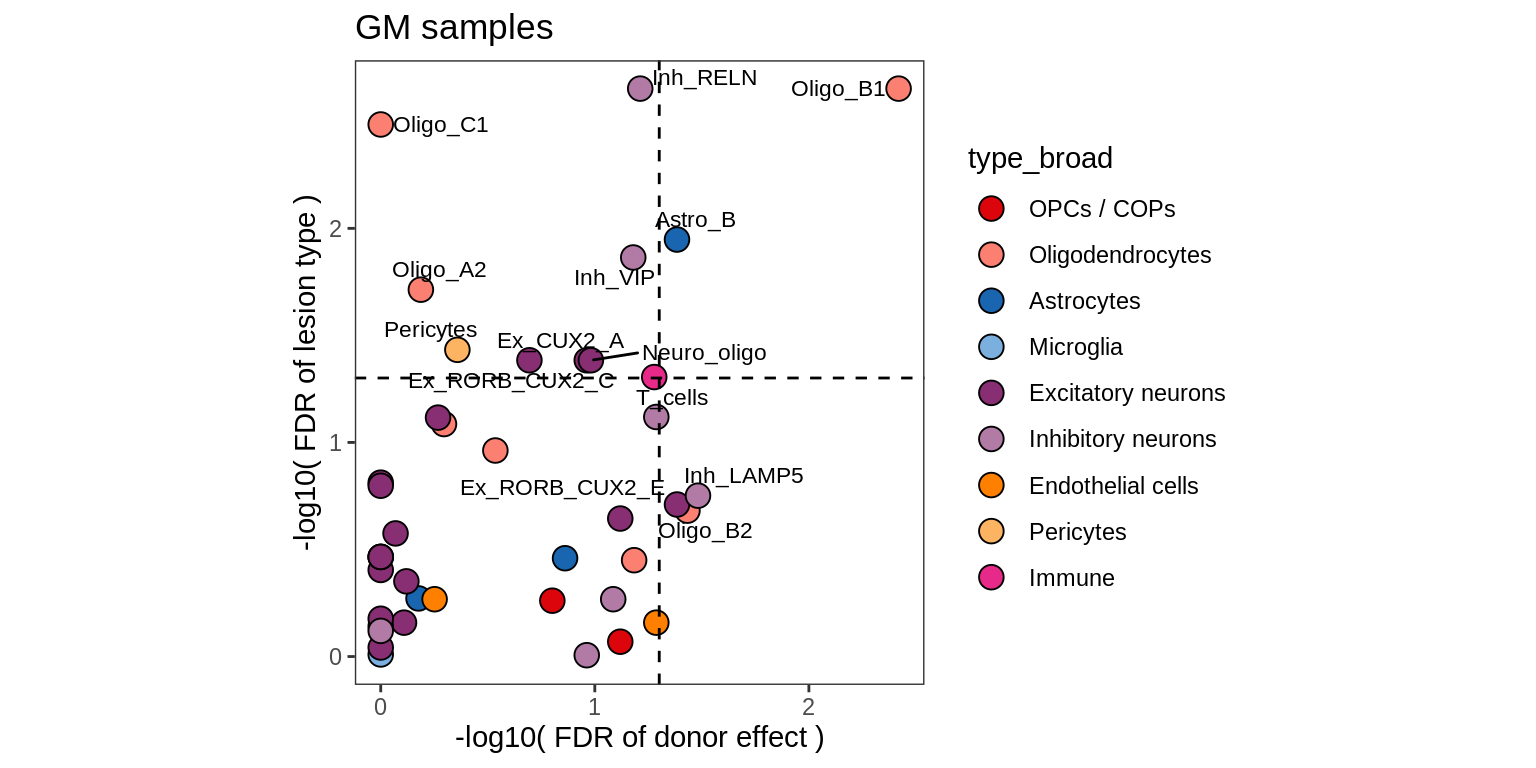
lesions_GM_1pcs

lesions_GM_2pcs
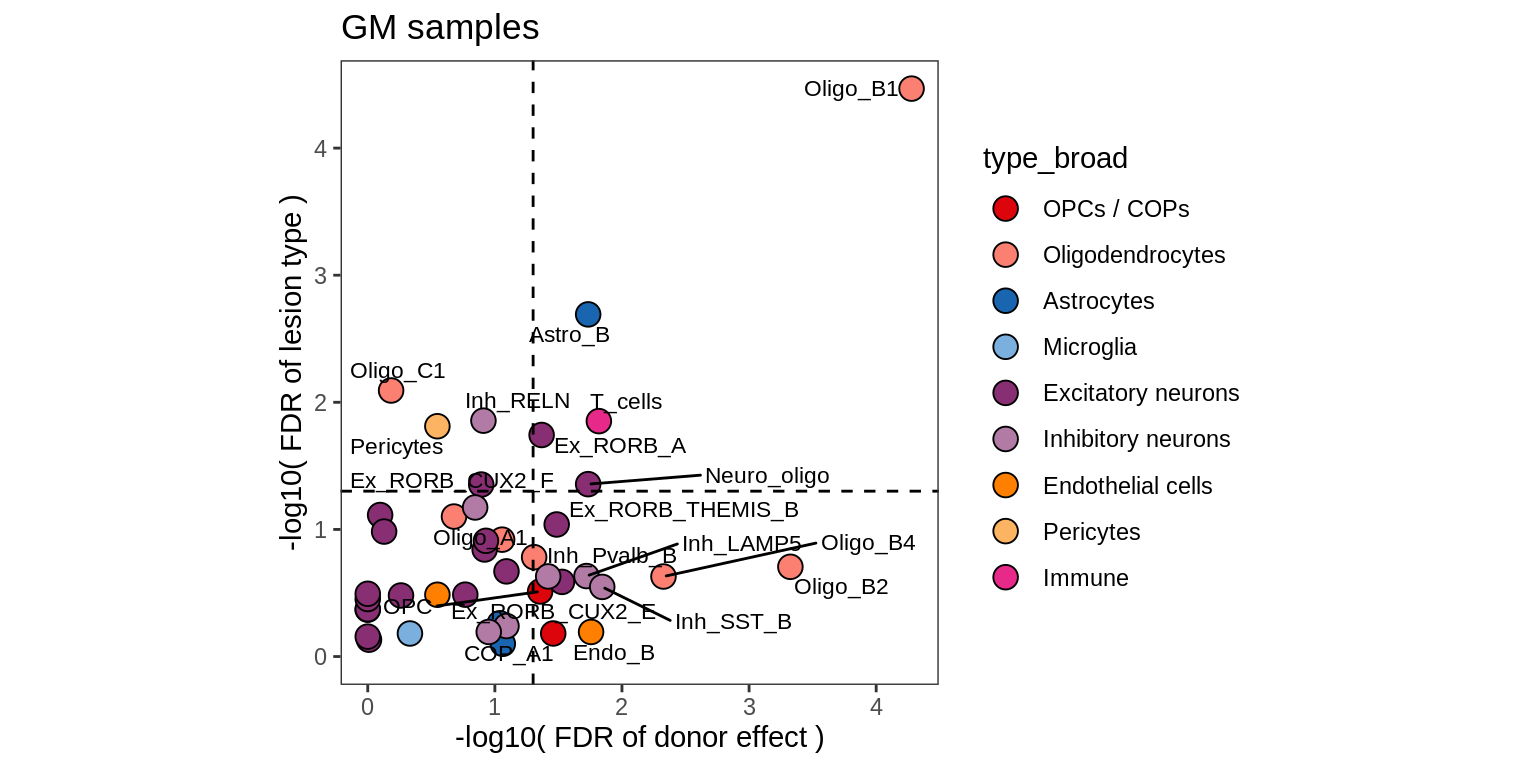
lesions_GM_3pcs
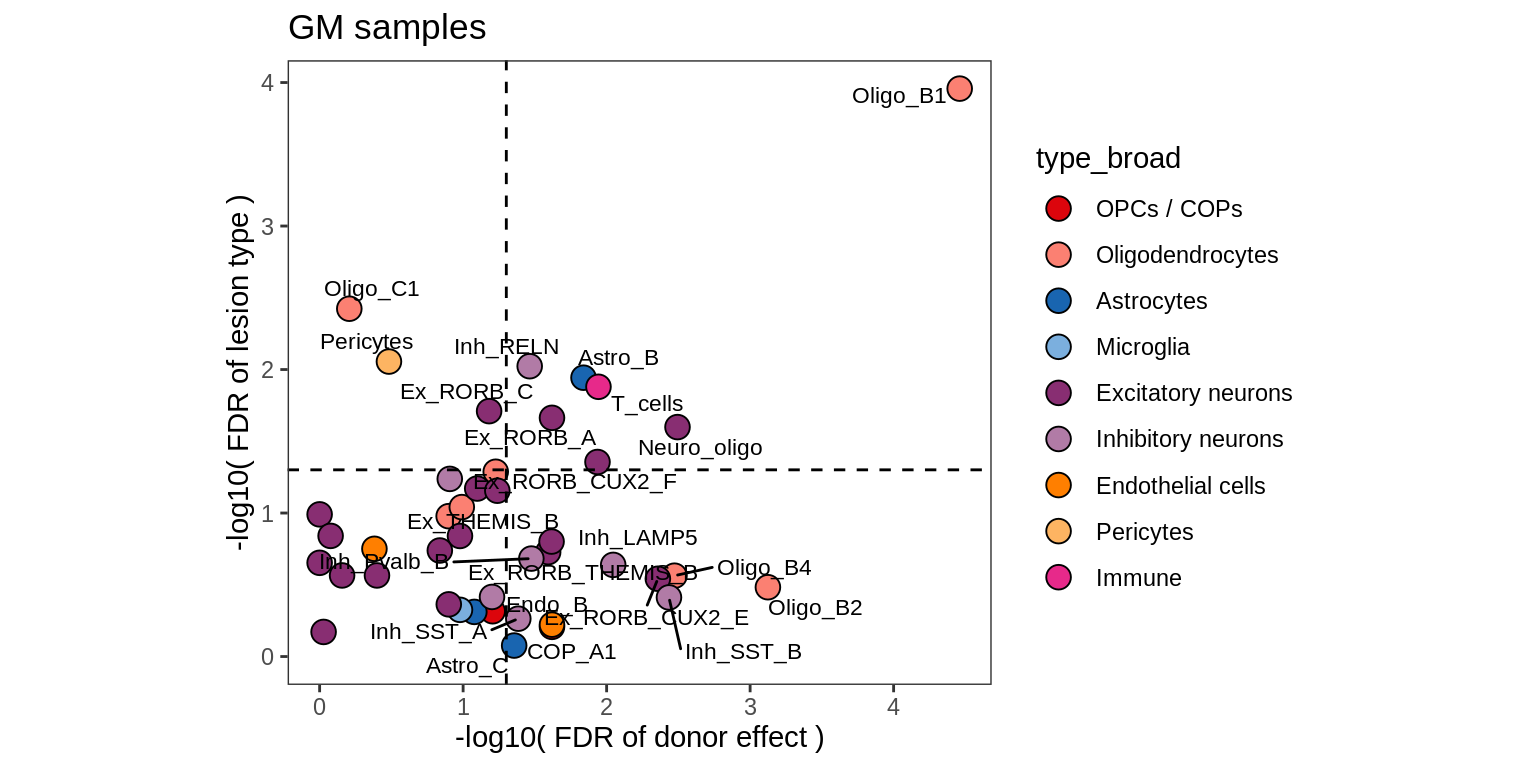
lesions_GM_4pcs
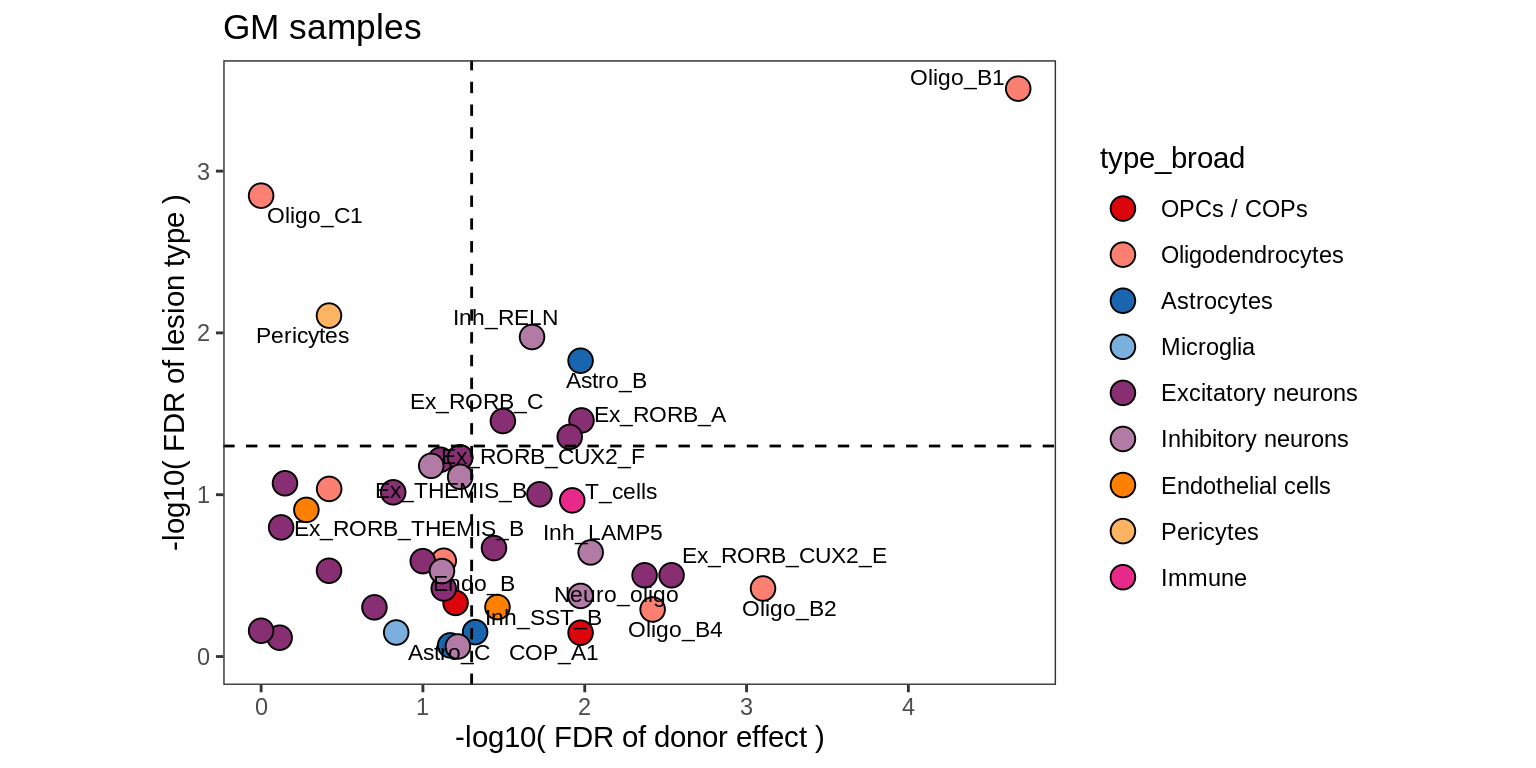
lesions_GM_5pcs

lesions_GM_6pcs
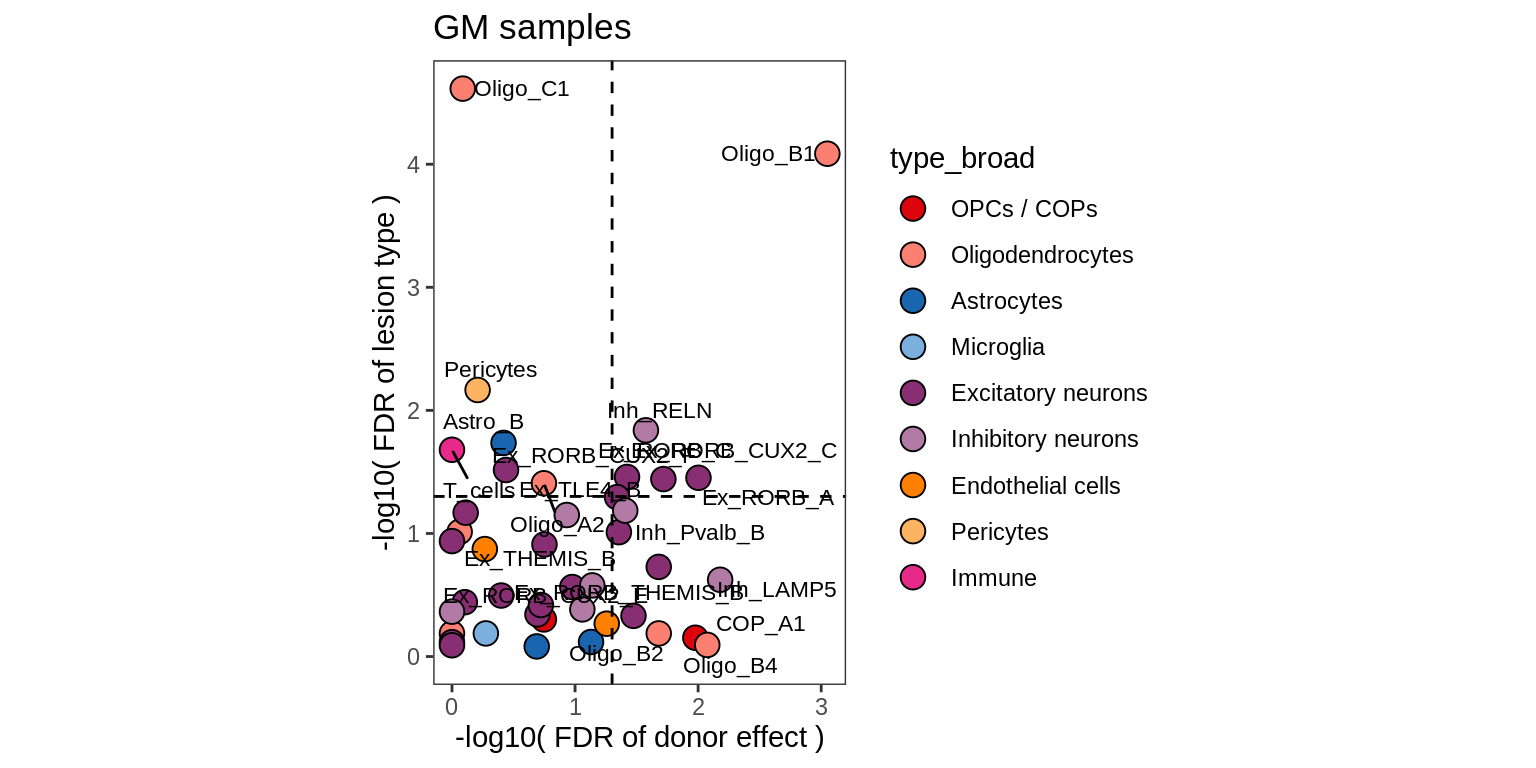
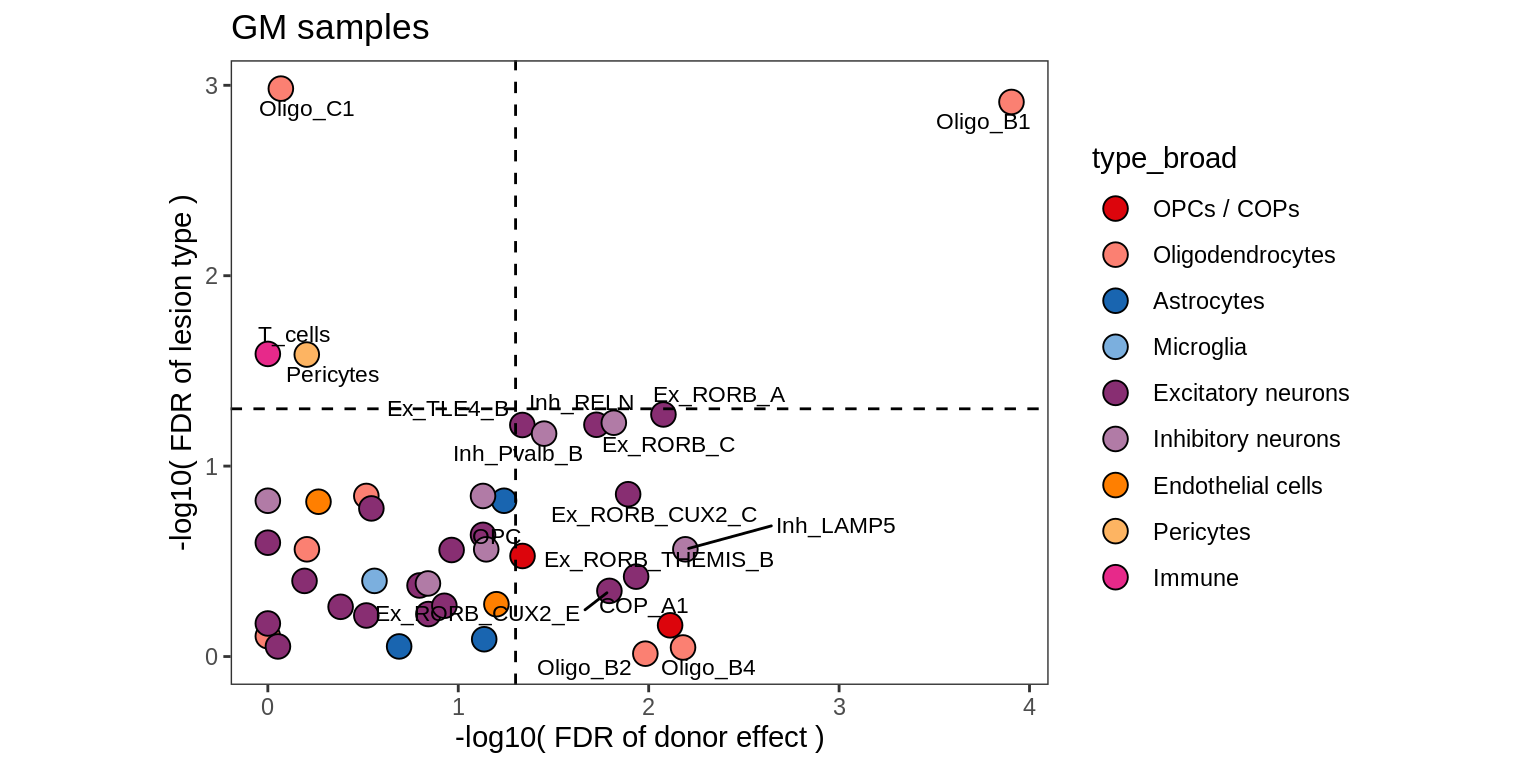
Abundance of GPR17-expressing cells in snRNAseq and IHC
g_seq = plot_raw_cell_counts_sel_gene(prop_fine_f, conos_dt[ matter == "WM" ], sel_g) +
ggtitle('snRNAseq nuclei counts')
g_ihc = plot_gpr17_ihc(gpr17_dt[ matter == "WM" ]) +
ggtitle('GPR17 IHC staining')
g = g_seq / g_ihc
print(g)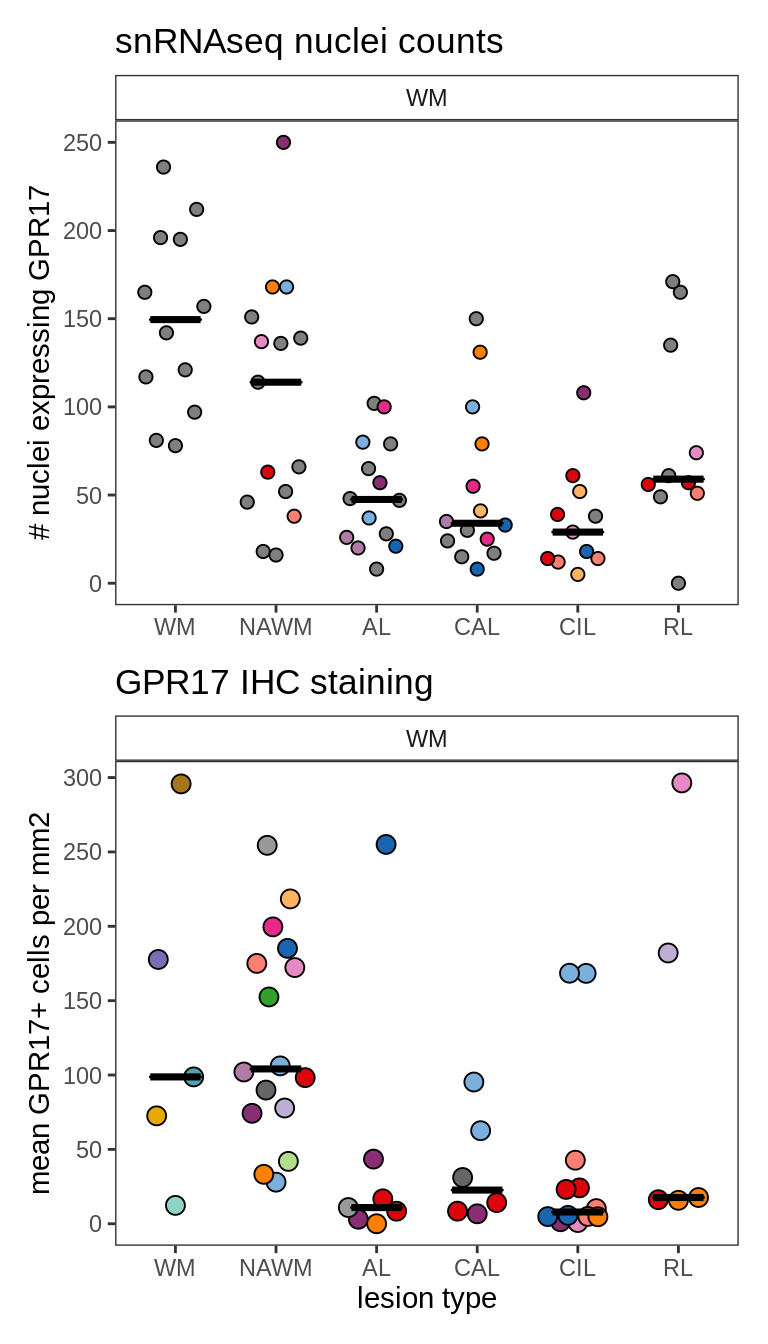
# define models
frm_full = "log(mean_per_mm2 + 0.1) ~ lesion_type + (1 | donor_id )"
frm_fix = "log(mean_per_mm2 + 0.1) ~ lesion_type"
frm_null = "log(mean_per_mm2 + 0.1) ~ 1"
message('evidence of lesion and donor effects in GPR17 IHC data\n')evidence of lesion and donor effects in GPR17 IHC datafor (this_m in c("WM", "GM")) {
# restrict data
data_dt = gpr17_dt[ matter == this_m ] %>%
.[, lesion_type := fct_drop(lesion_type) ]
# fit models
fit_full = glmmTMB::glmmTMB( as.formula(frm_full),
data = data_dt, family = 'gaussian')
fit_fix = update(fit_full, frm_fix)
fit_null = update(fit_full, frm_null)
# compare
message(' in ', this_m, ":")
print(anova(fit_full, fit_fix, fit_null))
} in WM:Data: data_dt
Models:
fit_null: log(mean_per_mm2 + 0.1) ~ 1, zi=~0, disp=~1
fit_fix: log(mean_per_mm2 + 0.1) ~ lesion_type, zi=~0, disp=~1
fit_full: log(mean_per_mm2 + 0.1) ~ lesion_type + (1 | donor_id), zi=~0, disp=~1
Df AIC BIC logLik deviance Chisq Chi Df Pr(>Chisq)
fit_null 2 201.87 205.73 -98.933 197.87
fit_fix 7 189.31 202.83 -87.655 175.31 22.5563 5 0.0004103 ***
fit_full 8 189.50 204.96 -86.752 173.50 1.8056 1 0.1790317
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1 in GM:Data: data_dt
Models:
fit_null: log(mean_per_mm2 + 0.1) ~ 1, zi=~0, disp=~1
fit_fix: log(mean_per_mm2 + 0.1) ~ lesion_type, zi=~0, disp=~1
fit_full: log(mean_per_mm2 + 0.1) ~ lesion_type + (1 | donor_id), zi=~0, disp=~1
Df AIC BIC logLik deviance Chisq Chi Df Pr(>Chisq)
fit_null 2 83.980 86.644 -39.990 79.980
fit_fix 4 61.778 67.106 -26.889 53.778 26.2024 2 2.043e-06 ***
fit_full 5 61.911 68.572 -25.955 51.911 1.8667 1 0.1719
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Compositional grouping heatmaps
Compositional groupings of samples (CLR)
# plot CLR heatmaps
cat("#### WM\n")WM
props_wm = calc_props_dt(conos_dt[ type_broad %in% wm_types ], sample_vars) %>%
.[ matter == "WM" ]
hm_wm = plot_clr_heatmap(props_wm, cluster_rows = TRUE, n_clusters = 5, what = 'clr')
hm_wm = draw(hm_wm, row_dend_width = unit(0.5, "in"), merge_legend = TRUE)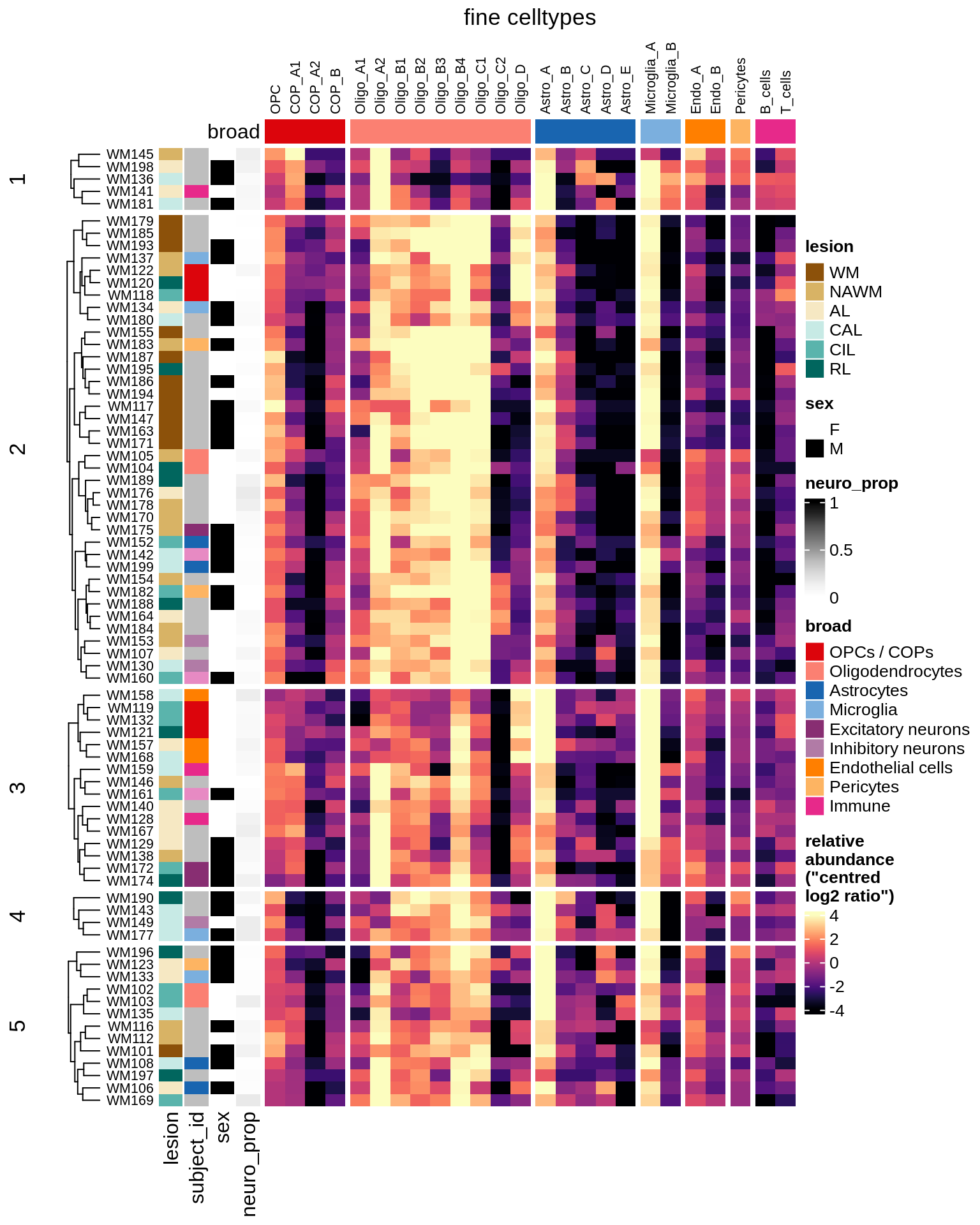
cat('\n\n')cat("#### GM\n")GM
props_gm = calc_props_dt(conos_dt[ type_broad %in% gm_types ], sample_vars) %>%
.[ matter == "GM" ]
hm_gm = plot_clr_heatmap(props_gm, cluster_rows = TRUE, n_clusters = 5, what = 'clr')
hm_gm = draw(hm_gm, row_dend_width = unit(0.5, "in"), merge_legend = TRUE)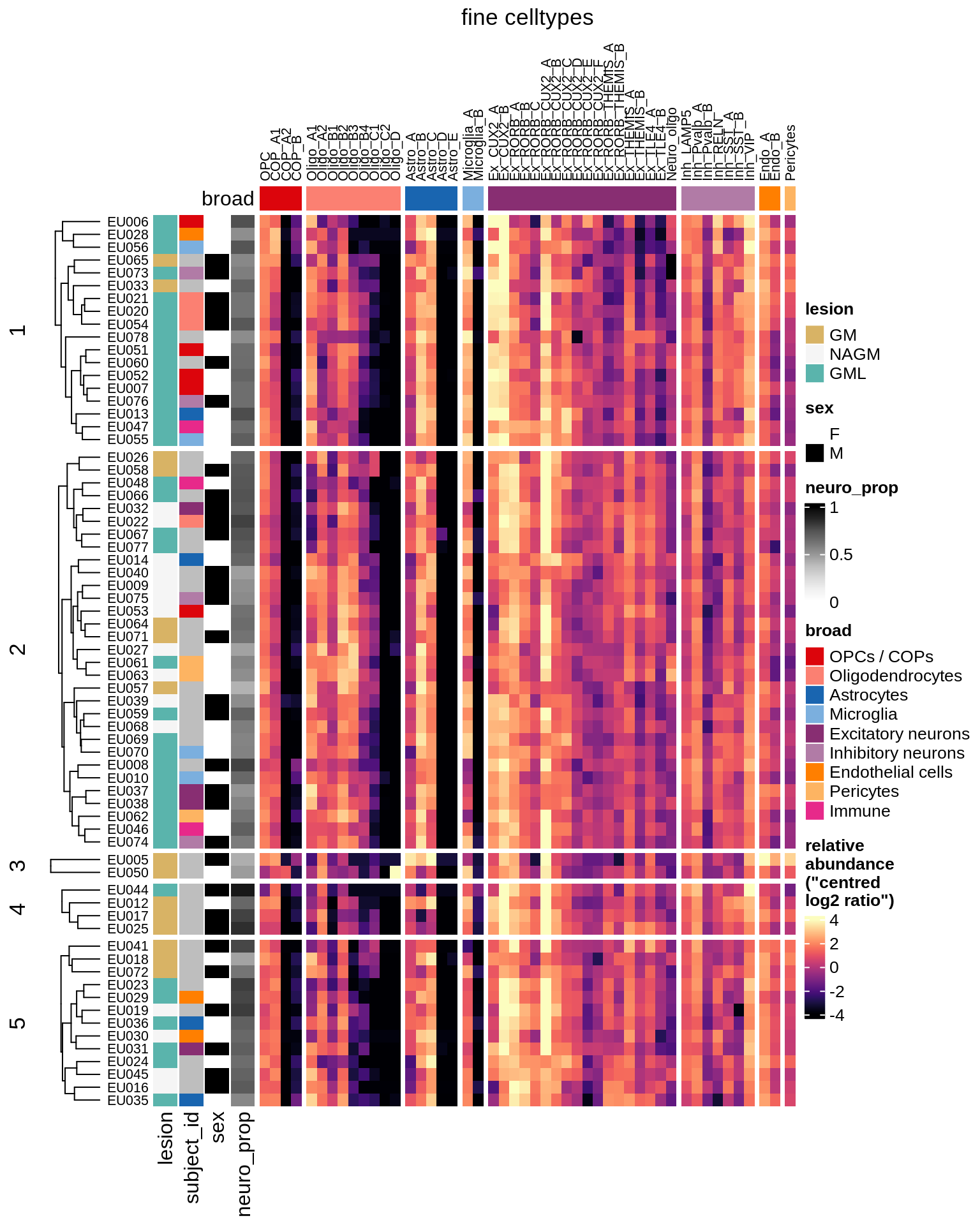
cat('\n\n')# save clusters
save_heatmap_clusters(hm_wm@ht_list[[1]], sprintf(clr_pat, "WM", date_tag))
save_heatmap_clusters(hm_gm@ht_list[[1]], sprintf(clr_pat, "GM", date_tag))Compositional groupings of samples (log proportions)
cat("#### WM\n")WM
props_wm = calc_props_dt(conos_dt[ type_broad %in% wm_types ], sample_vars) %>%
.[ matter == "WM" ] # & neuro_ok == TRUE]
draw(plot_clr_heatmap(props_wm, cluster_rows = FALSE, what = 'log_p'),
row_dend_width = unit(0.5, "in"), merge_legend = TRUE)
cat('\n\n')cat("#### GM\n")GM
props_gm = calc_props_dt(conos_dt[ type_broad %in% gm_types ], sample_vars) %>%
.[ matter == "GM" ] # & neuro_ok == TRUE]
draw(plot_clr_heatmap(props_gm, cluster_rows = FALSE, what = 'log_p'),
row_dend_width = unit(0.5, "in"), merge_legend = TRUE)
cat('\n\n')Compositional groupings of samples (log proportions, oligodendroglia only)
cat("#### WM\n")WM
props_wm = calc_props_dt(conos_dt[ type_broad %in% olg_types ], sample_vars) %>%
.[ matter == "WM" ] # & neuro_ok == TRUE]
draw(plot_clr_heatmap(props_wm, cluster_rows = FALSE, what = 'log_p'),
row_dend_width = unit(1, "in"), merge_legend = TRUE)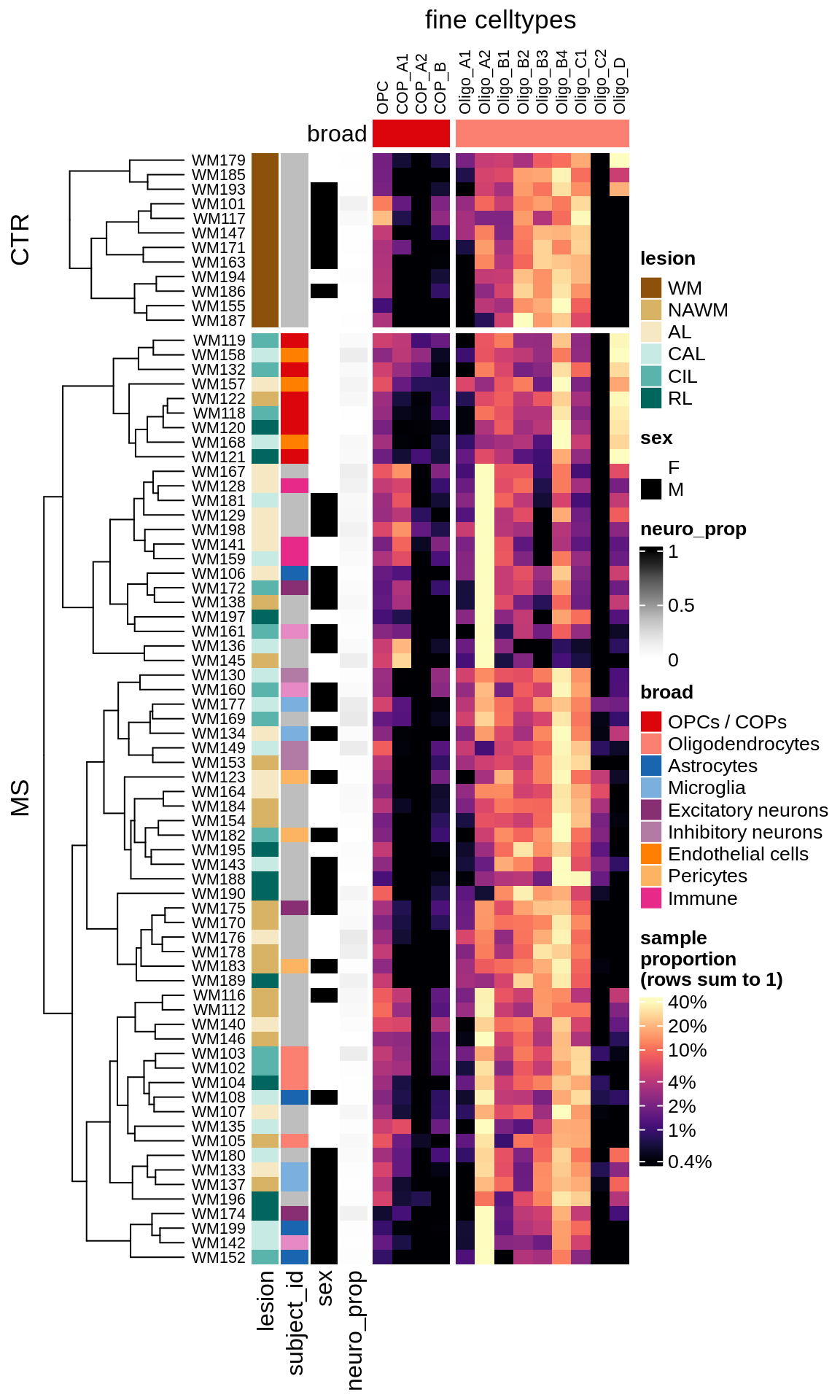
cat('\n\n')cat("#### GM\n")GM
props_gm = calc_props_dt(conos_dt[ type_broad %in% olg_types ], sample_vars) %>%
.[ matter == "GM" ] # & neuro_ok == TRUE]
draw(plot_clr_heatmap(props_gm, cluster_rows = FALSE, what = 'log_p'),
row_dend_width = unit(1, "in"), merge_legend = TRUE)
cat('\n\n')Compositional groupings of samples (log proportions, oligodendroglia only, ordered by patient)
cat("#### WM\n")WM
props_wm = calc_props_dt(conos_dt[ type_broad %in% olg_types ], sample_vars) %>%
.[ matter == "WM" ] # & neuro_ok == TRUE]
draw(plot_clr_heatmap(props_wm, cluster_rows = FALSE, what = 'log_p',
order_subj = TRUE), row_dend_width = unit(1, "in"), merge_legend = TRUE)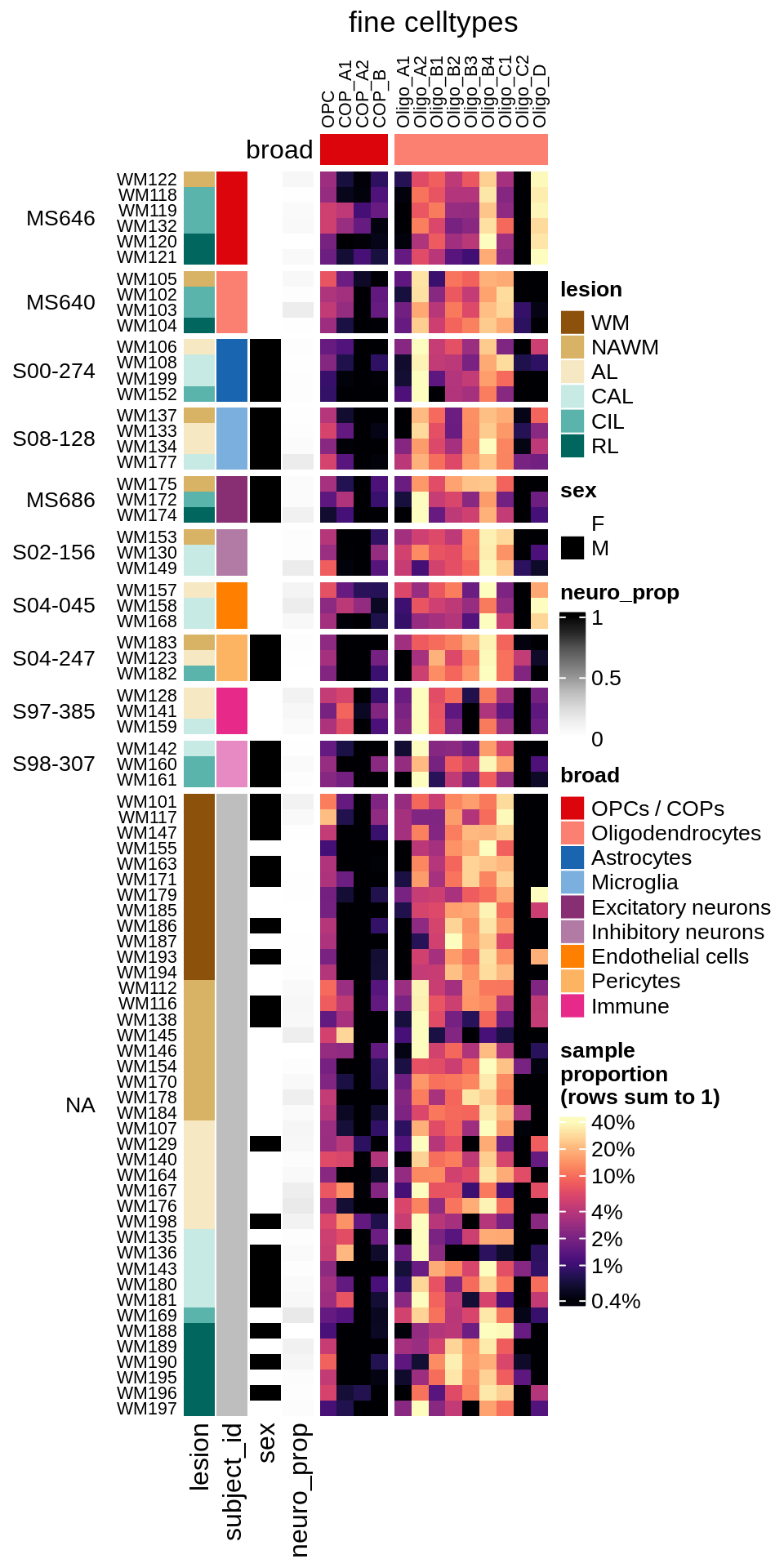
cat('\n\n')cat("#### GM\n")GM
props_gm = calc_props_dt(conos_dt[ type_broad %in% olg_types ], sample_vars) %>%
.[ matter == "GM" ] # & neuro_ok == TRUE]
draw(plot_clr_heatmap(props_gm, cluster_rows = FALSE, what = 'log_p',
order_subj = TRUE), row_dend_width = unit(1, "in"), merge_legend = TRUE)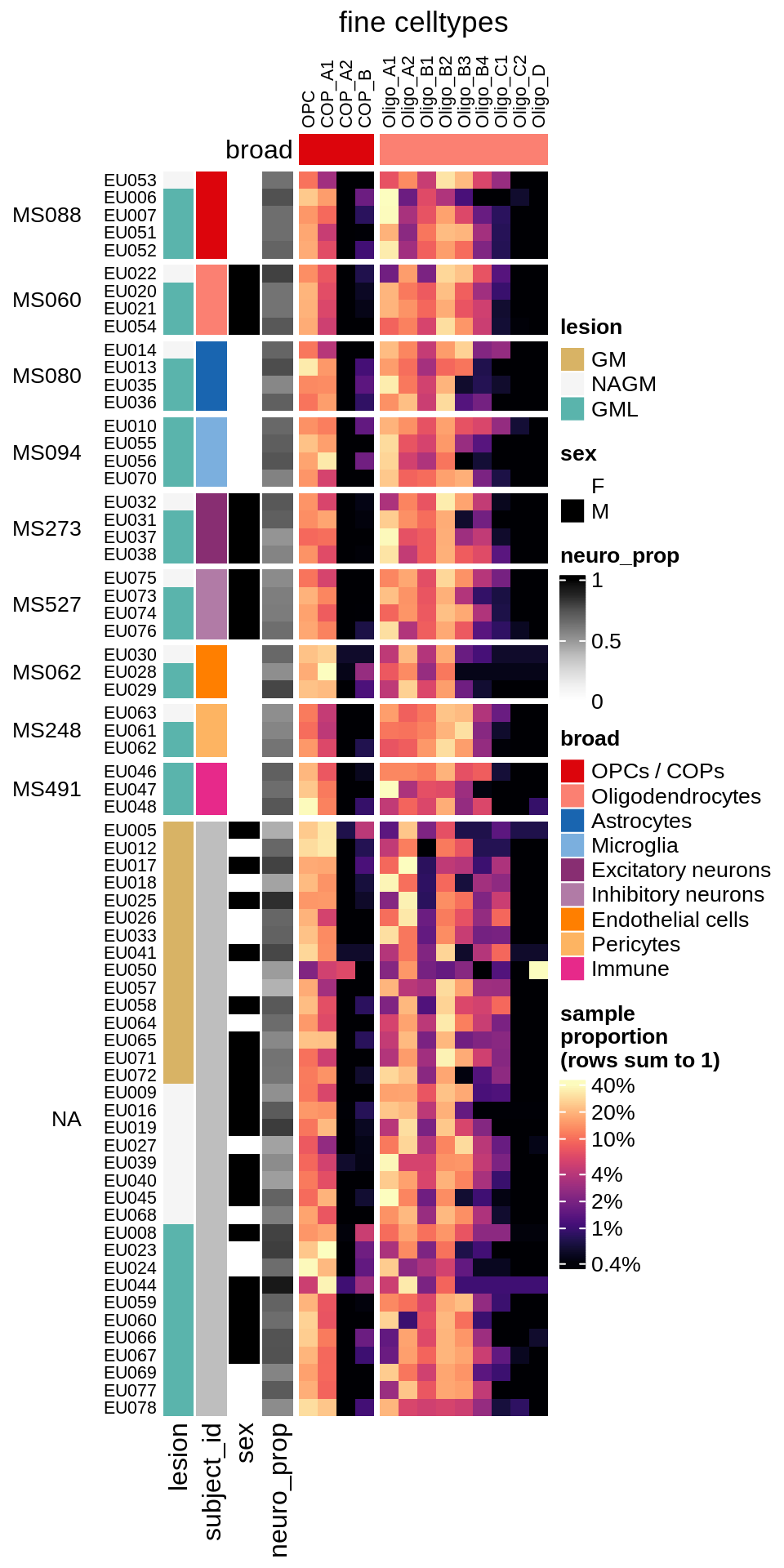
cat('\n\n')Compositional groupings of samples (GM, CLR, neurons only)
props_gm = calc_props_dt(conos_dt[ type_broad %in% neu_types ], sample_vars) %>%
.[ matter == "GM" ] %>%
merge(wide_neu[, .(sample_id, ctrl_PC01, ctrl_PC02, ctrl_PC03, ctrl_PC04)],
by = 'sample_id')
draw(plot_clr_heatmap(props_gm, cluster_rows = FALSE, what = 'clr'),
row_dend_width = unit(1, "in"), merge_legend = TRUE)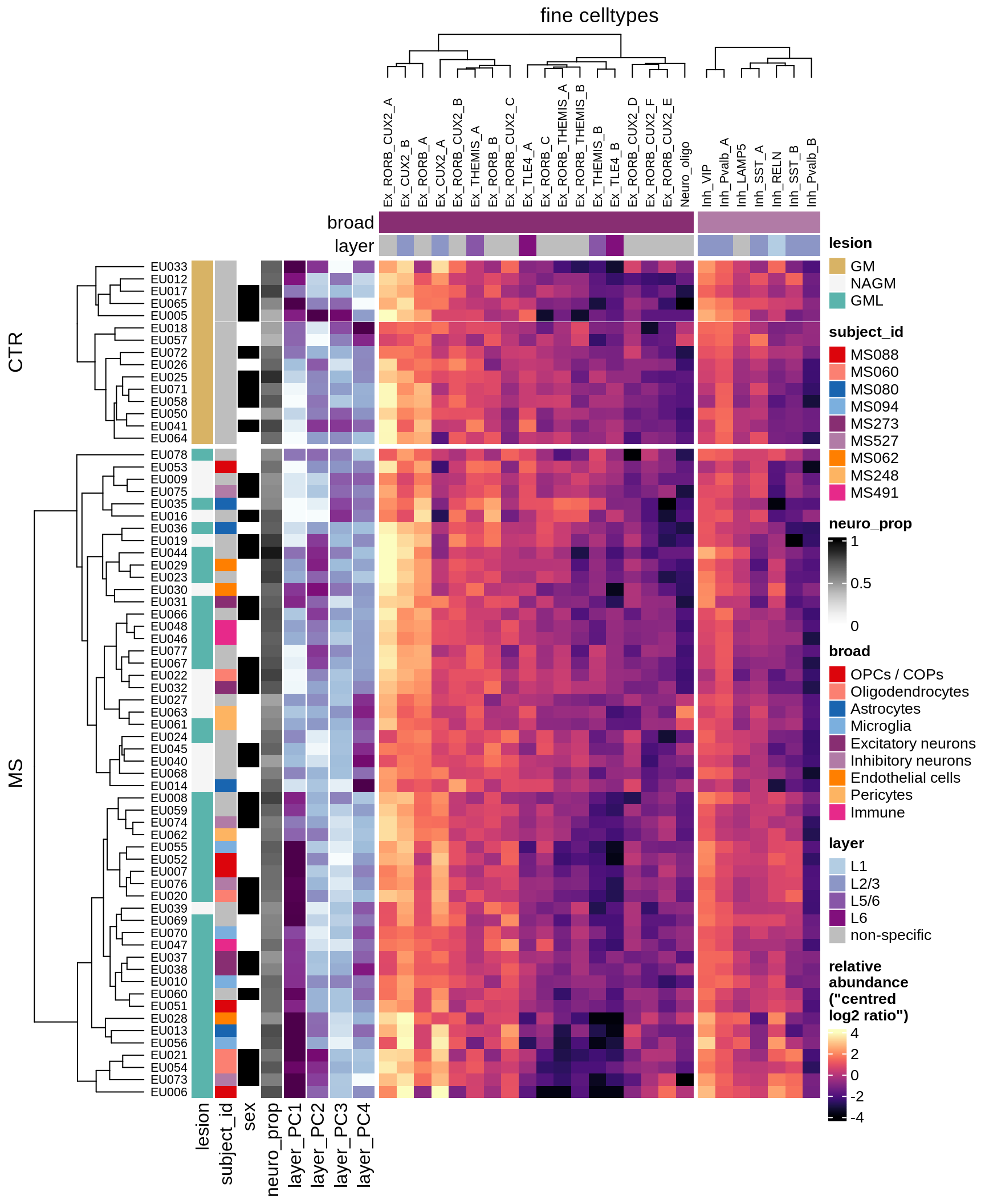
Barplots of WM oligodendroglia proportions
m = 'WM'
cat('### By hand\n')By hand
print(plot_sample_propn_barplots(conos_dt[matter == m], olg_grps_dt,
types = olg_types, show_broad = FALSE))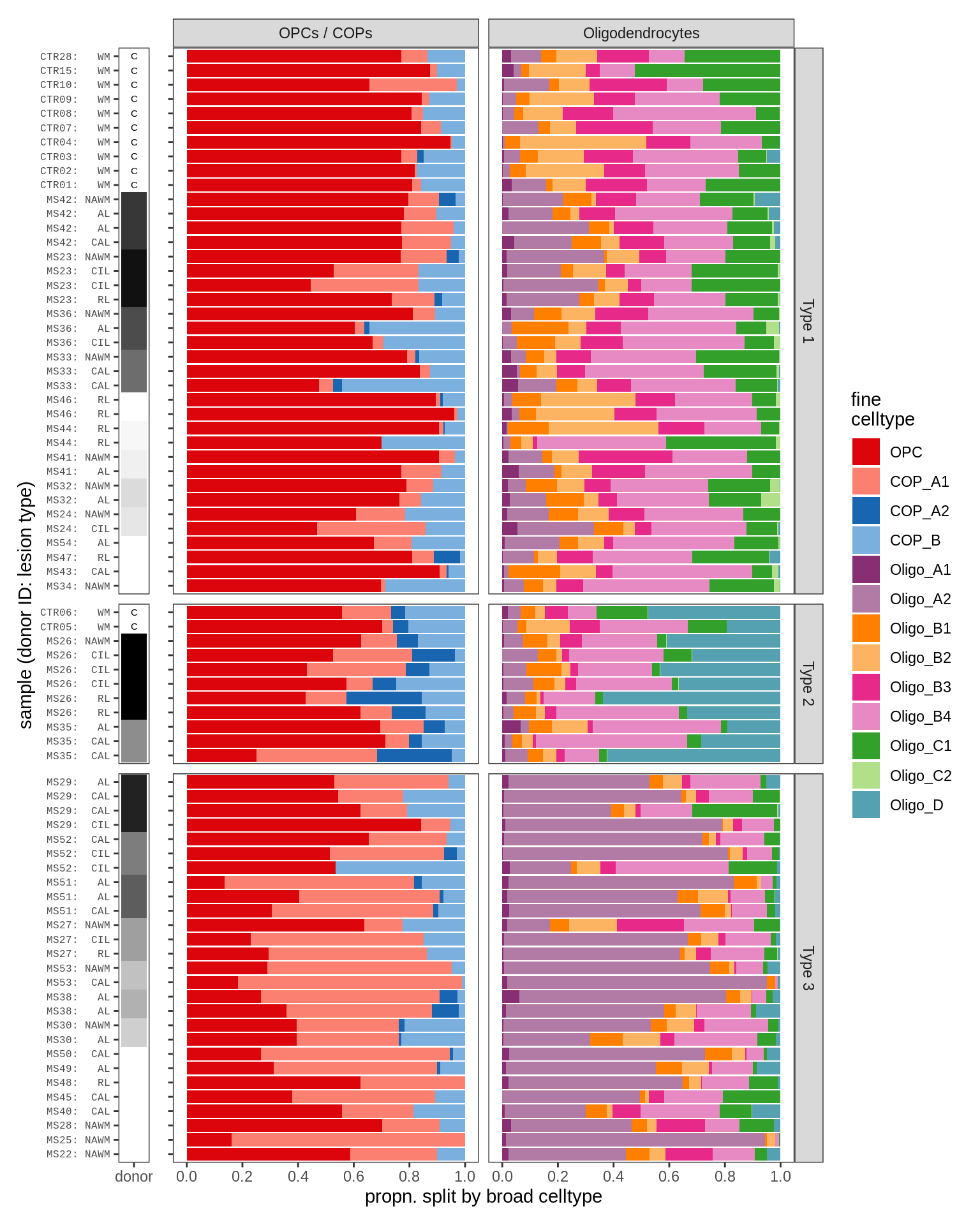
cat('\n\n')cat('### CLR clusters\n')CLR clusters
print(plot_sample_propn_barplots(conos_dt[matter == m], olg_clust_dt,
types = olg_types, show_broad = FALSE))
cat('\n\n')cat('### By hand, no OPC/Oligo split\n')By hand, no OPC/Oligo split
print(plot_sample_propn_barplots(conos_dt[matter == m], olg_grps_dt,
types = olg_types, show_broad = FALSE, split_broad = FALSE))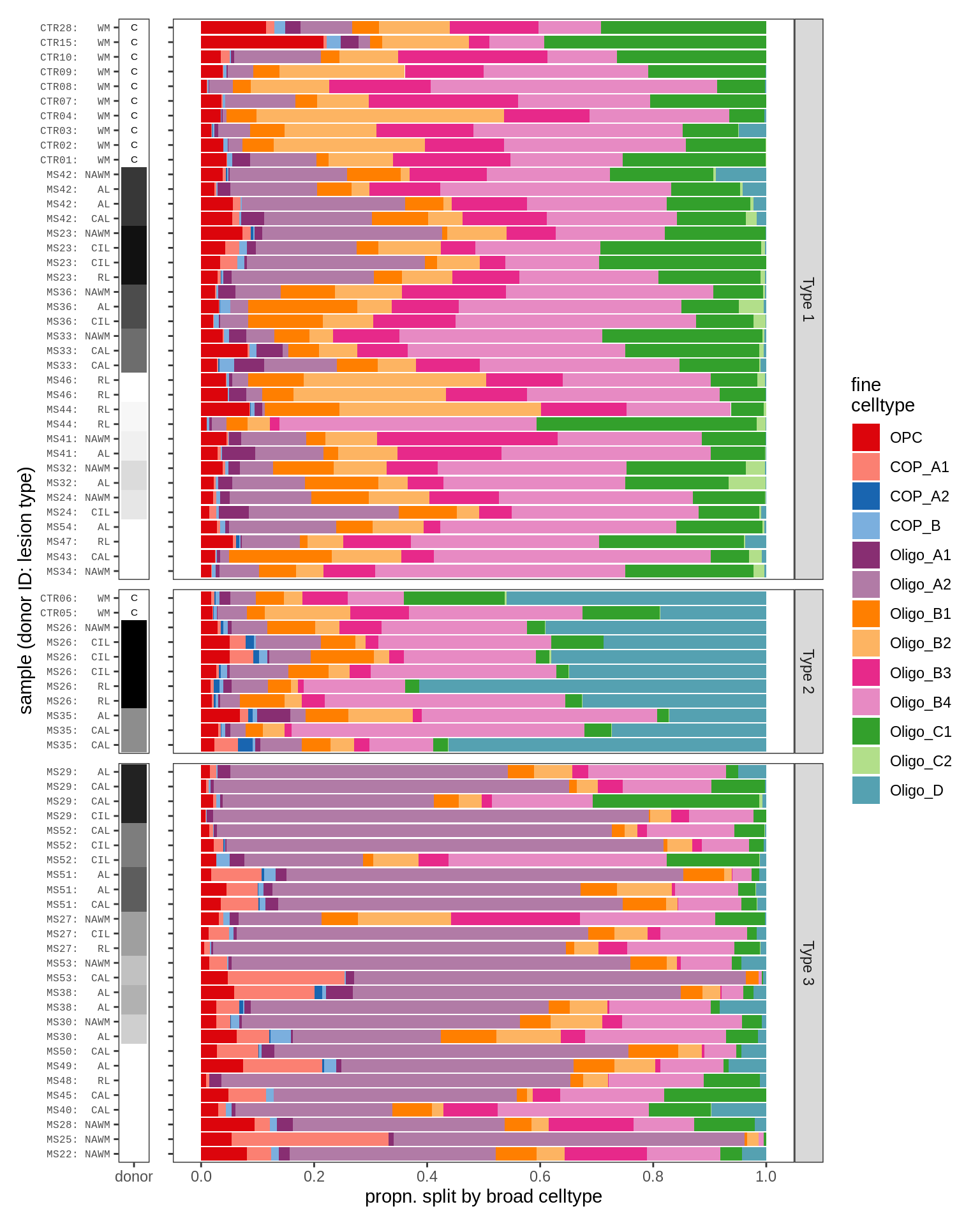
cat('\n\n')CLR plots of oligodendroglia
# set up what we want
names_ls = c("WM", "GM_all", "GM_all_resid", "GM_out", "GM_out_resid")
title_ls = c("WM", "GM", "GM (w/o layers)", "GM (SD016/13 excluded)",
"GM (w/o layers, no SD016/13)") %>% setNames(names_ls)
matter_ls = c("WM", "GM", "GM", "GM", "GM") %>% setNames(names_ls)
outs_ls = c(FALSE, FALSE, FALSE, TRUE, TRUE) %>% setNames(names_ls)
layers_ls = c(FALSE, FALSE, TRUE, FALSE, TRUE) %>% setNames(names_ls)
outlier_ls = "SD016/13"
sel_pcs = c("ctrl_PC01", "ctrl_PC02", "ctrl_PC03", "ctrl_PC04")
# run through
for (nn in names_ls) {
# do title
cat('### ', title_ls[[ nn ]], '\n')
# get data
input_dt = conos_dt %>%
.[(matter == matter_ls[[ nn ]]) & (type_broad %in% olg_types)]
# remove outliers?
if (outs_ls[[nn]])
input_dt = input_dt[ !(subject_id %in% outlier_ls) ]
# restrict to samples with min. no. of cells
input_dt = input_dt[, n_type := .N, by = type_fine ] %>%
.[ n_type >= min_cells ] %>% .[ order(sample_id, type_fine) ]
# do either with or without layers
if (layers_ls[[ nn ]]) {
suppressWarnings(print(plot_sample_clrs_layers(input_dt,
all_pcs_dt[, c("sample_id", sel_pcs), with = FALSE ])))
} else {
suppressWarnings(print(plot_sample_clrs(input_dt, prior_size = 1e3)))
}
cat('\n\n')
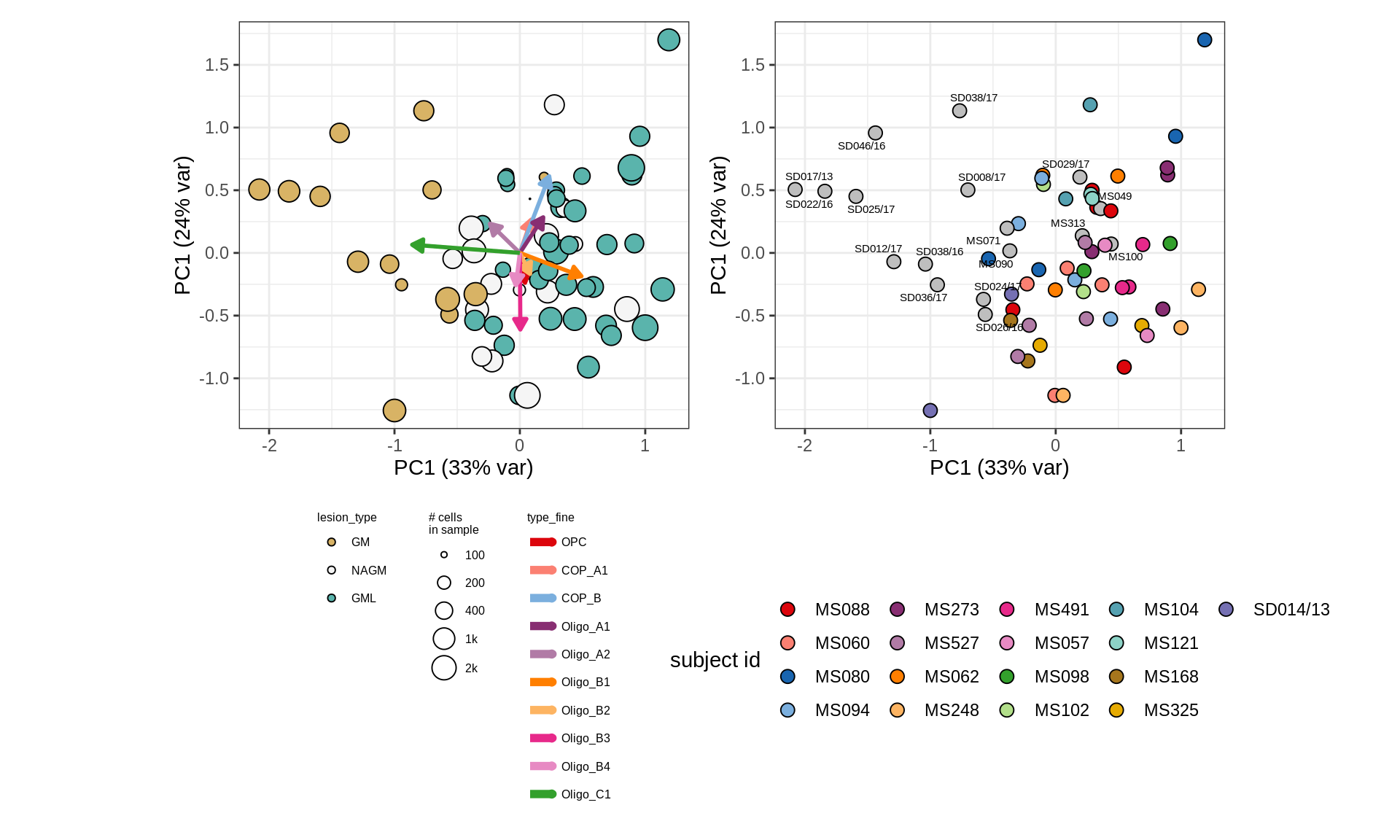
}WM
GM

GM (w/o layers)
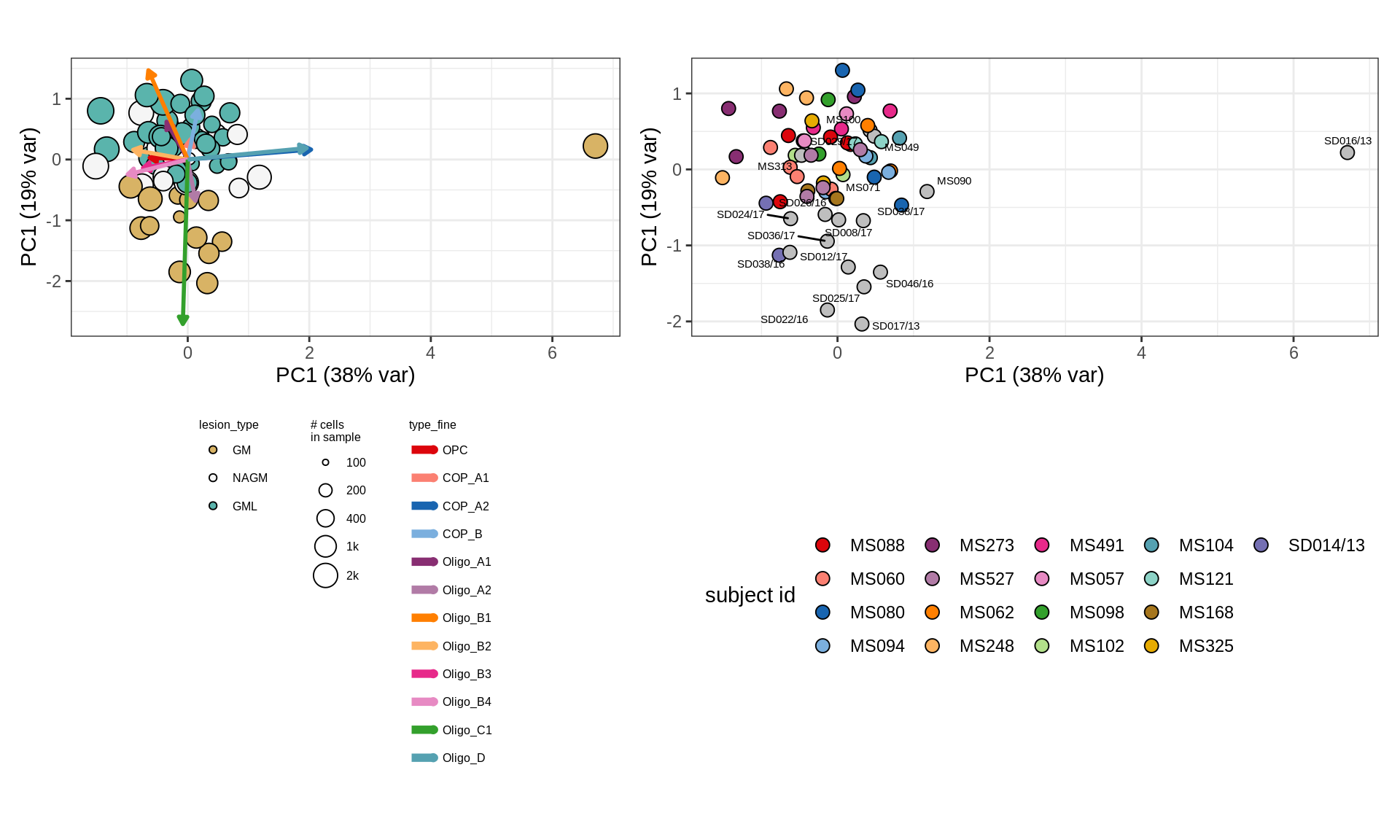
GM (SD016/13 excluded)
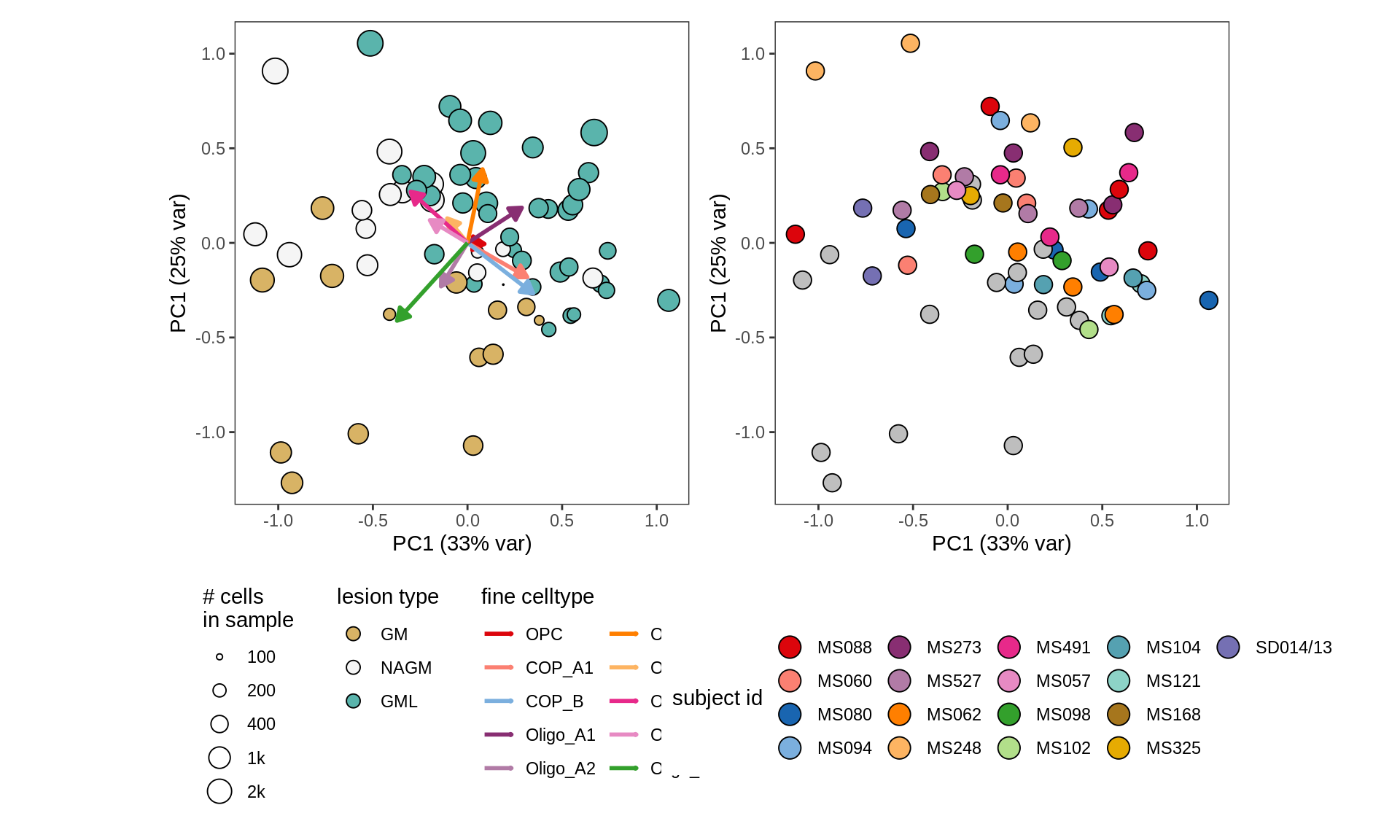
GM (w/o layers, no SD016/13)
# plot
cat('### ', 'WM w oligo grps by hand', '\n')WM w oligo grps by hand
# annotate with oligo groups
input_dt = conos_dt %>%
.[(matter == "WM") & (type_broad %in% olg_types)] %>%
merge(olg_grps_dt, by = "subject_id") %>%
.[, n_type := .N, by = type_fine ] %>%
.[ n_type >= min_cells ] %>% .[ order(sample_id, type_fine) ]
# plot
suppressWarnings(print(plot_sample_clrs(input_dt, prior_size = 1e3)))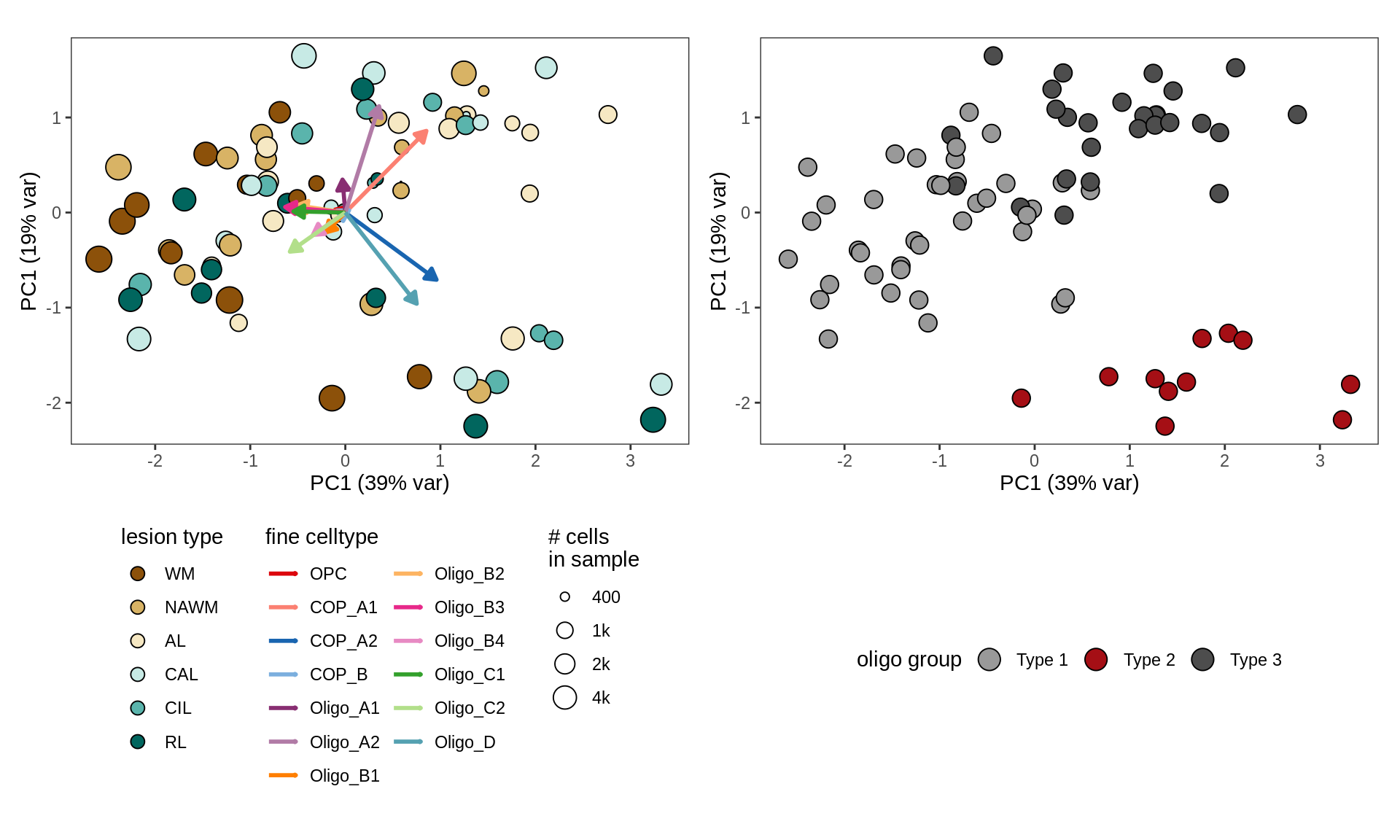
cat('\n\n')# add CLR clustering
cat('### ', 'WM w oligo grps from CLR clusters', '\n')WM w oligo grps from CLR clusters
# annotate with oligo groups
input_dt = conos_dt %>%
.[(matter == "WM") & (type_broad %in% olg_types)] %>%
merge(olg_clust_dt, by = "sample_id") %>%
.[, n_type := .N, by = type_fine ] %>%
.[ n_type >= min_cells ] %>% .[ order(sample_id, type_fine) ]
# plot
suppressWarnings(print(plot_sample_clrs(input_dt, prior_size = 1e3)))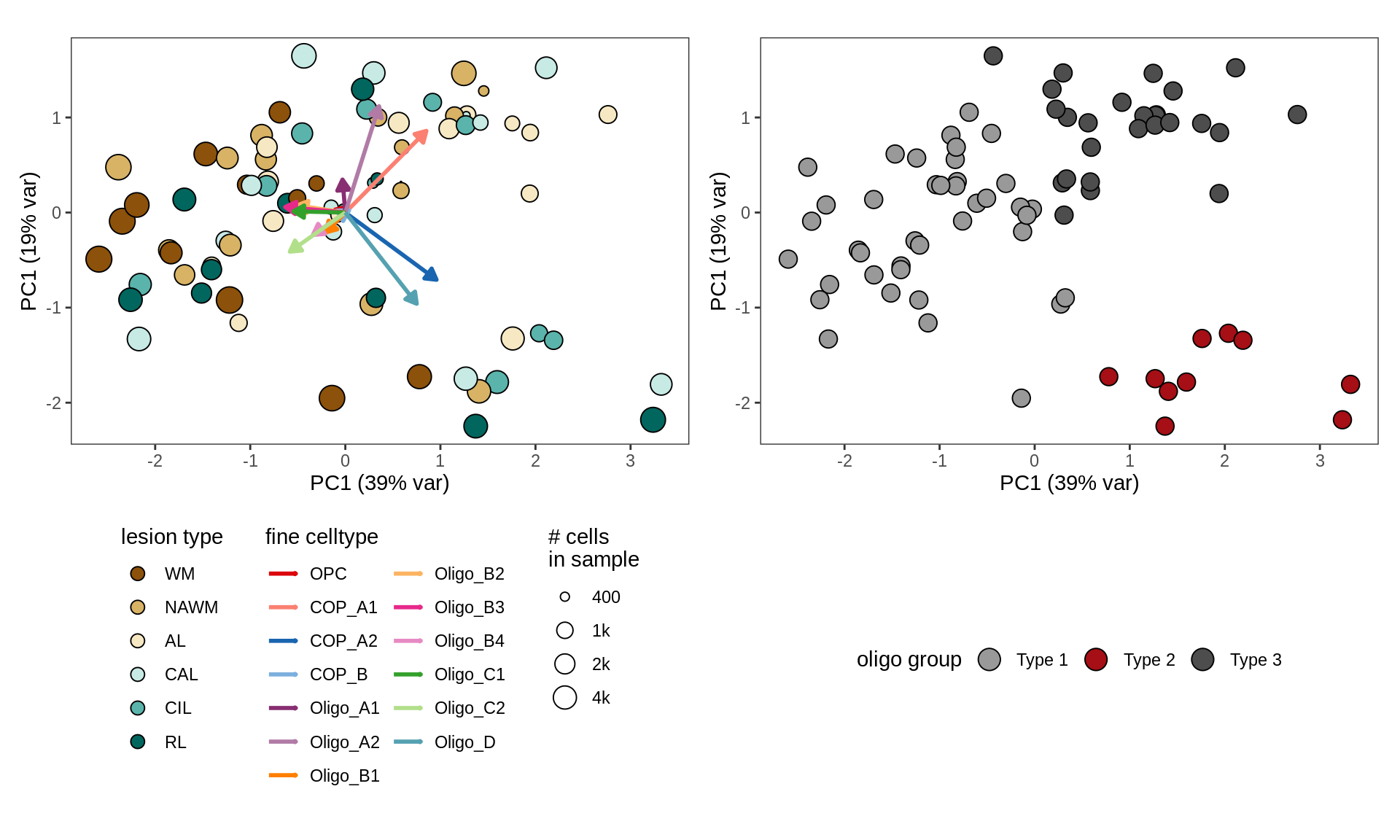
cat('\n\n')GM layers
Proportions with layers
(plot_propns_layers(props_dt[ matter == "GM" & str_detect(type_broad, 'neuron') ]))Warning: Transformation introduced infinite values in continuous y-axis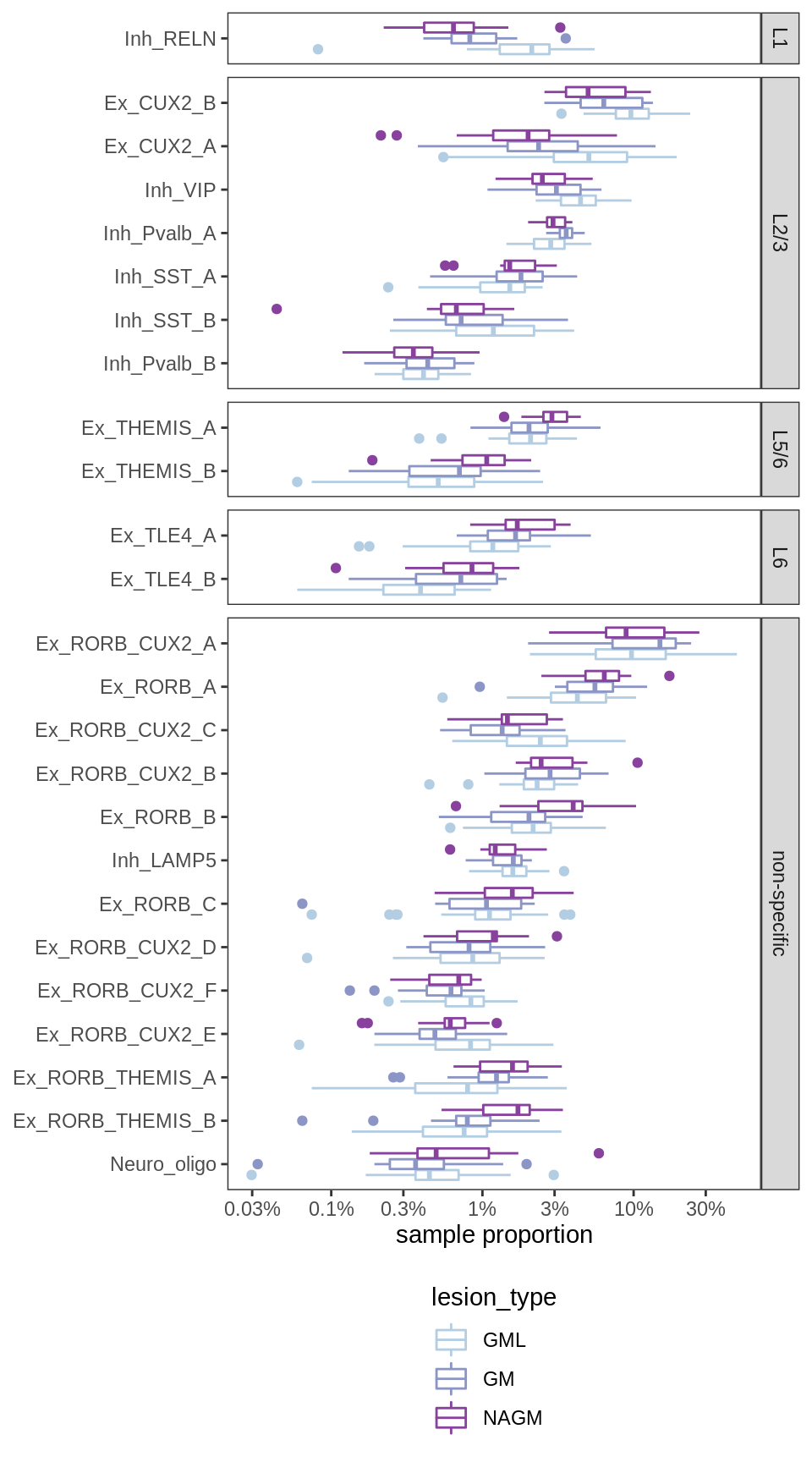
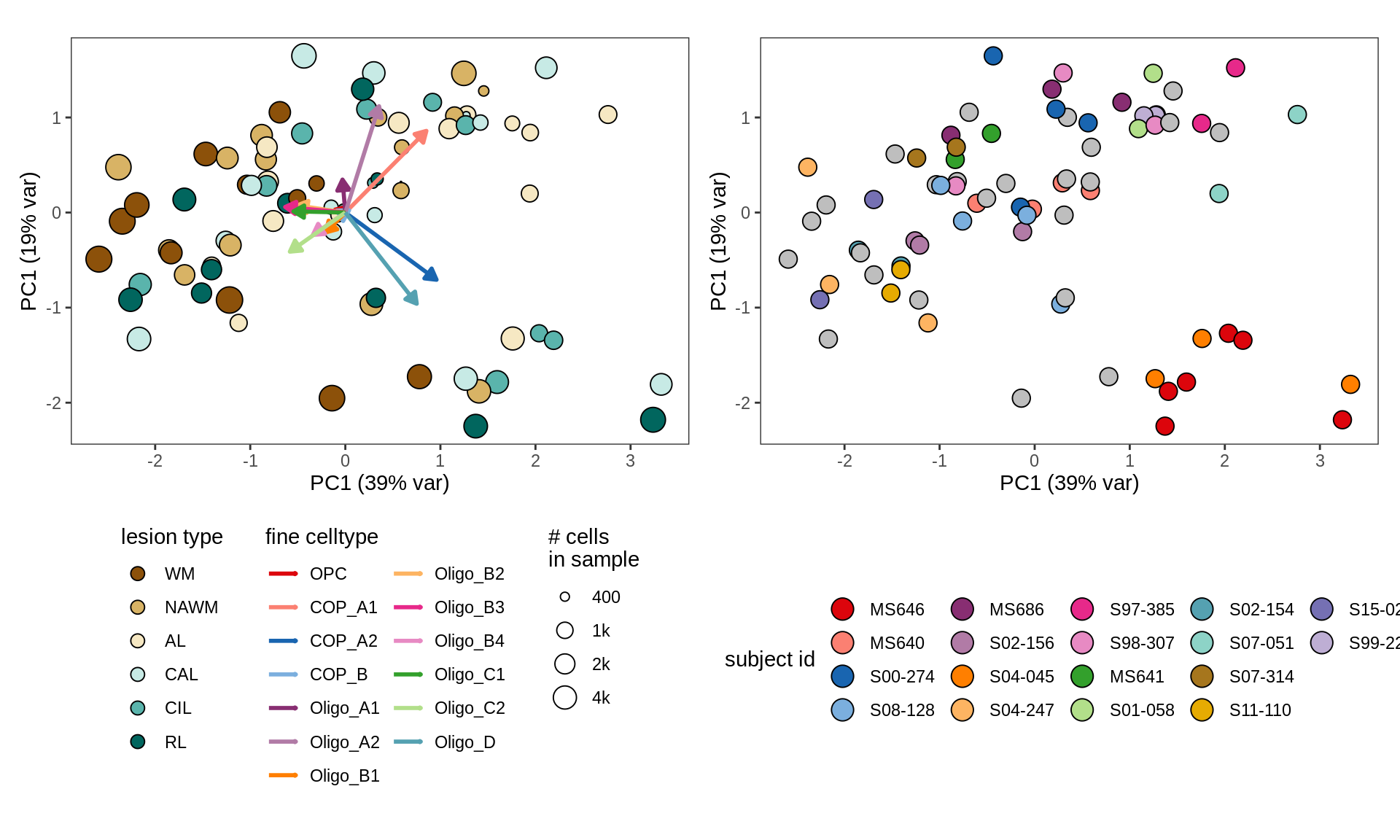
PCA of proportions, neurons only, all samples
(plot_pca_results(wide_neu, ctrl_pcs_dt, pc_vars, what = "proj"))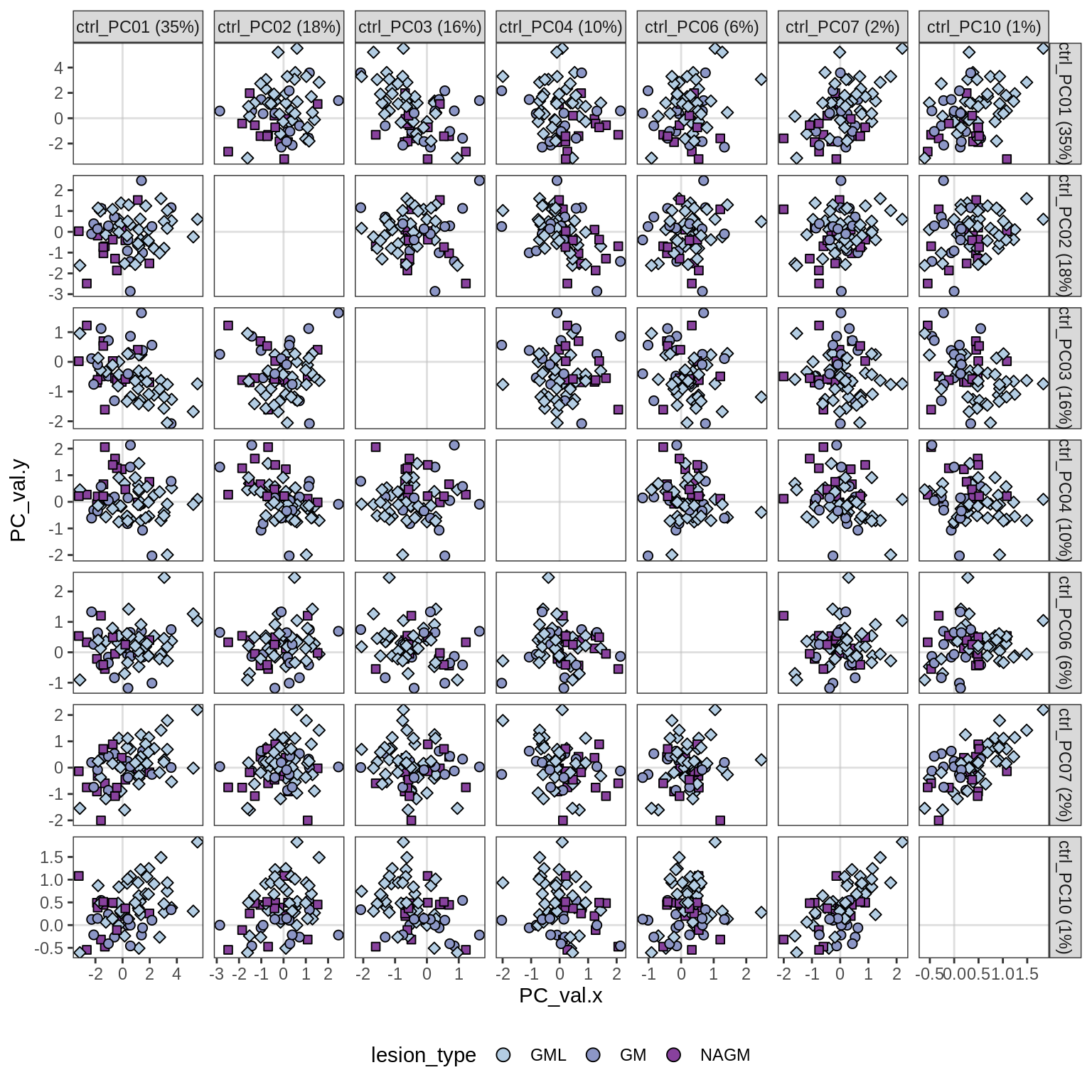
Patients over PC1
(plot_patients_over_pc(wide_neu, pc_vars))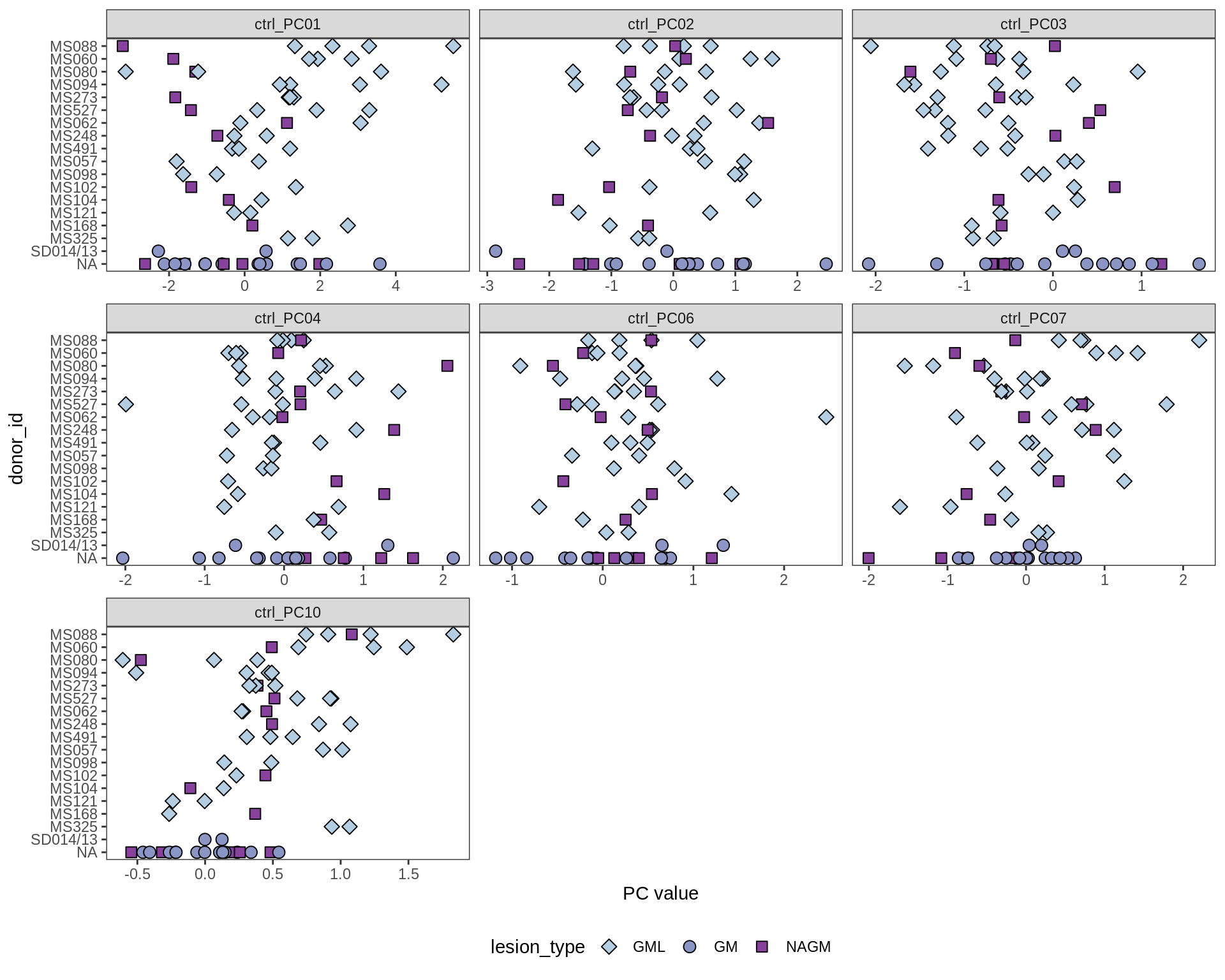
PC variance explained and layer correlations
(plot_pca_loadings(ctrl_pcs_dt, cut_var_exp = cut_var_exp,
cut_layer_cor = cut_layer_cor))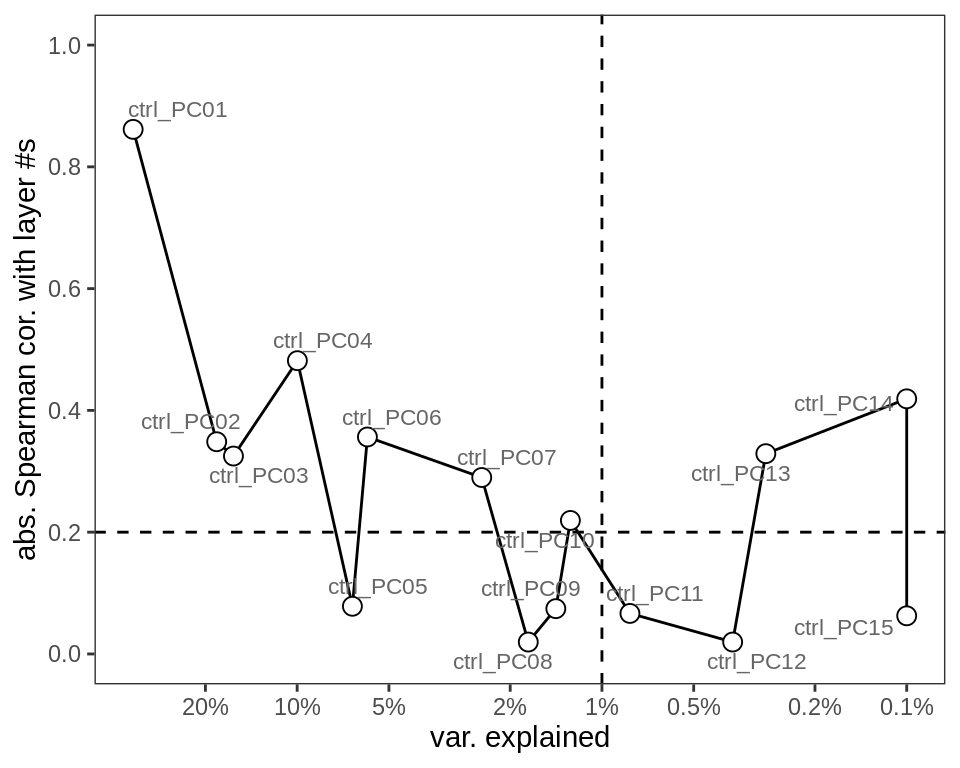
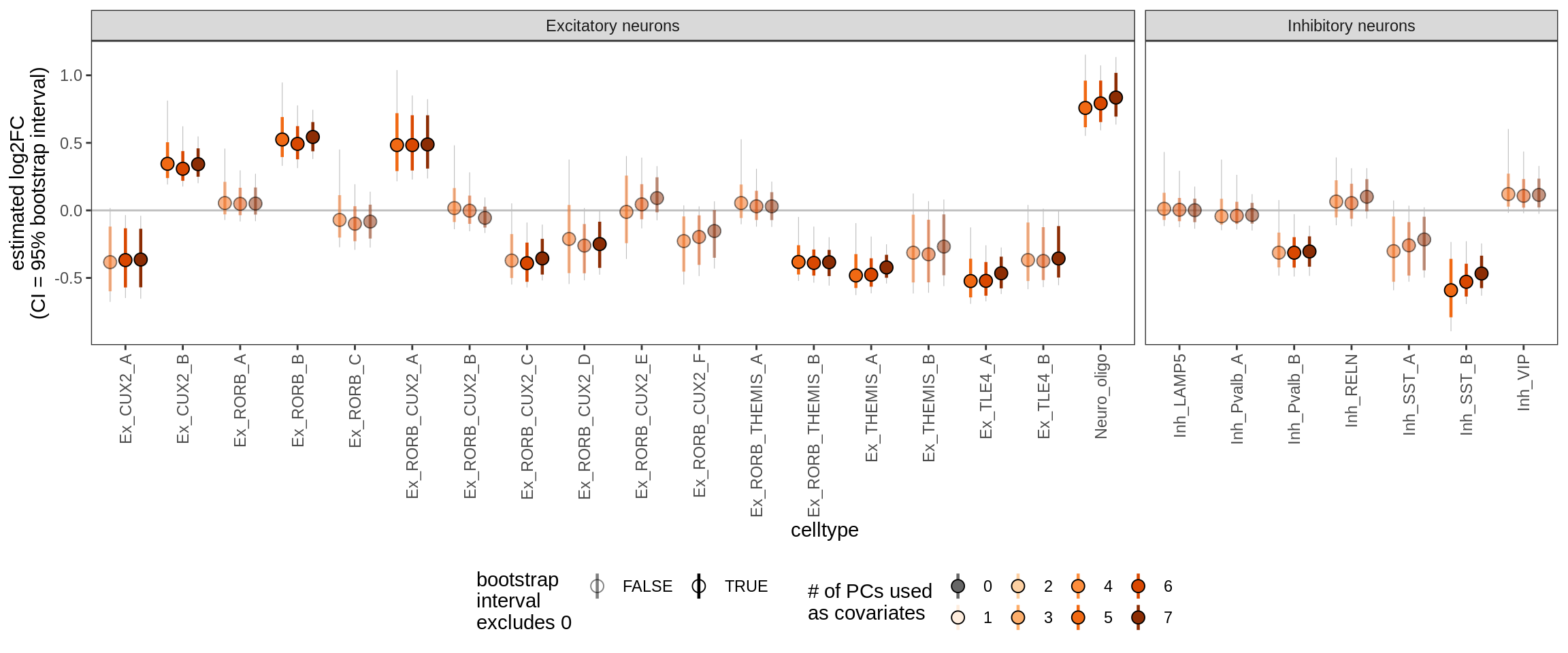
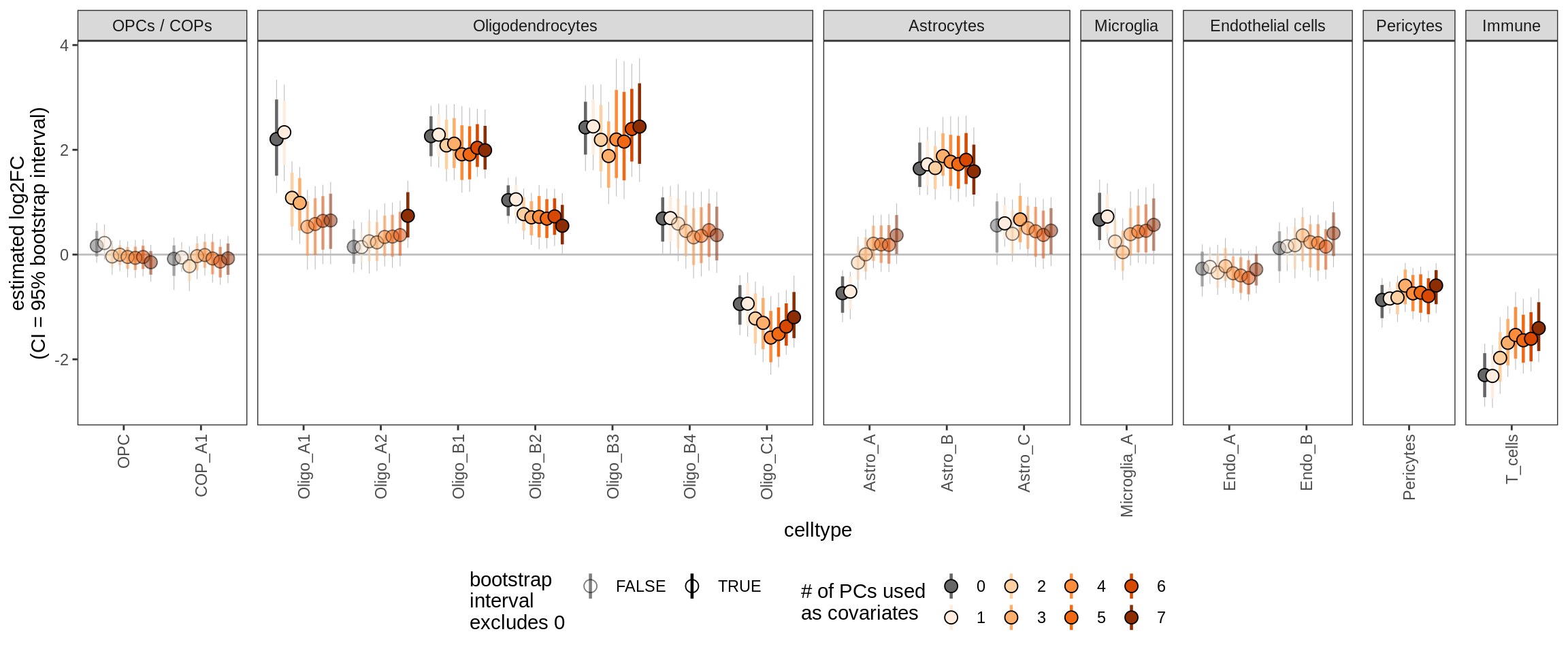
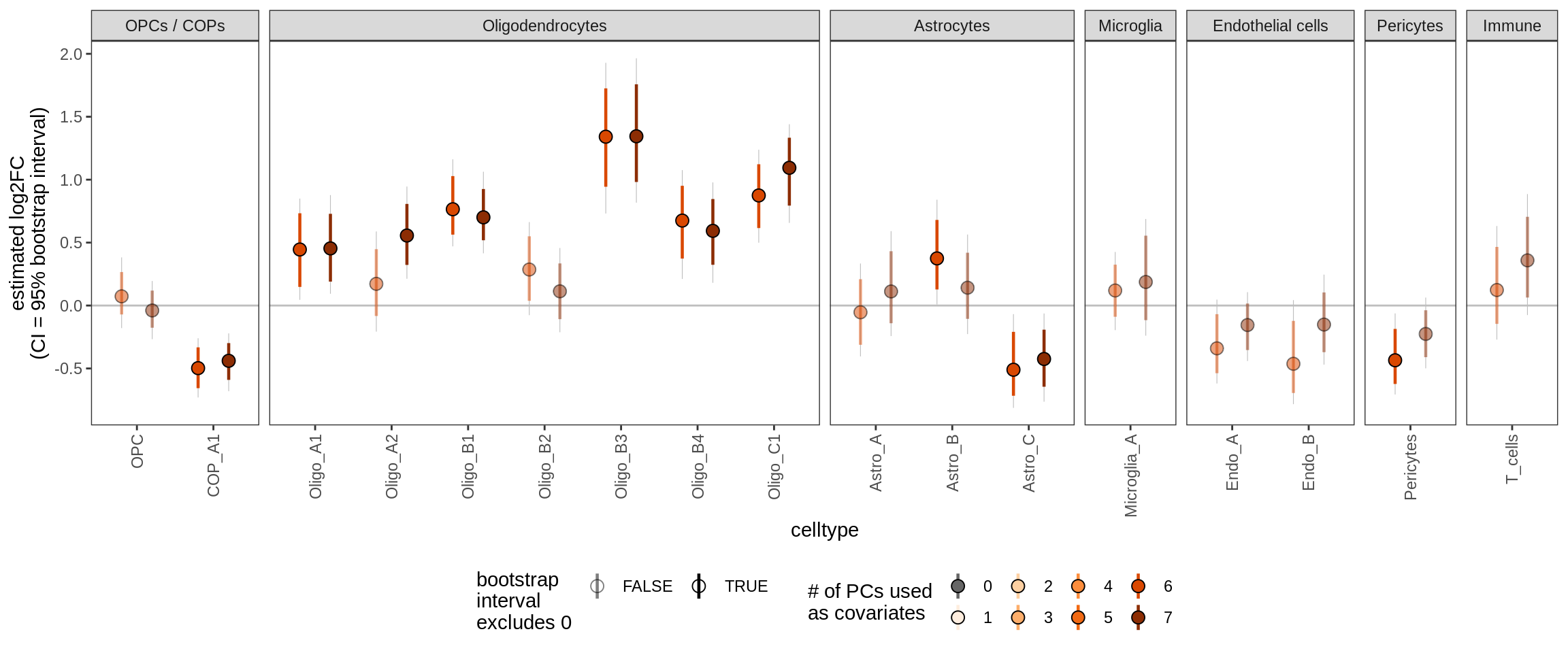
ANCOM-BC results
ANCOM-BC standard results
for (nn in names(ancom_ls)) {
cat('#### ', nn, '\n')
print(plot_ancombc_ci(ancom_ls[[nn]], q_cut = 0.05))
cat('\n\n')
}lesions_WM

lesions_GM
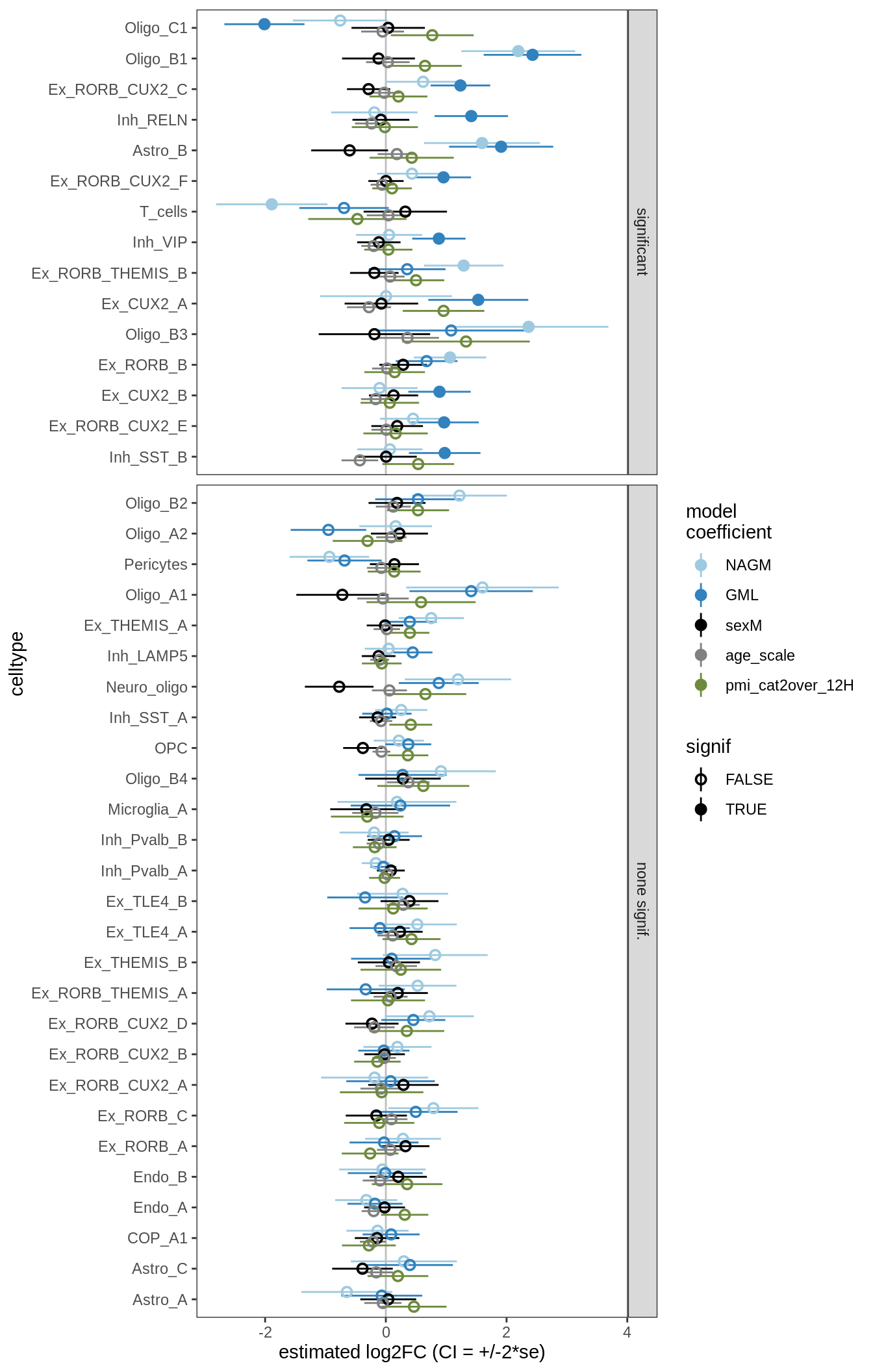
lesions_GM_1pcs
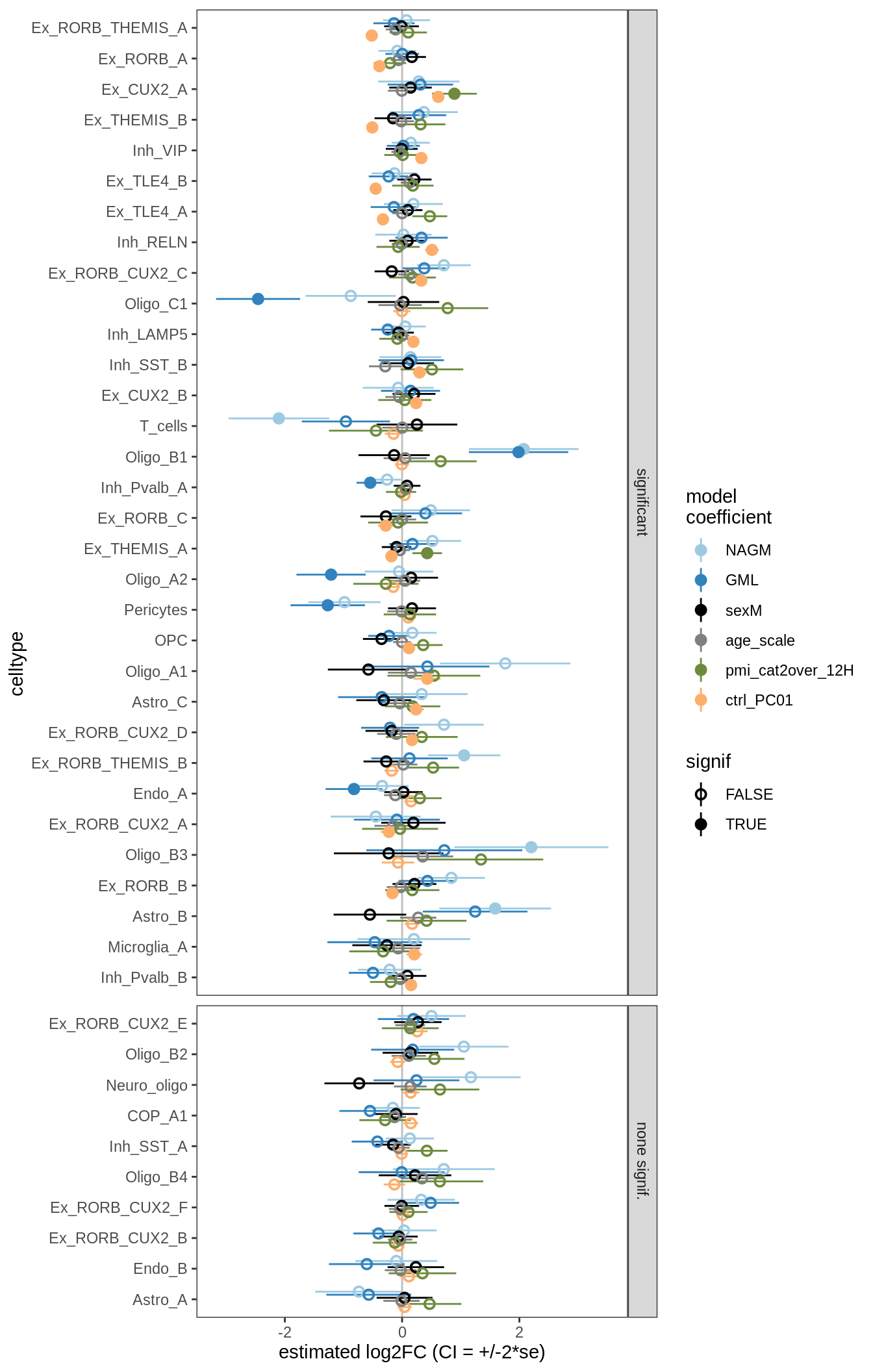
lesions_GM_2pcs
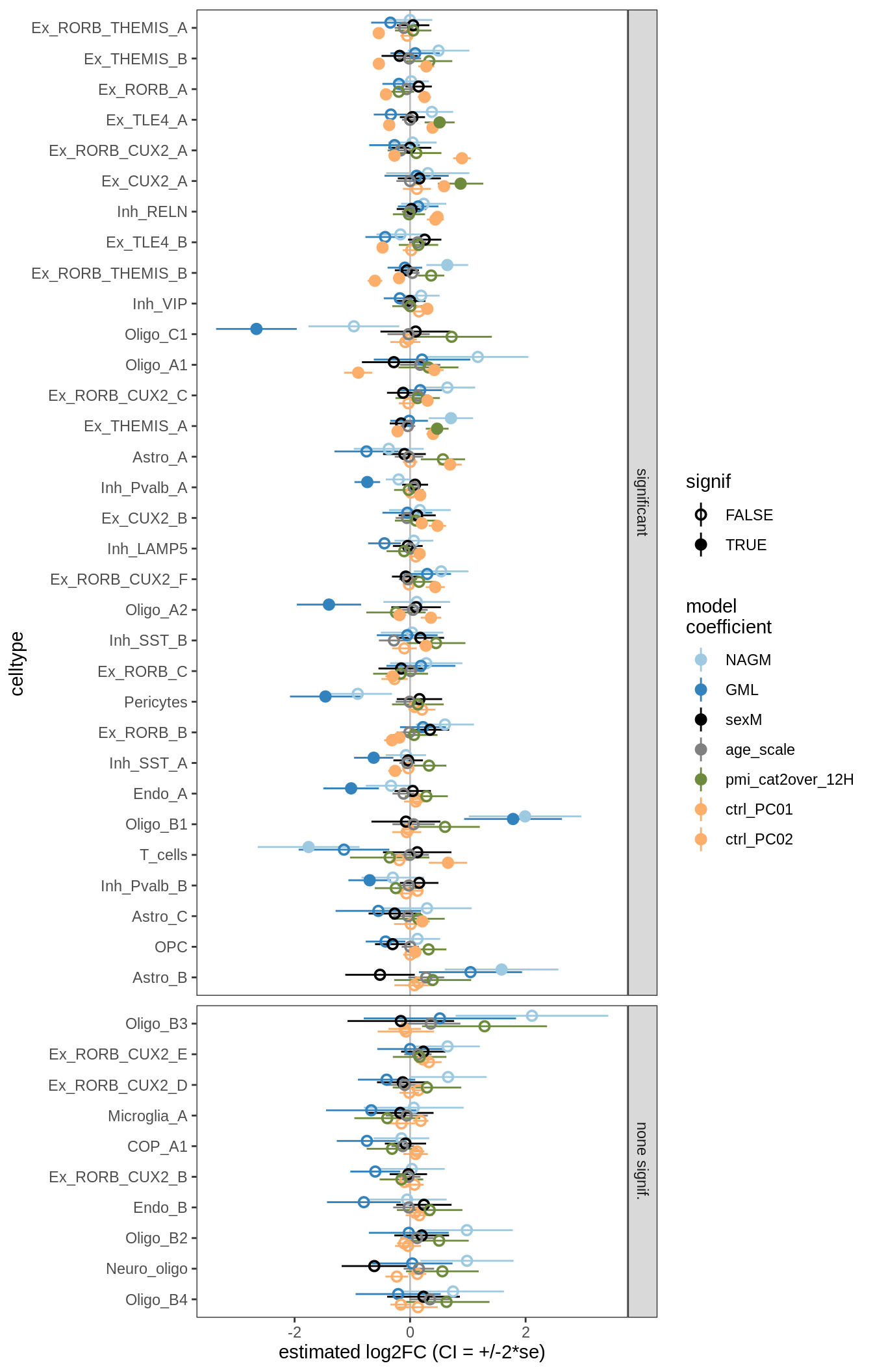
lesions_GM_3pcs
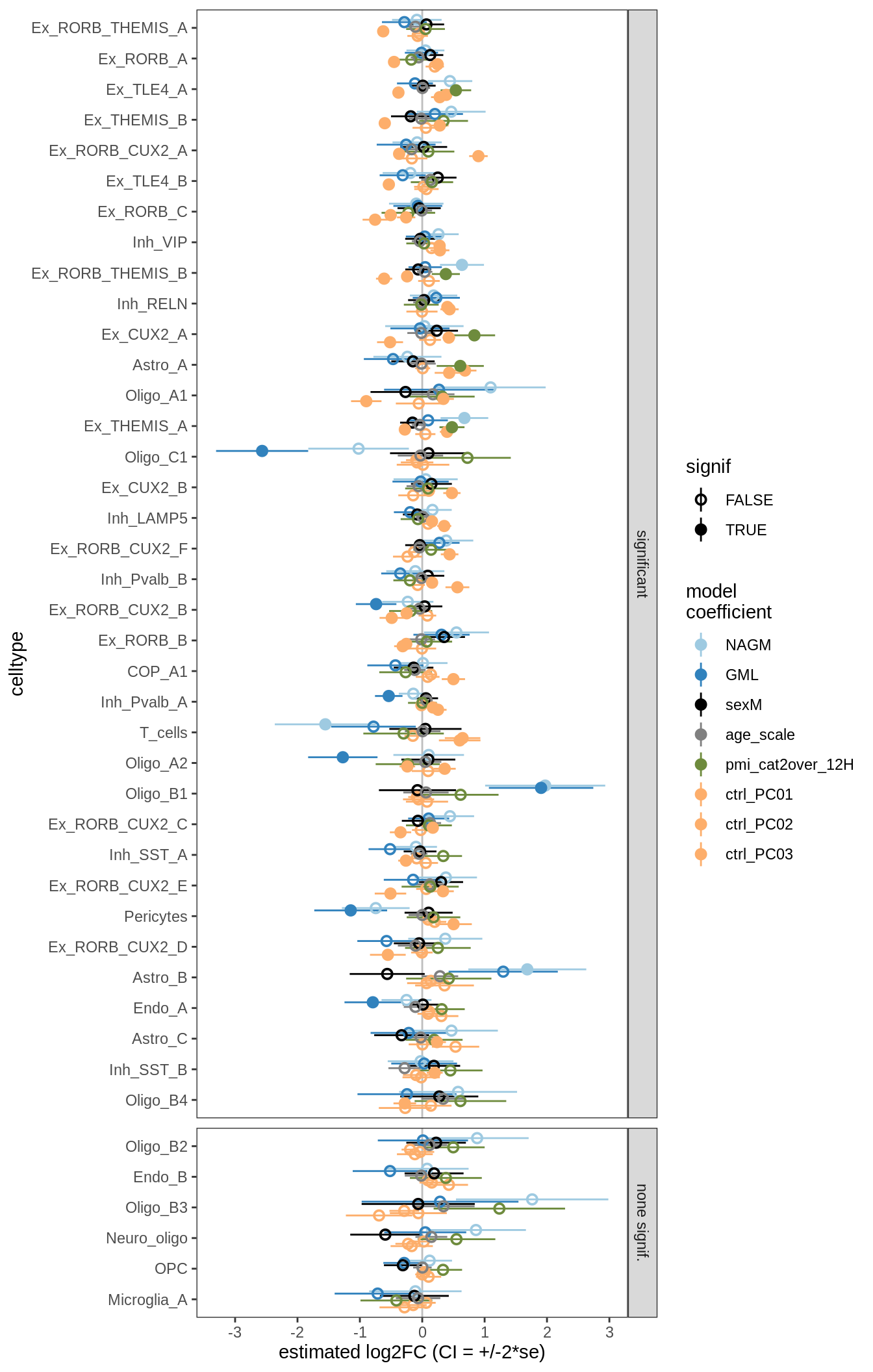
lesions_GM_4pcs
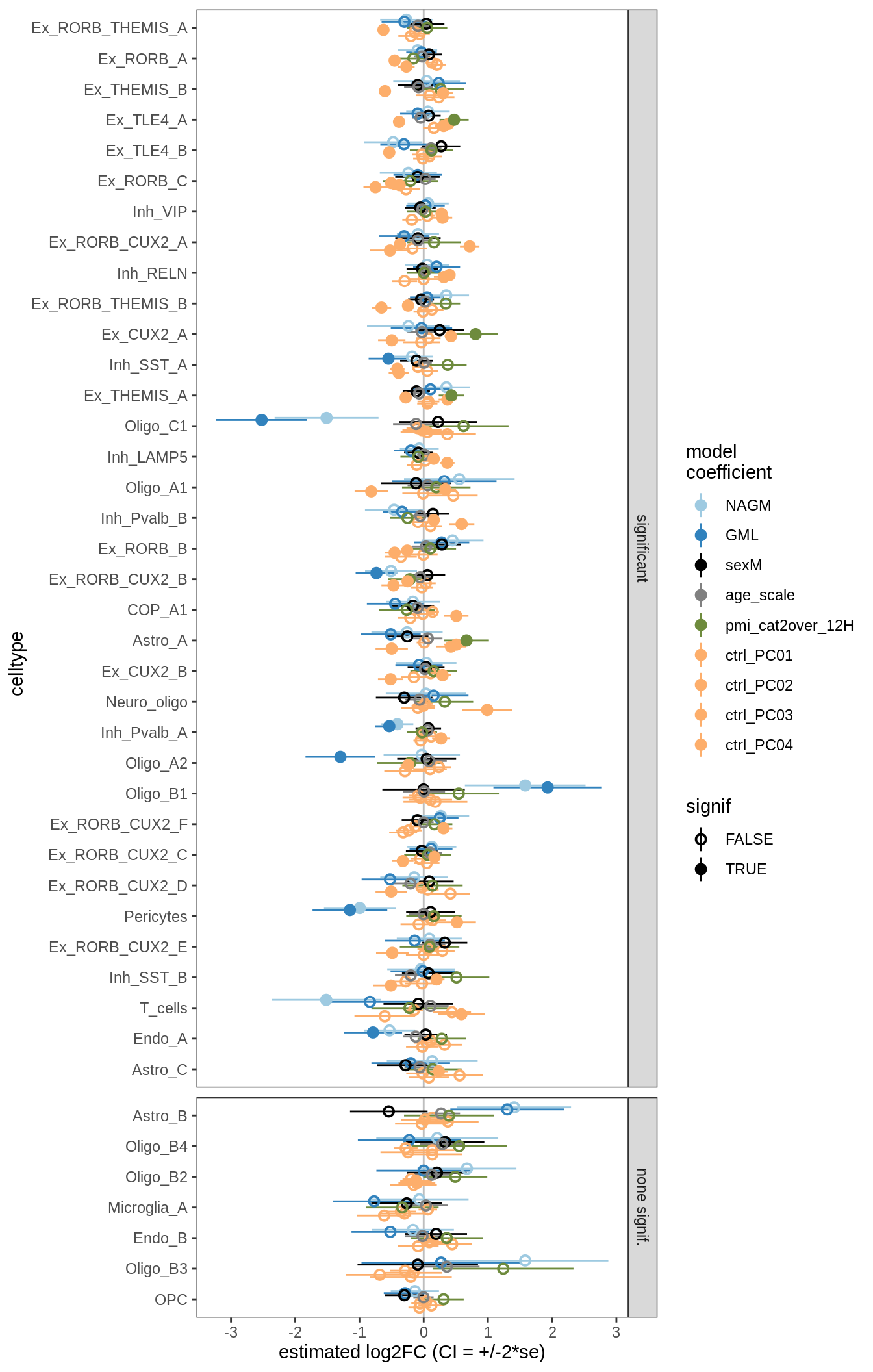
lesions_GM_5pcs
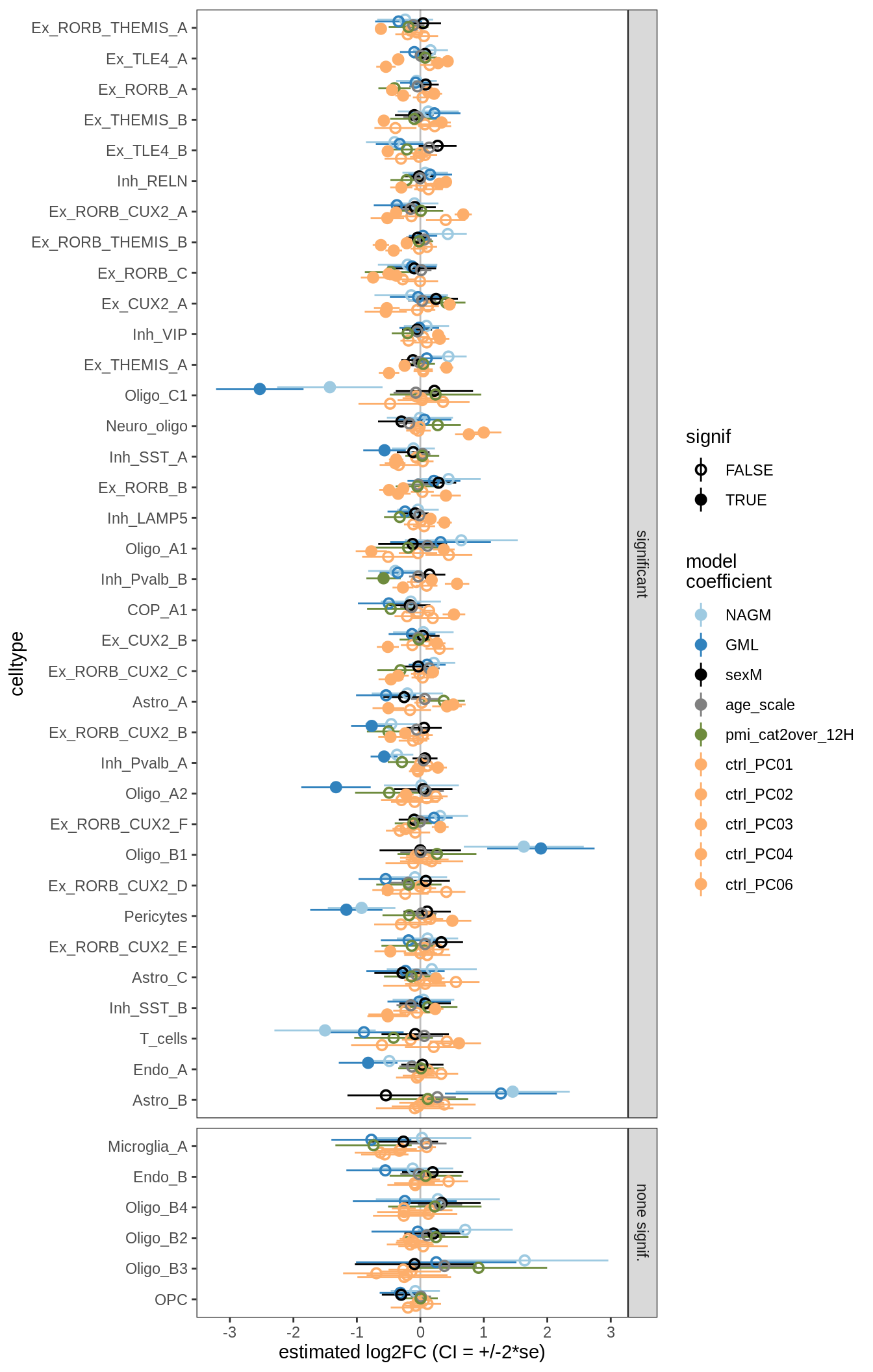
lesions_GM_6pcs

lesions_GM_7pcs
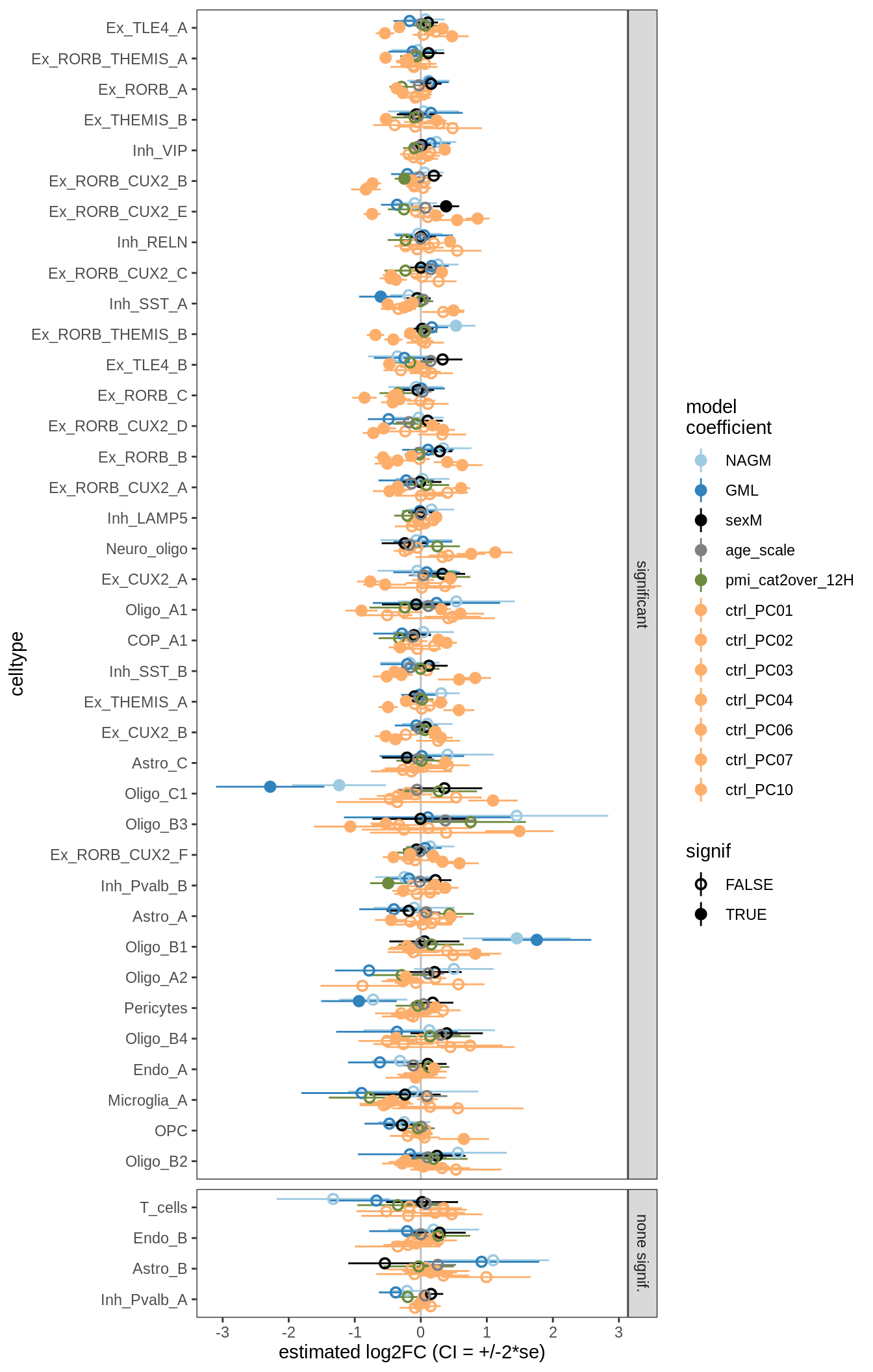
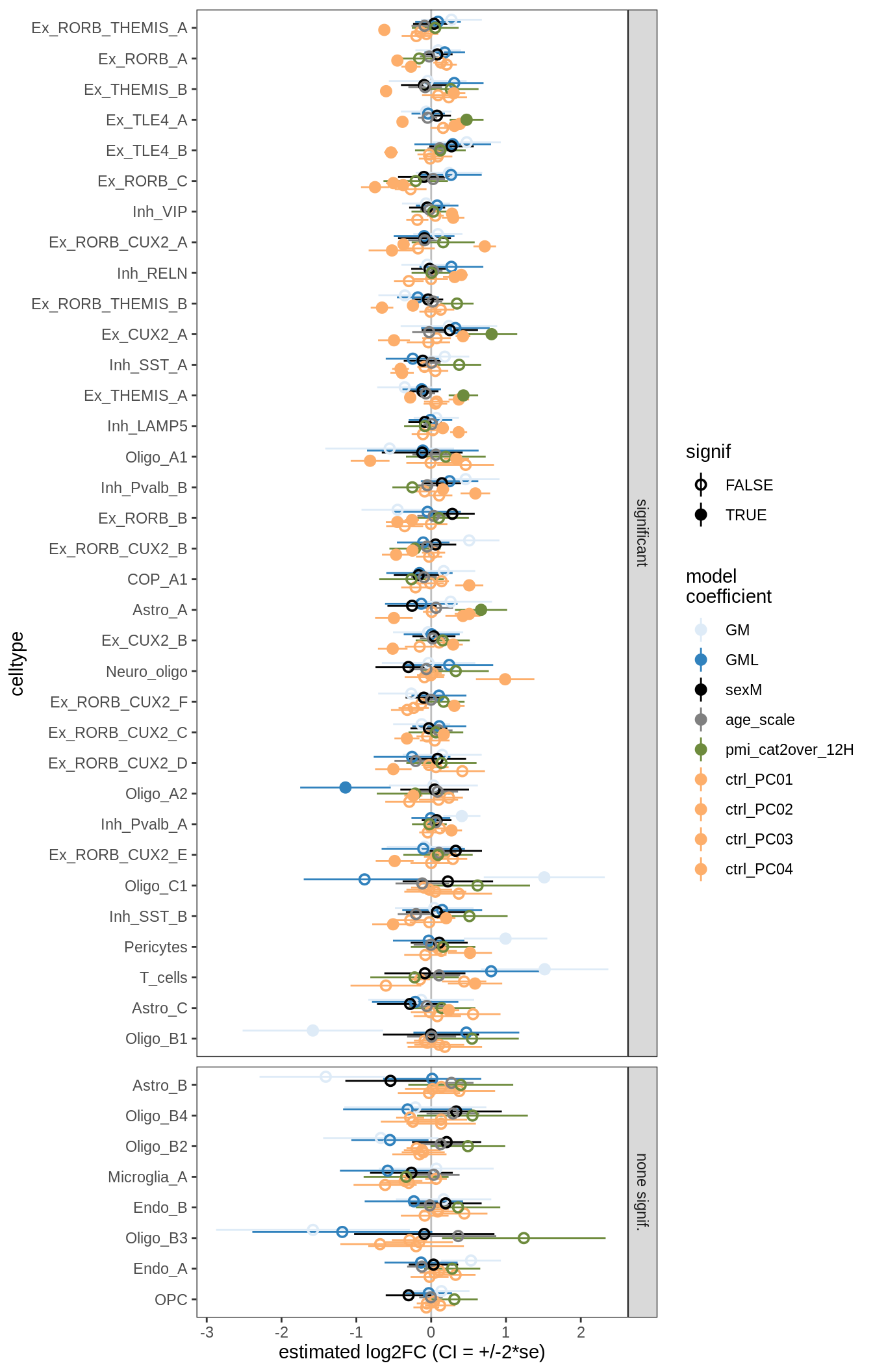
ANCOM-BC standard results, lesions only
for (nn in names(ancom_ls)) {
cat('#### ', nn, '\n')
print(plot_ancombc_ci(ancom_ls[[nn]], coef_filter = "lesion_type", q_cut = 0.05))
cat('\n\n')
}lesions_WM
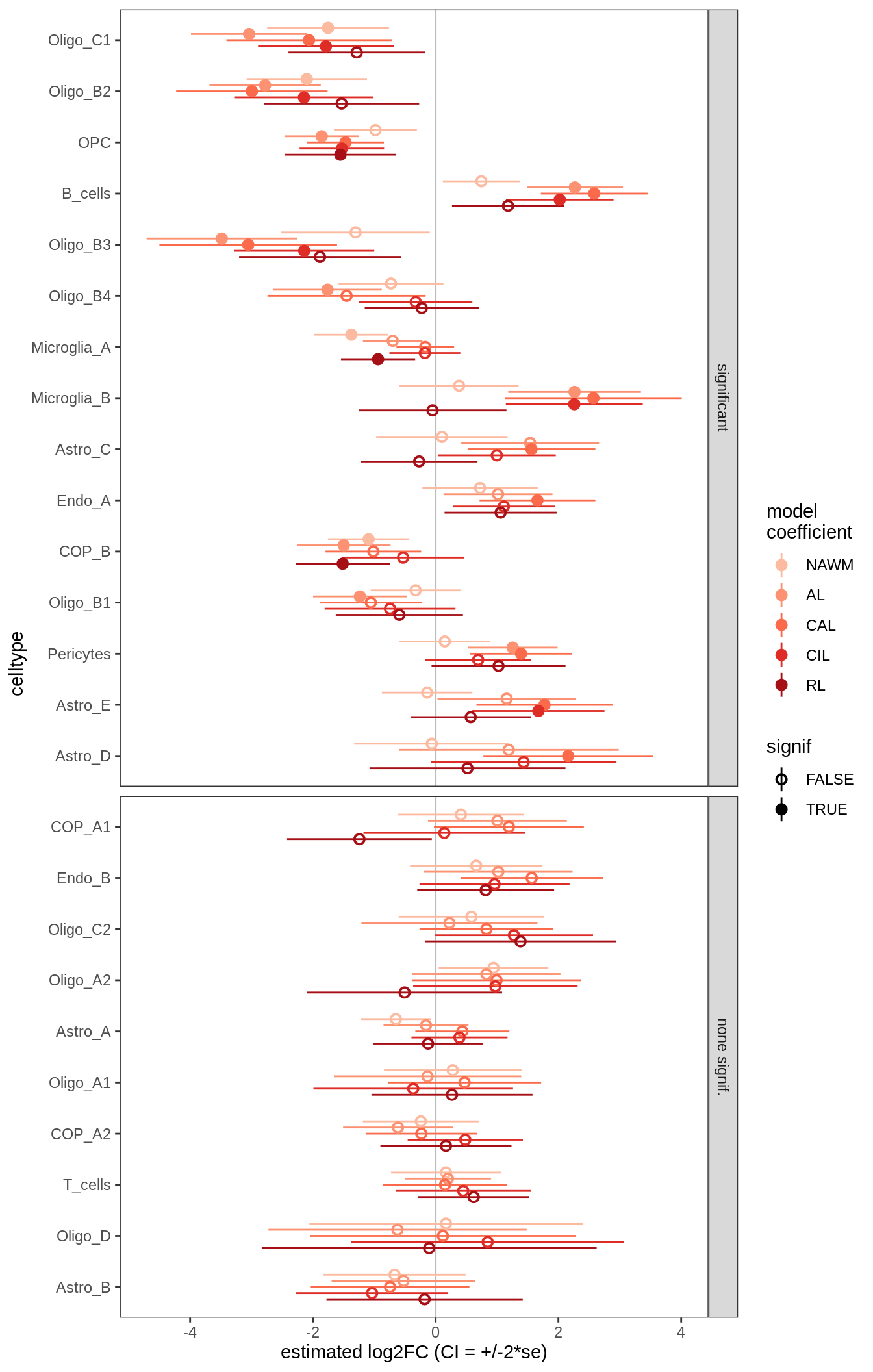
lesions_GM

lesions_GM_1pcs
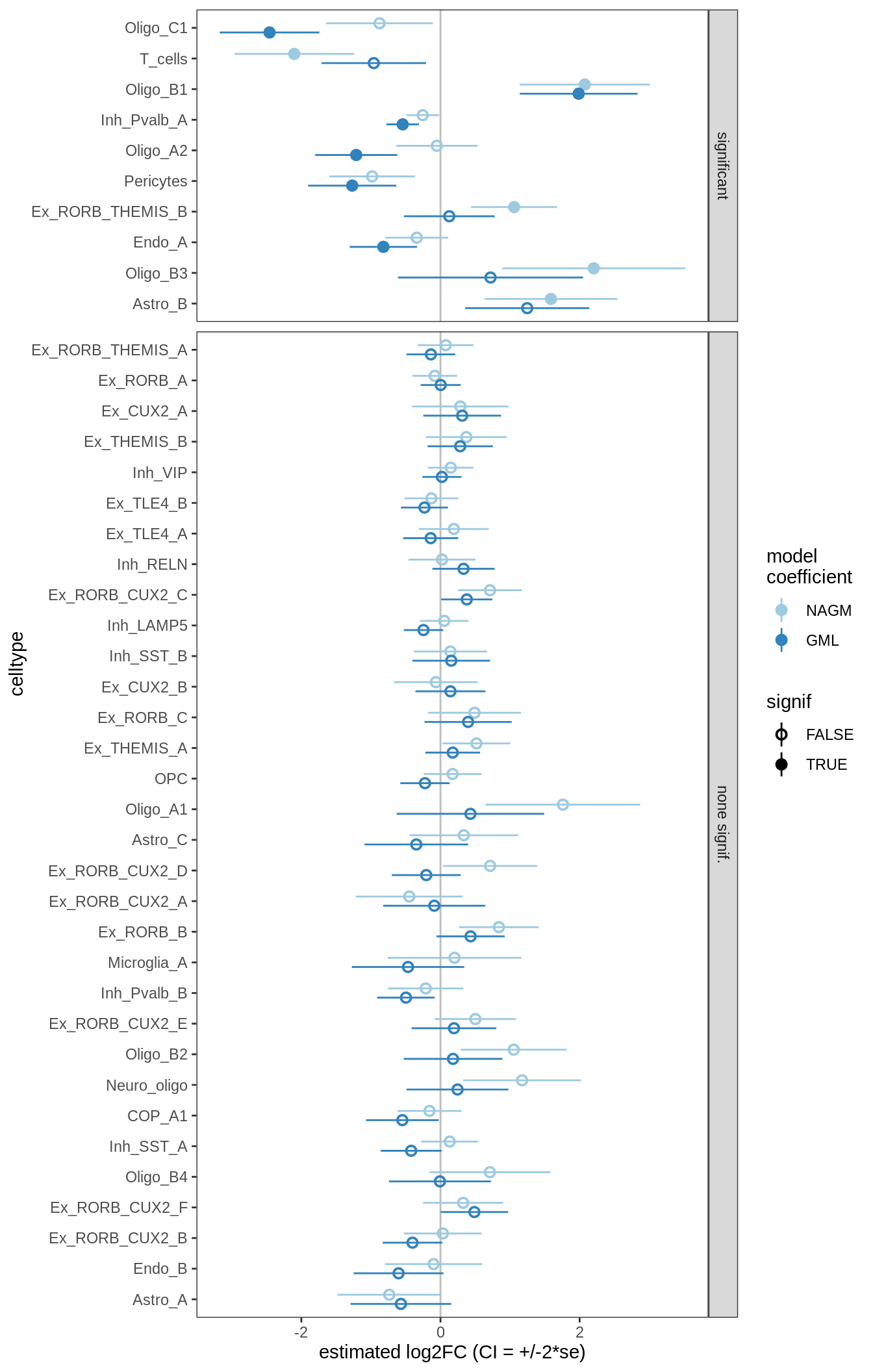
lesions_GM_2pcs
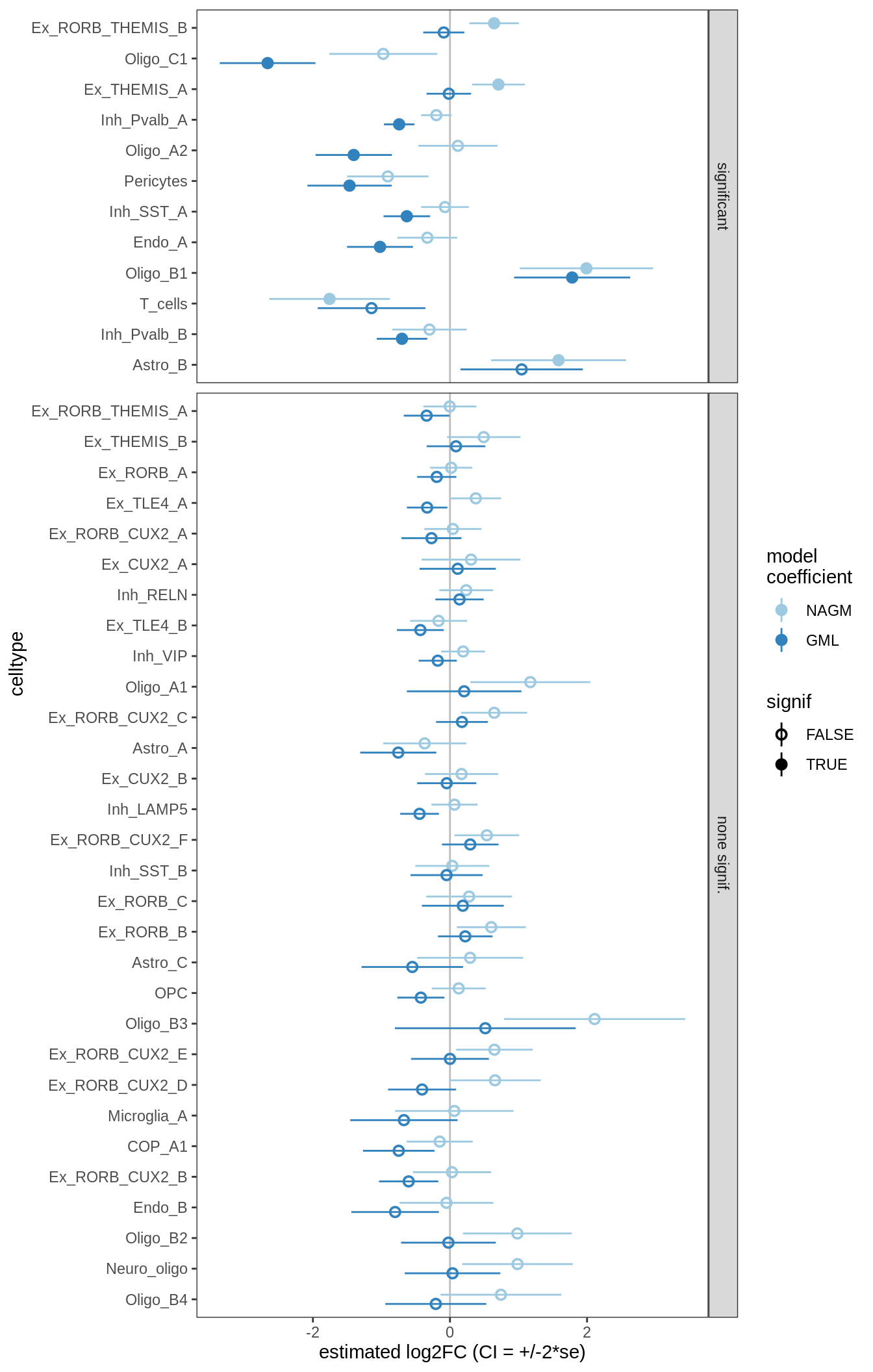
lesions_GM_3pcs

lesions_GM_4pcs

lesions_GM_5pcs

lesions_GM_6pcs
lesions_GM_7pcs

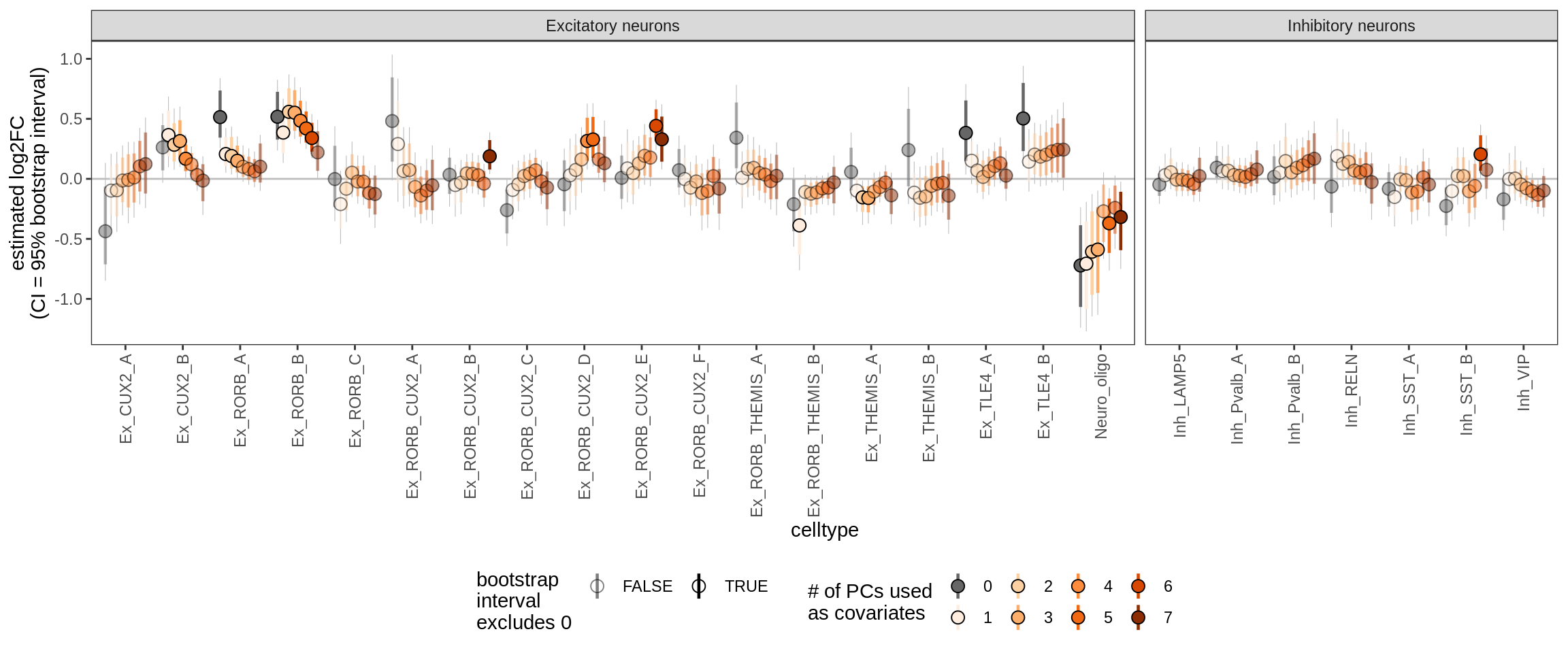
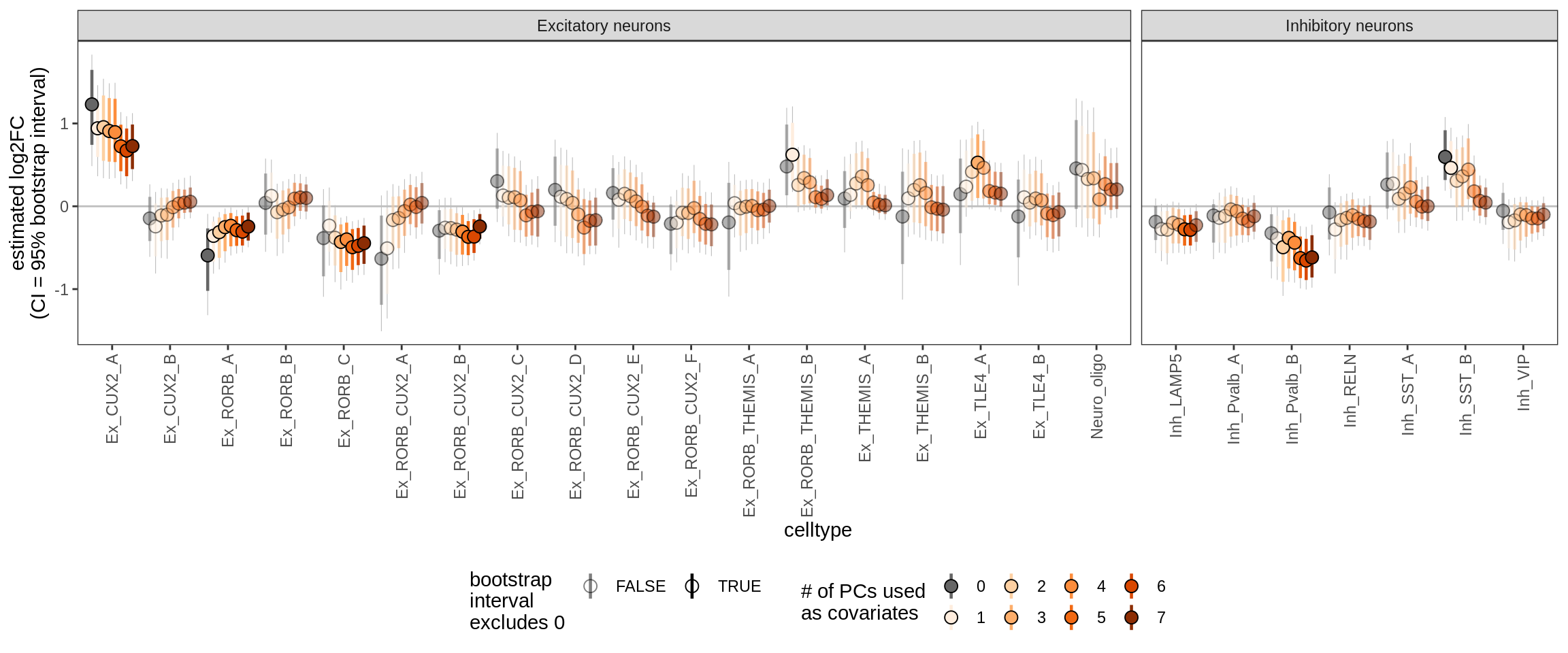
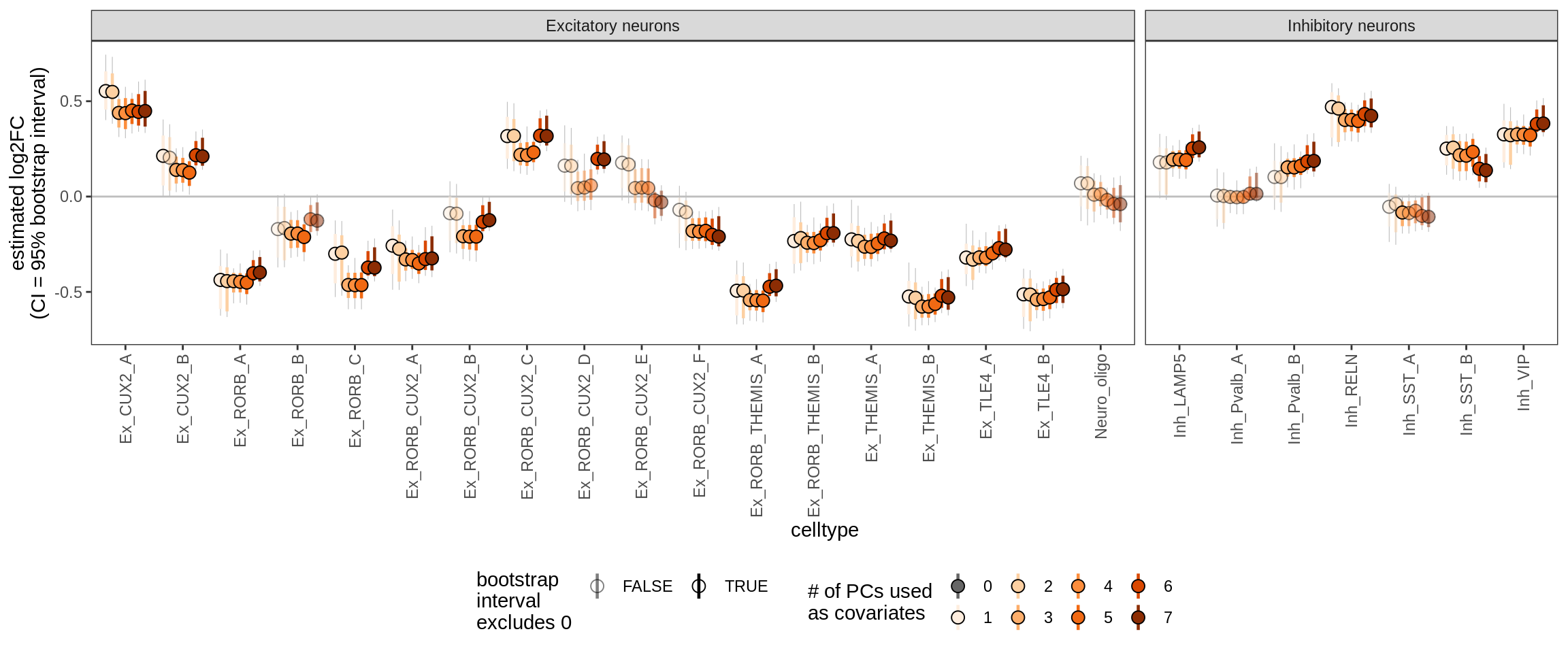
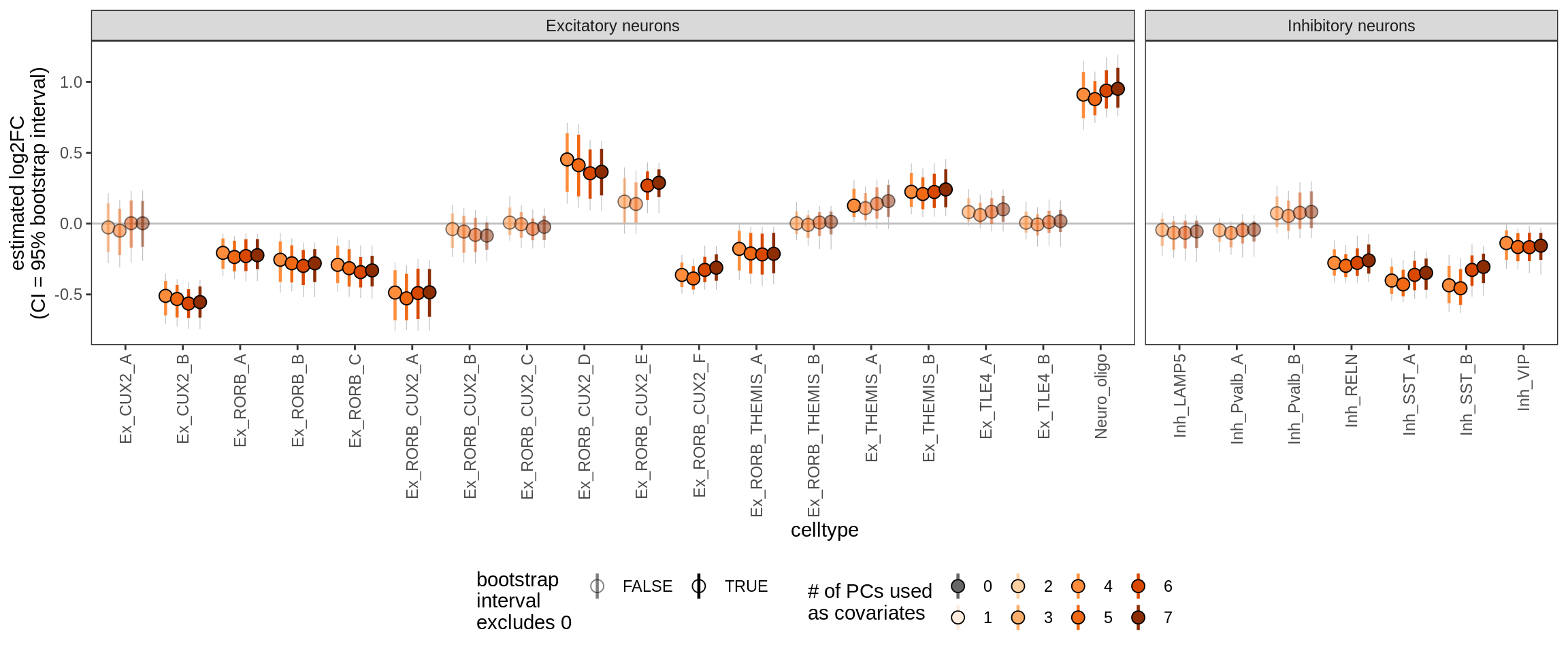
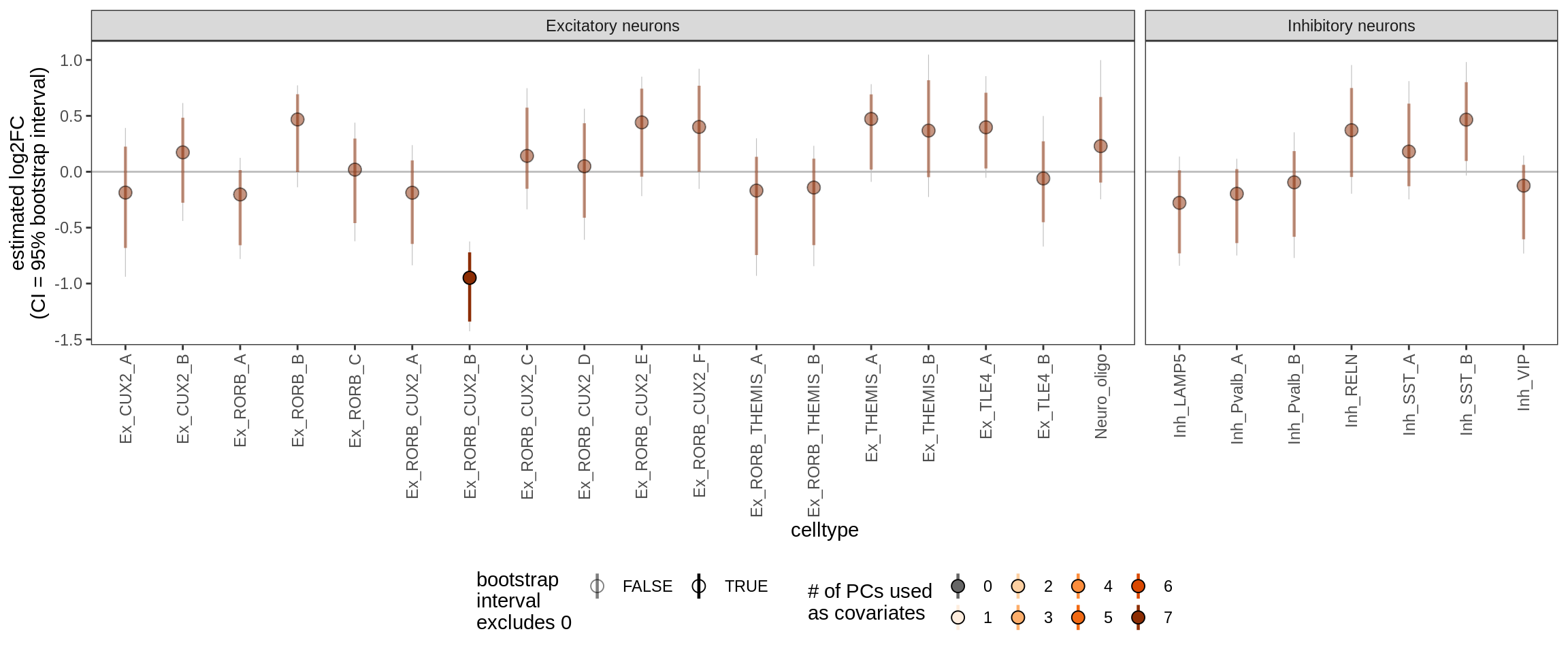
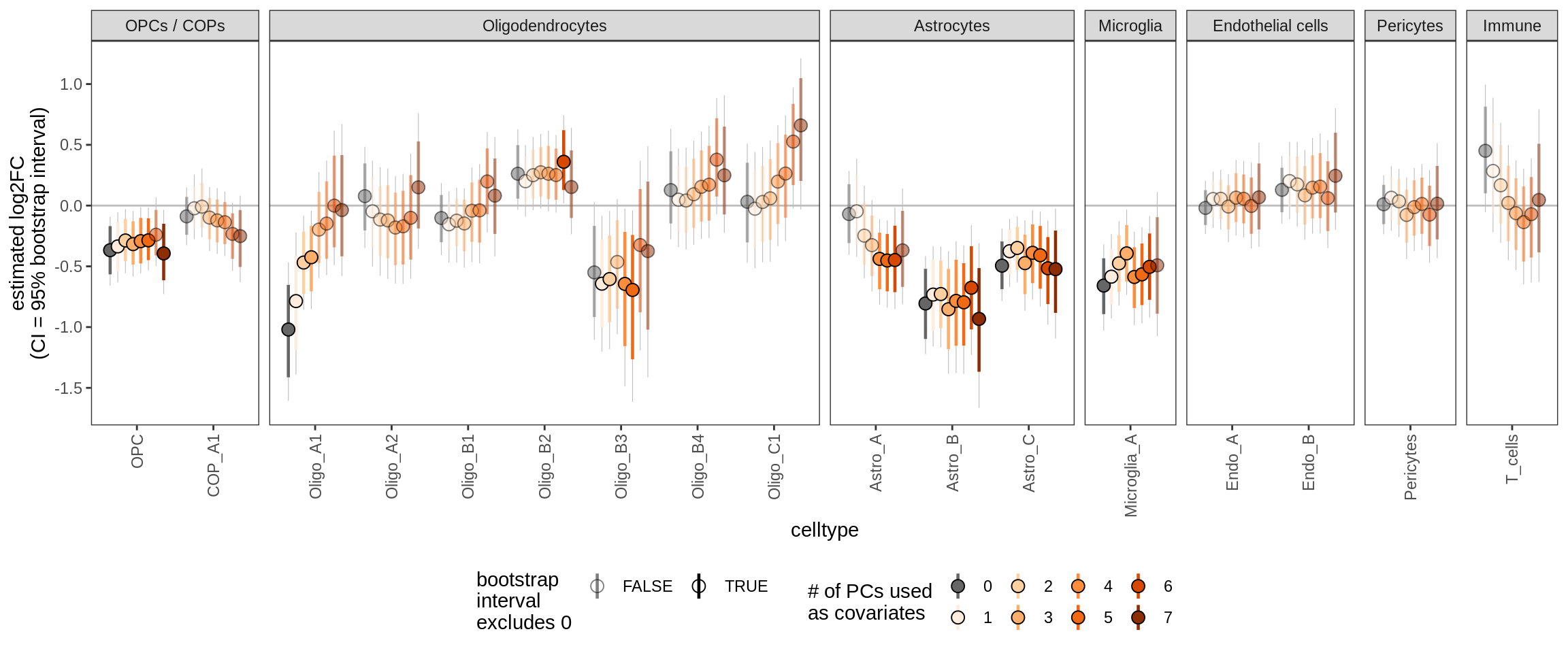
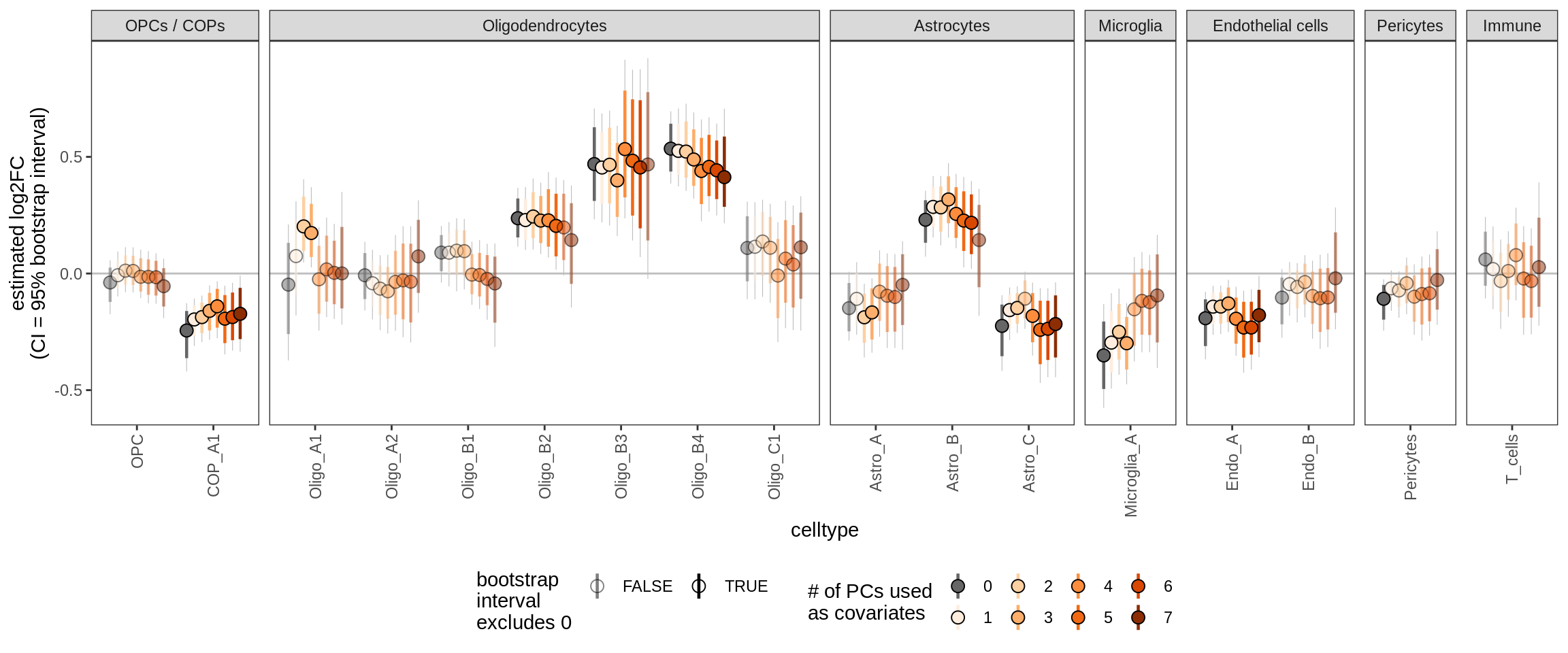
ANCOM-BC bootstrap results
for (nn in names(boots_ls)) {
cat('#### ', nn, '\n')
print(plot_boots_dt(boots_ls[[nn]]))
cat('\n\n')
}lesions_WM
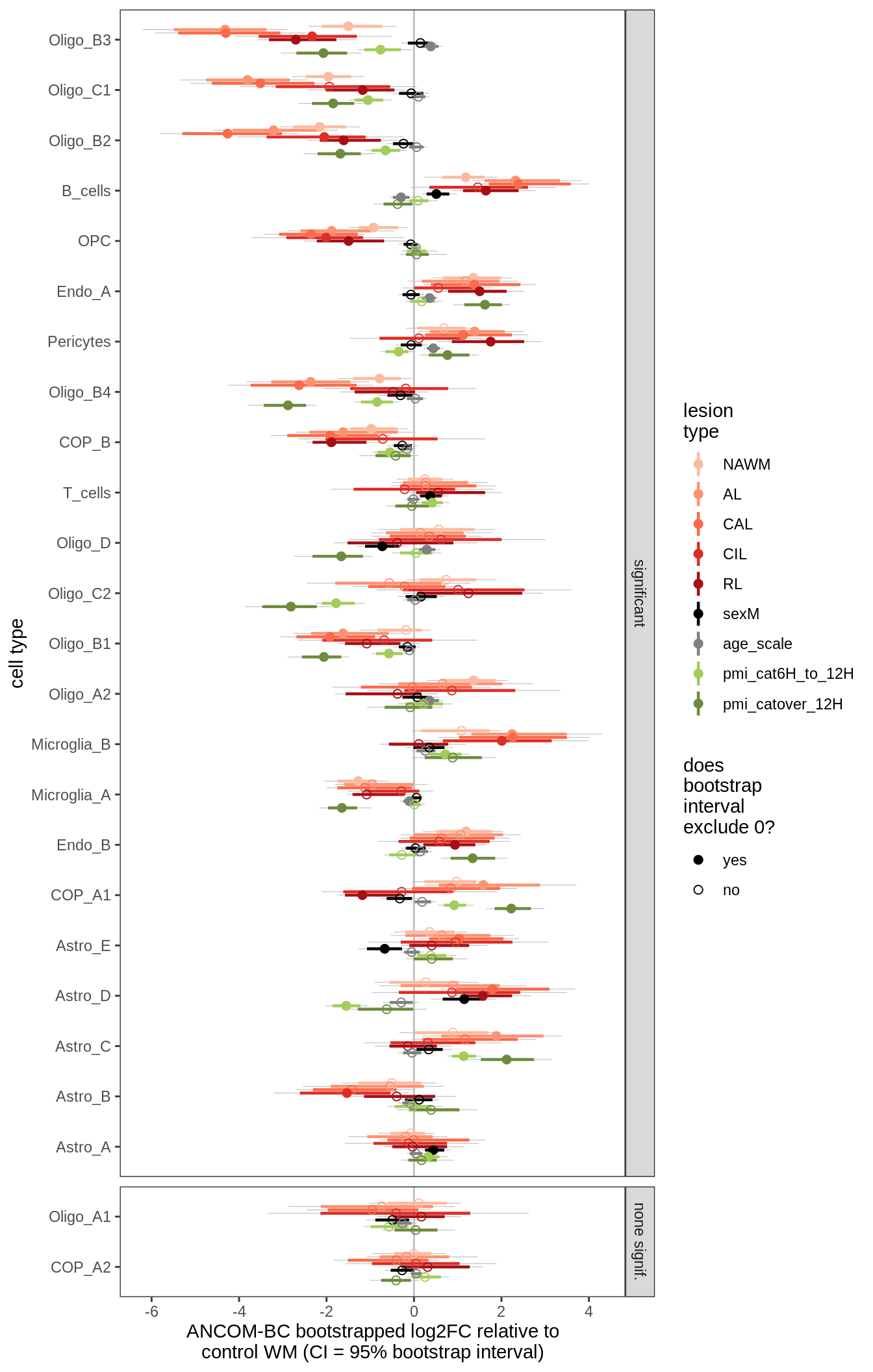
lesions_GM

lesions_GM_1pcs
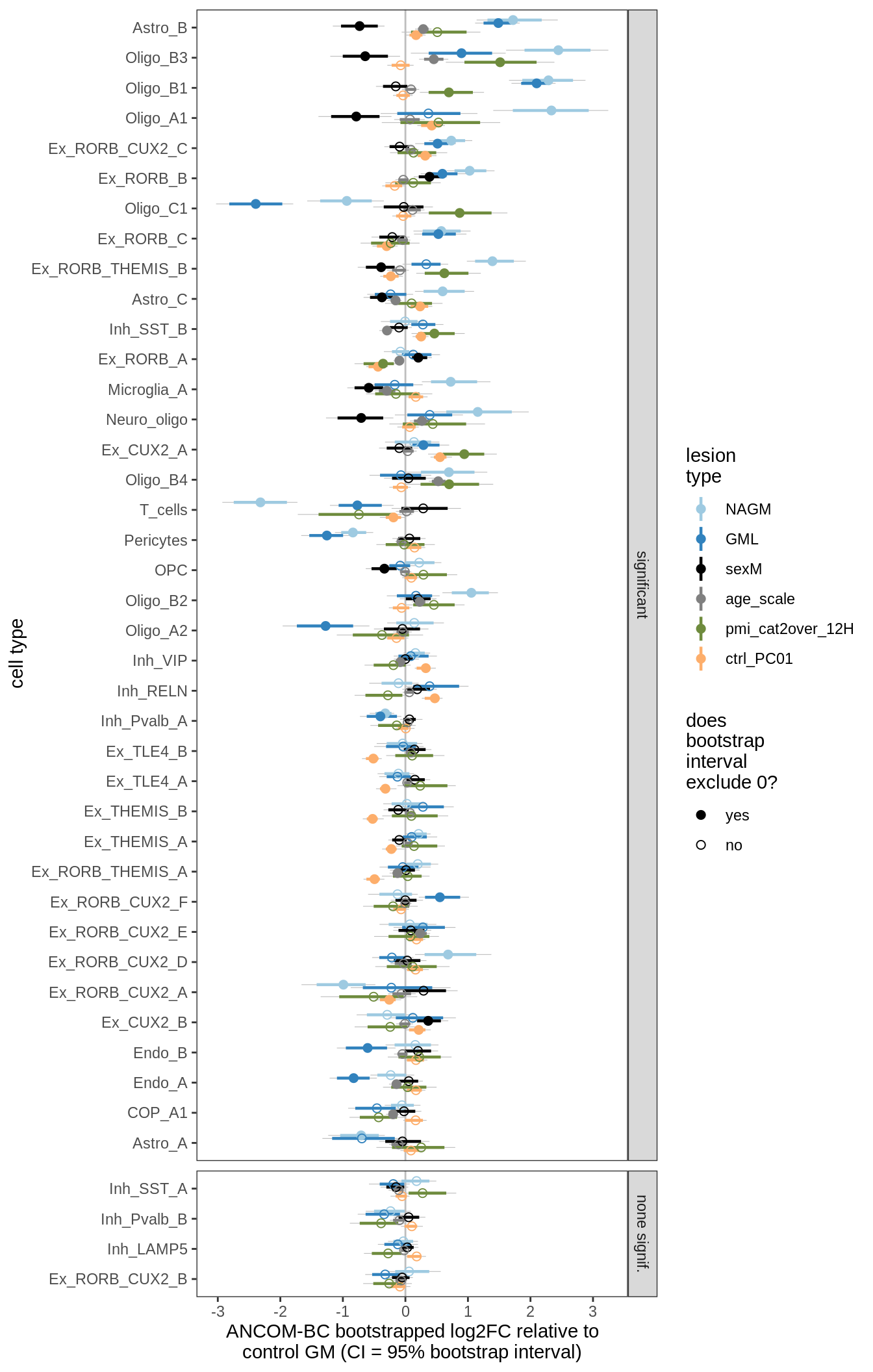
lesions_GM_2pcs
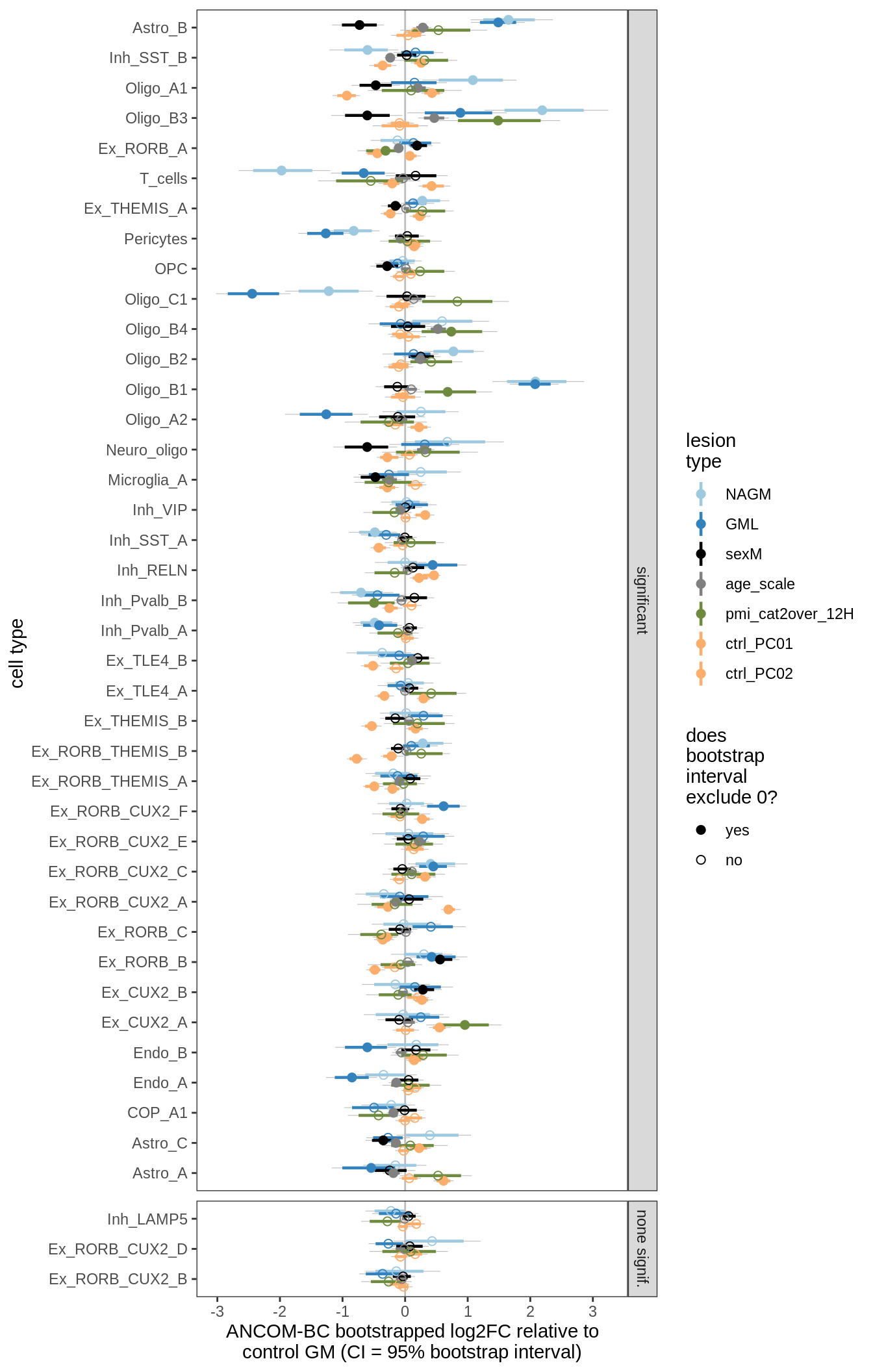
lesions_GM_3pcs
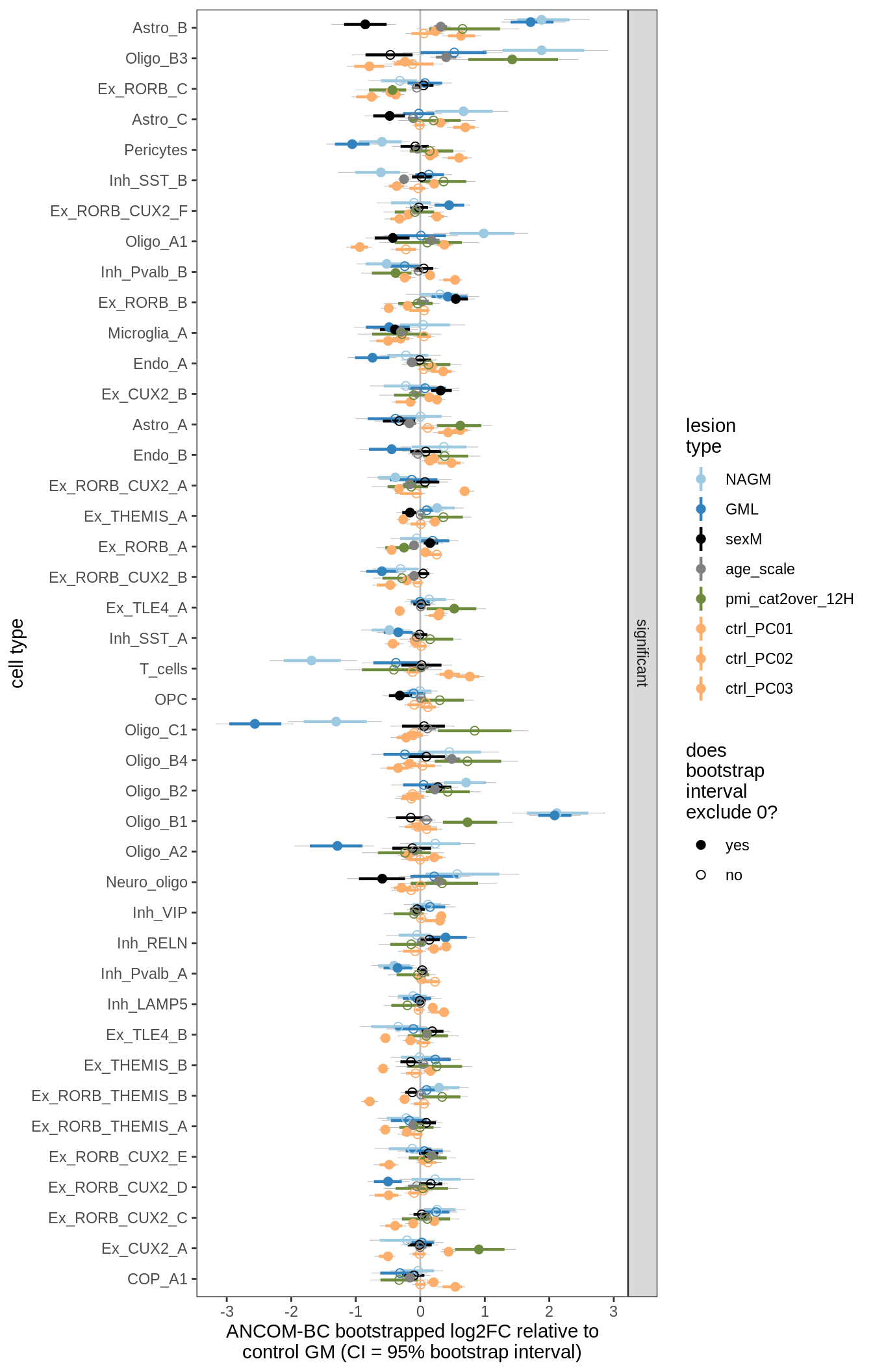
lesions_GM_4pcs
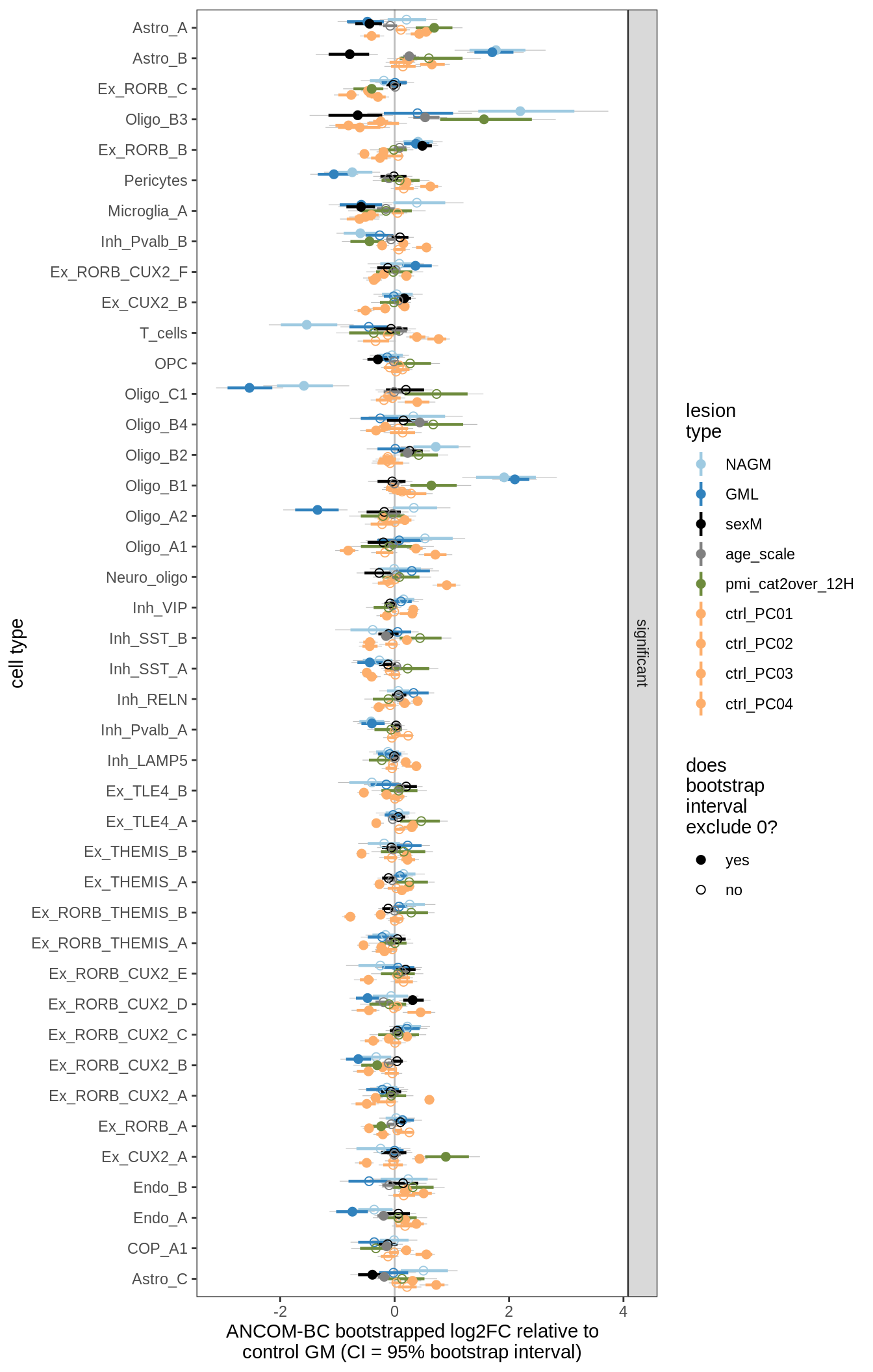
lesions_GM_5pcs
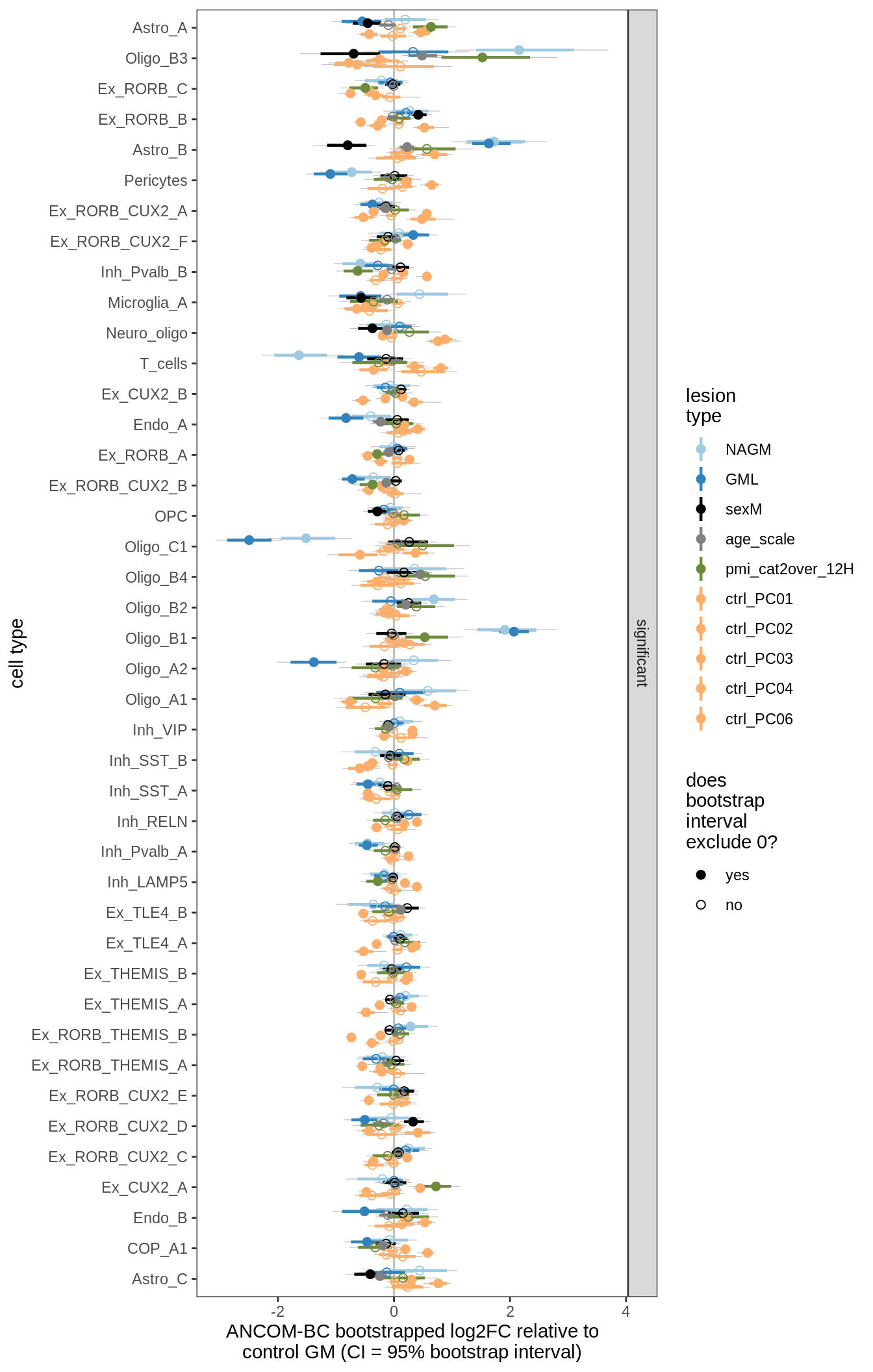
lesions_GM_6pcs
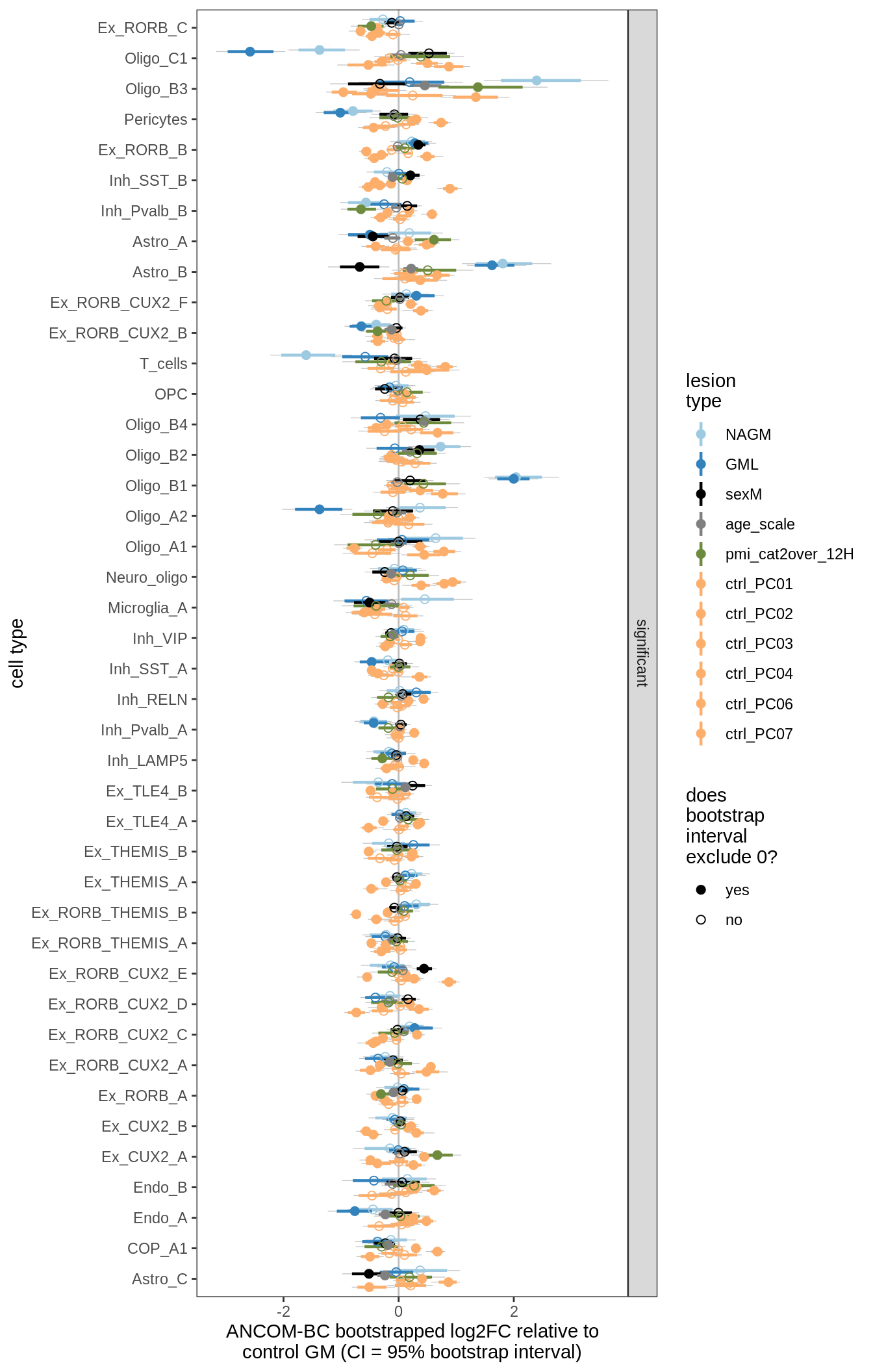
lesions_GM_7pcs
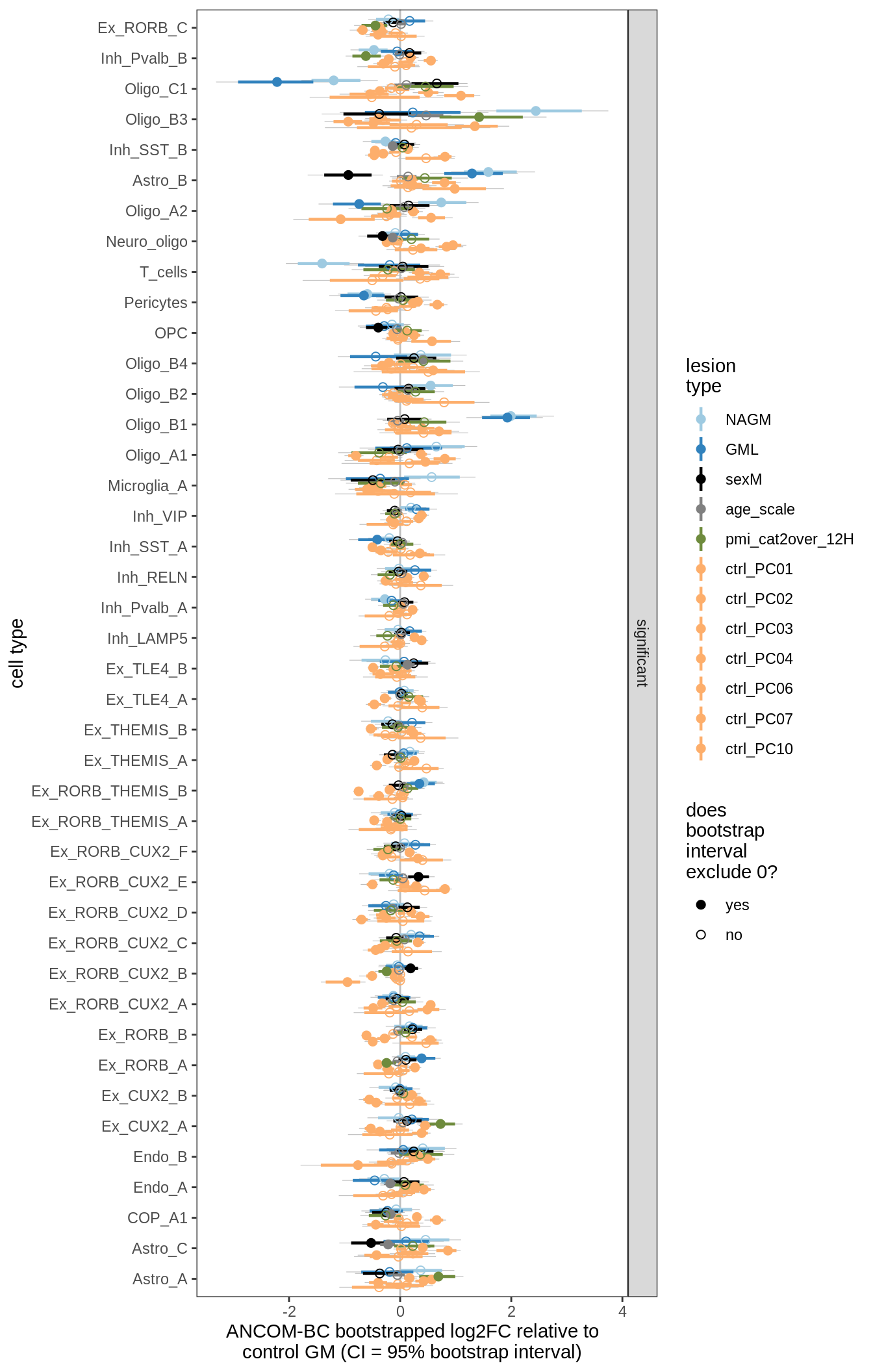
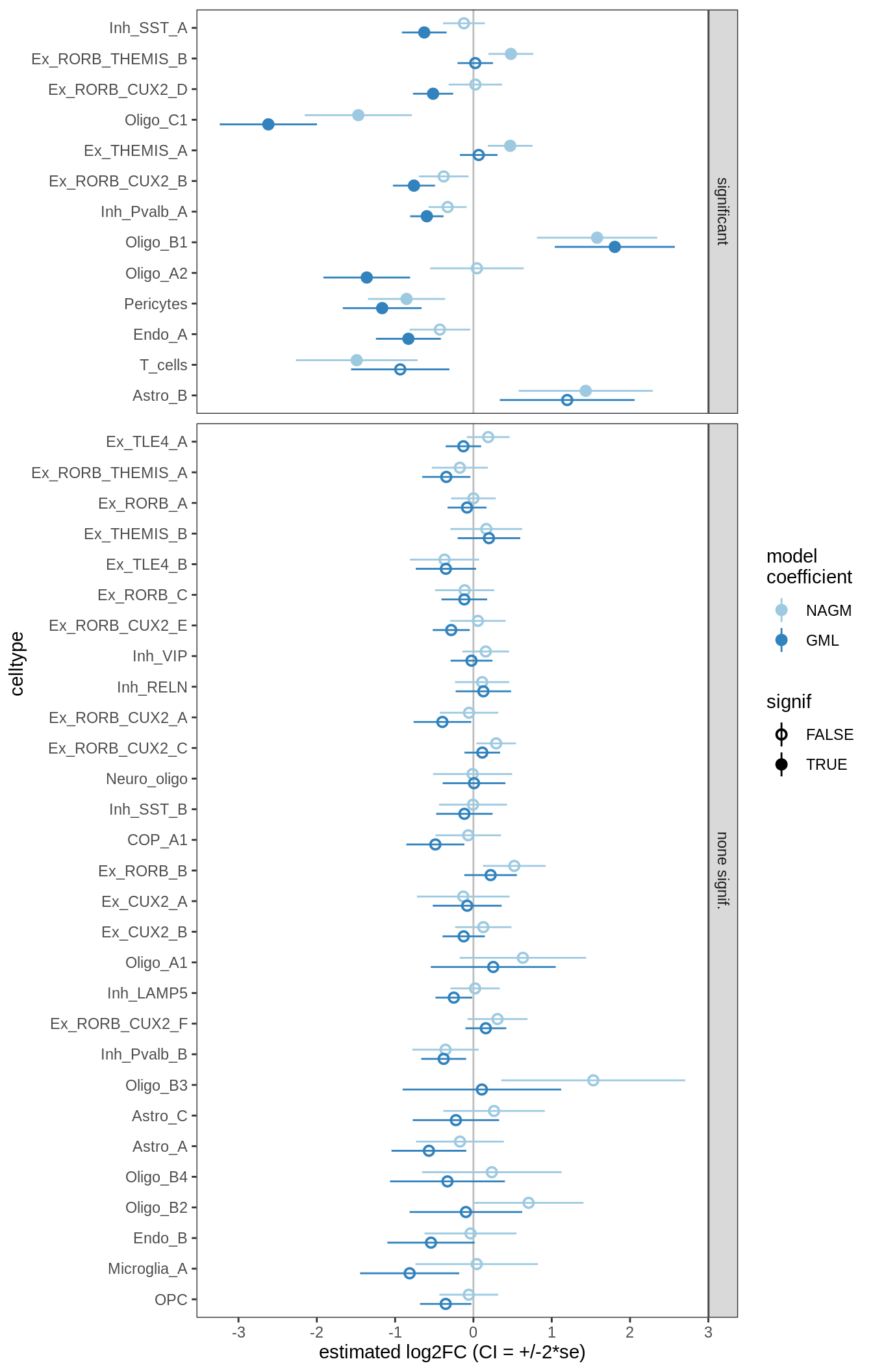
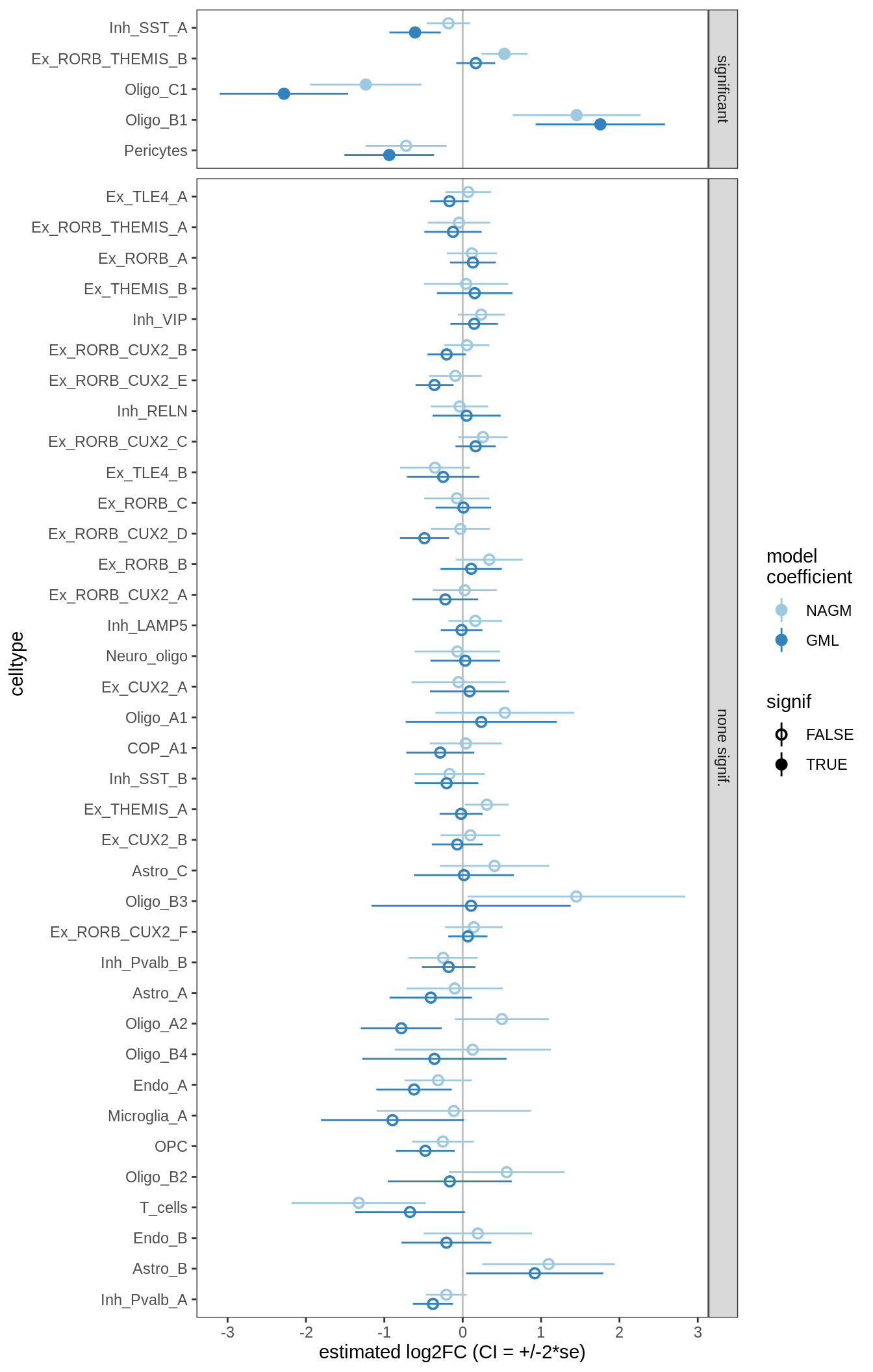
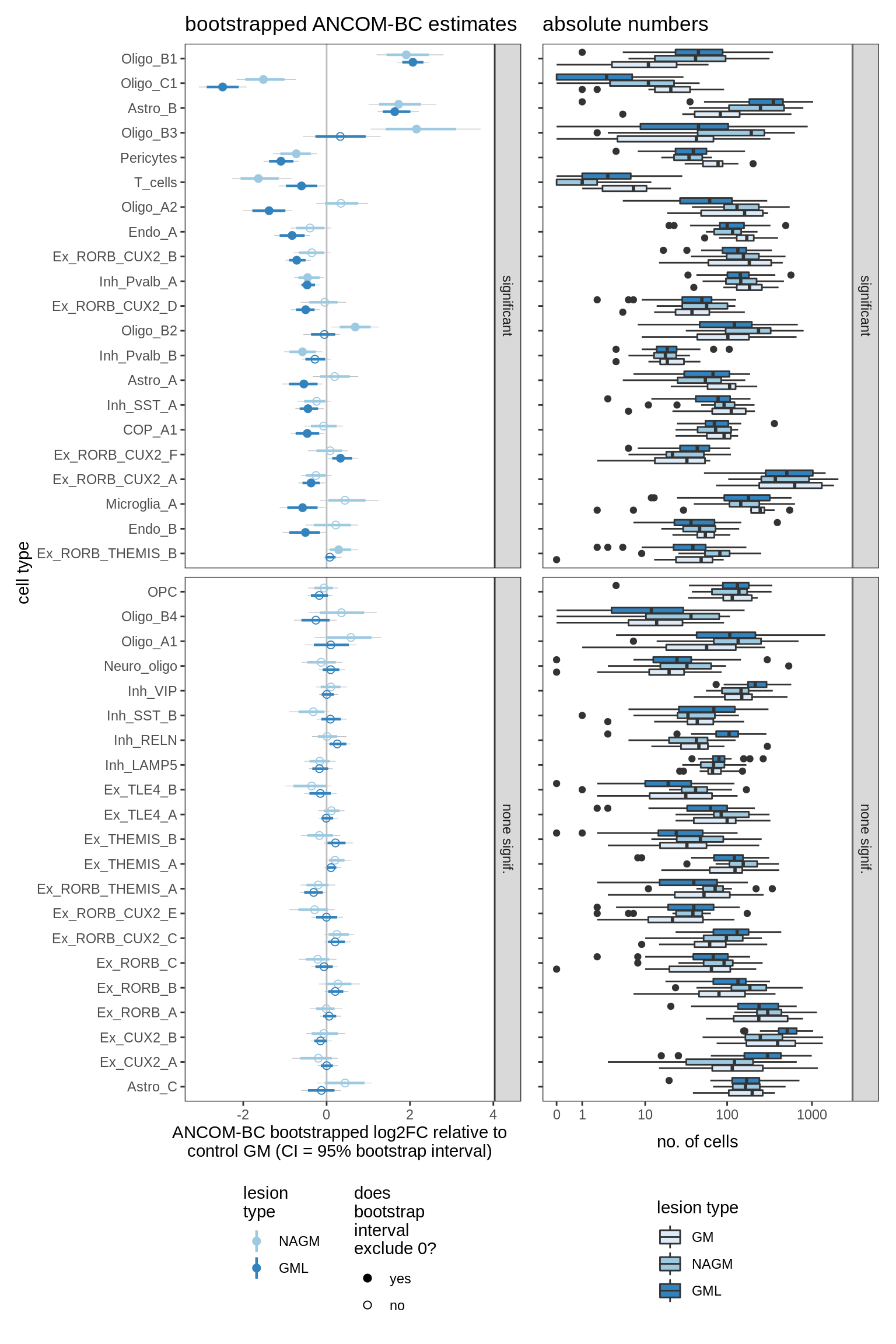
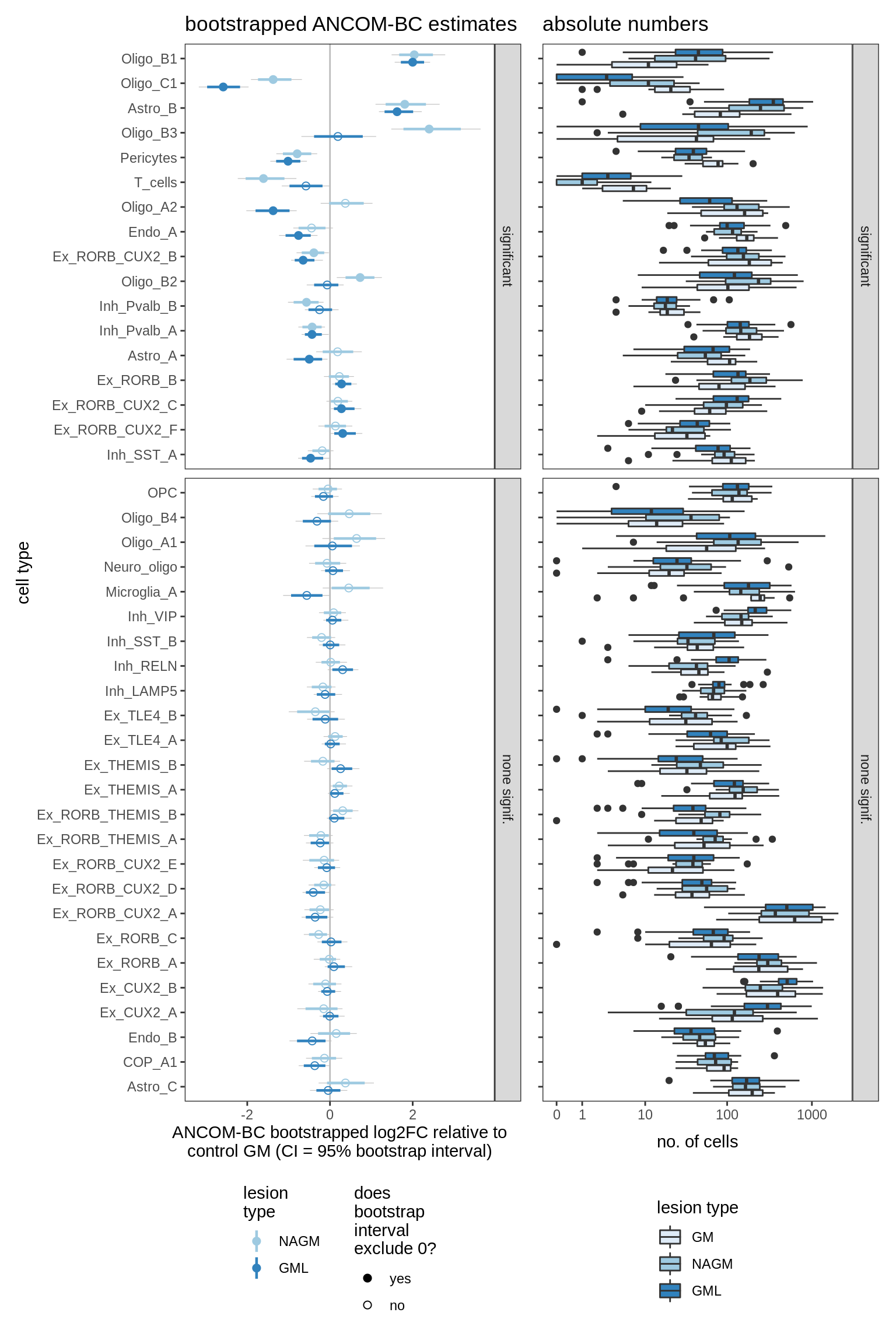
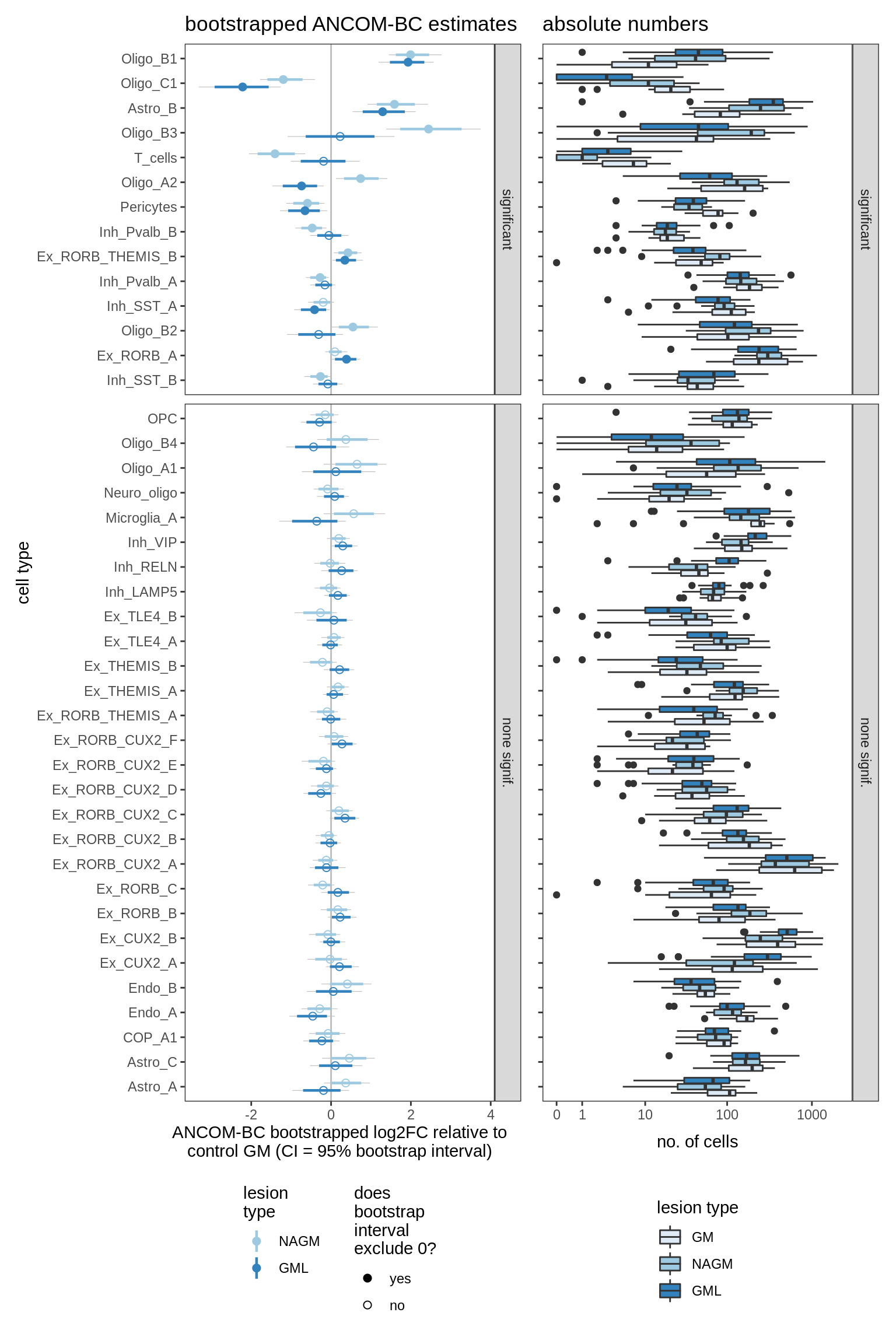
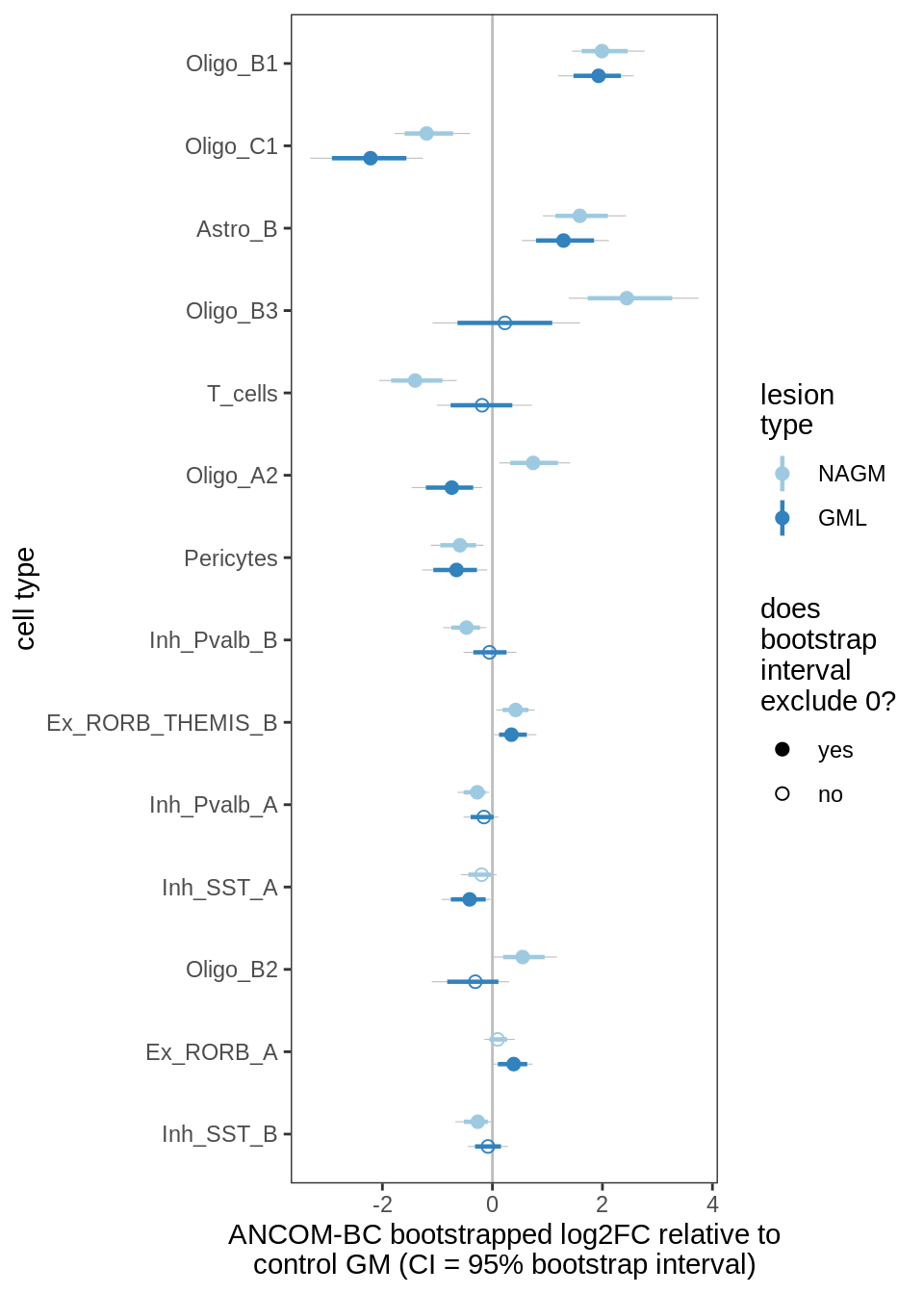
ANCOM-BC bootstrap results, lesions only
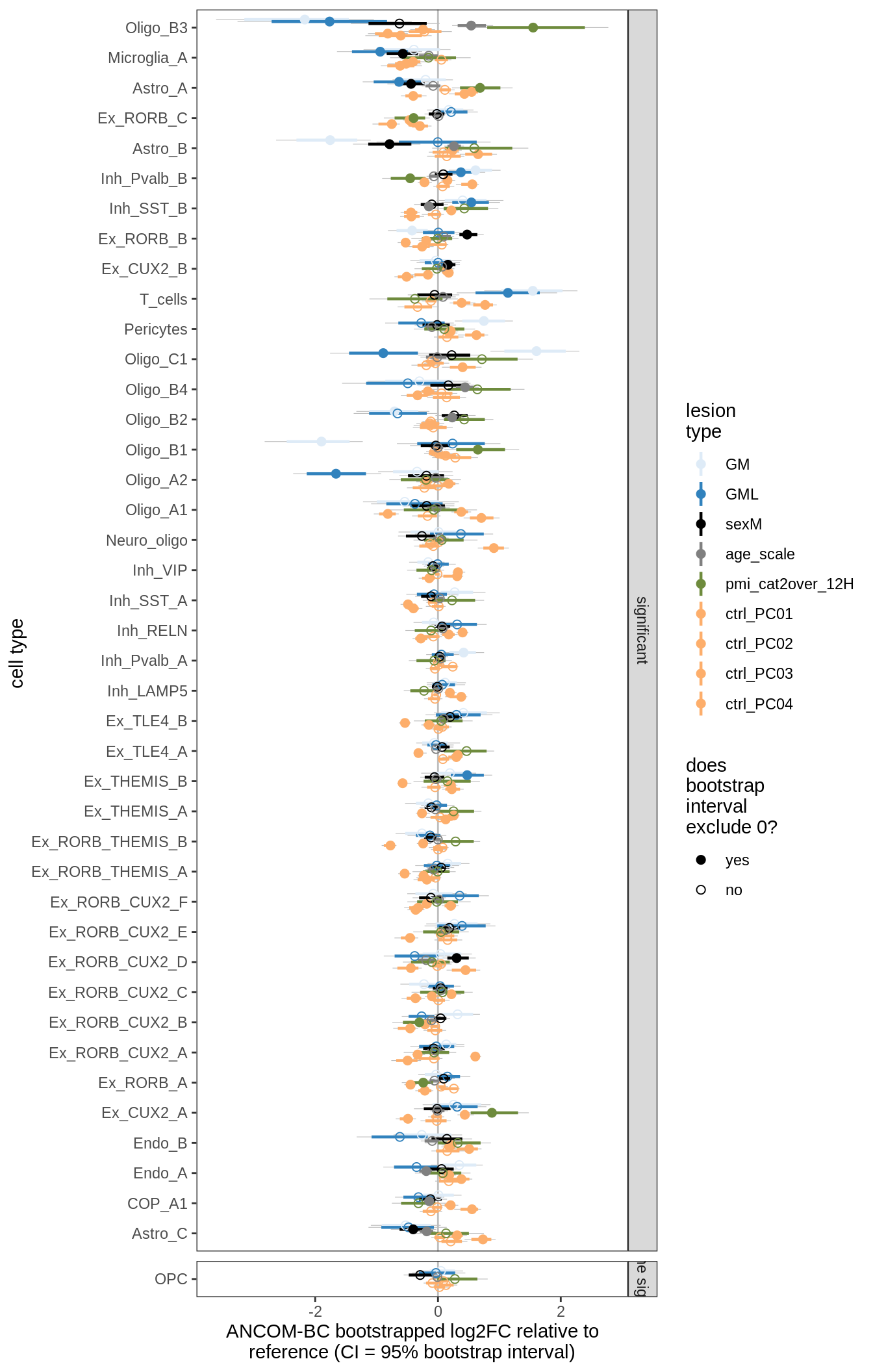
for (nn in names(boots_ls)) {
cat('#### ', nn, '\n')
print(plot_boots_dt(boots_ls[[nn]], coef_filter = "lesion_type",
ancom_obj = ancom_ls[[nn]], meta_dt = meta_dt))
cat('\n\n')
}lesions_WM
lesions_GM

lesions_GM_1pcs
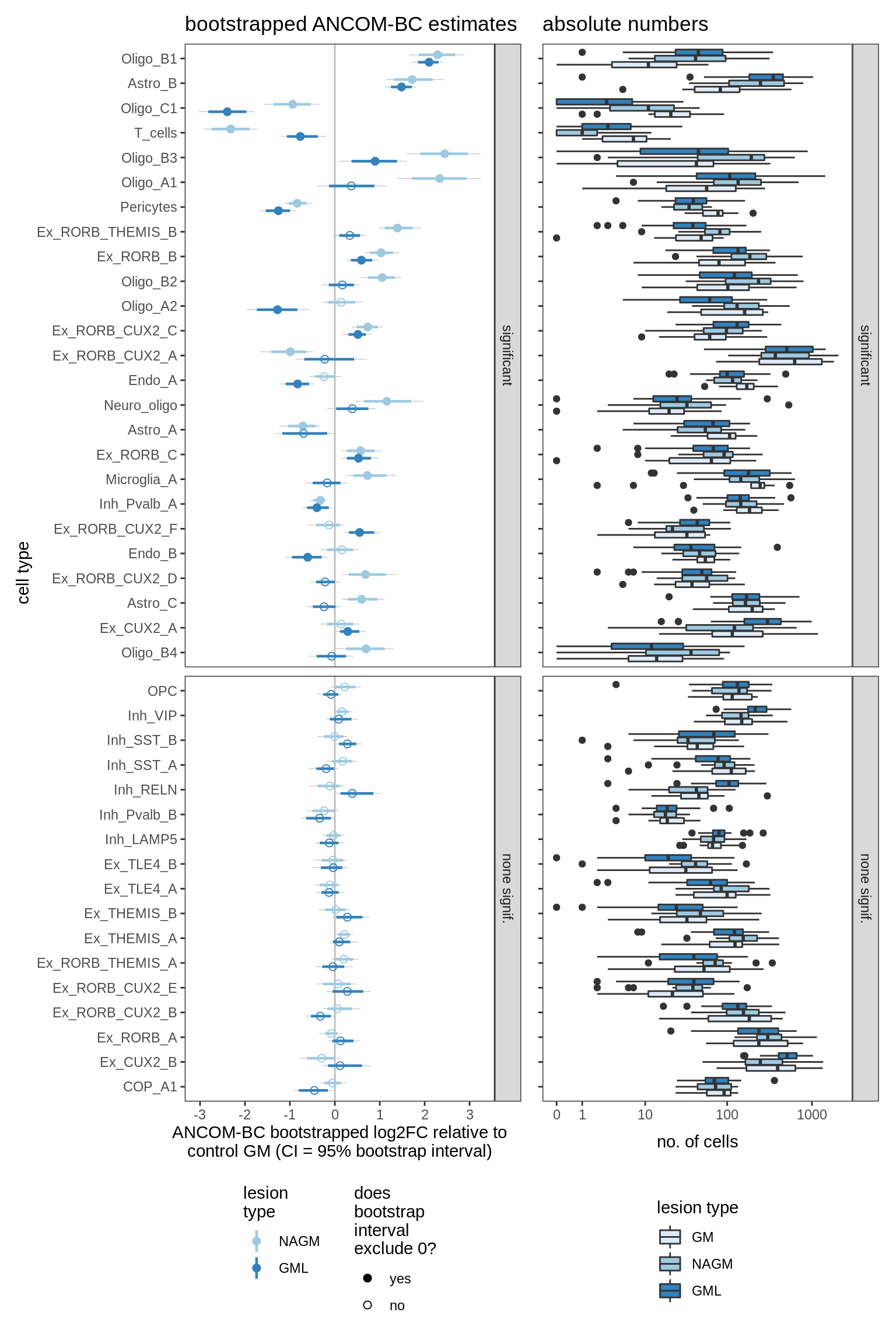
lesions_GM_2pcs
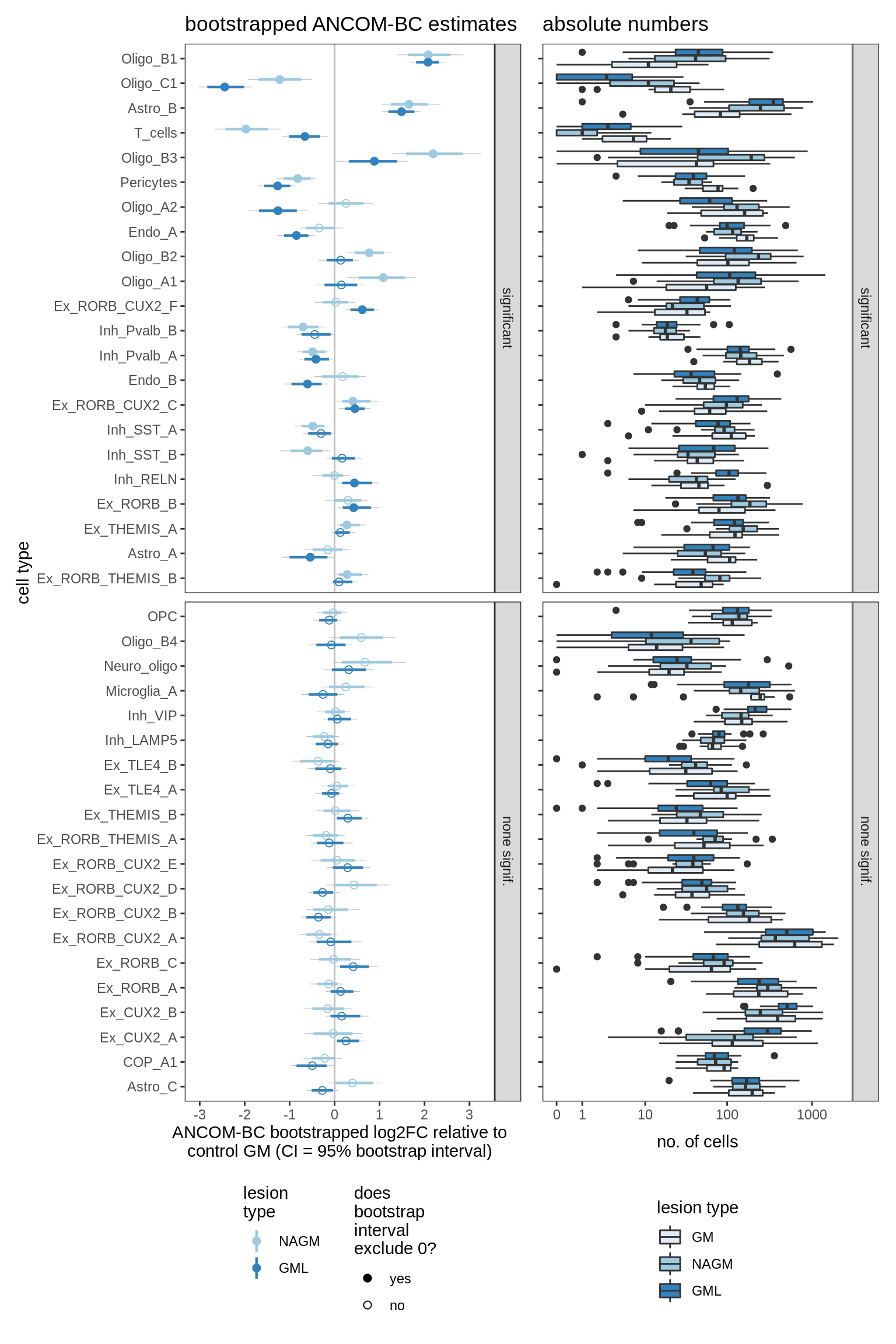
lesions_GM_3pcs
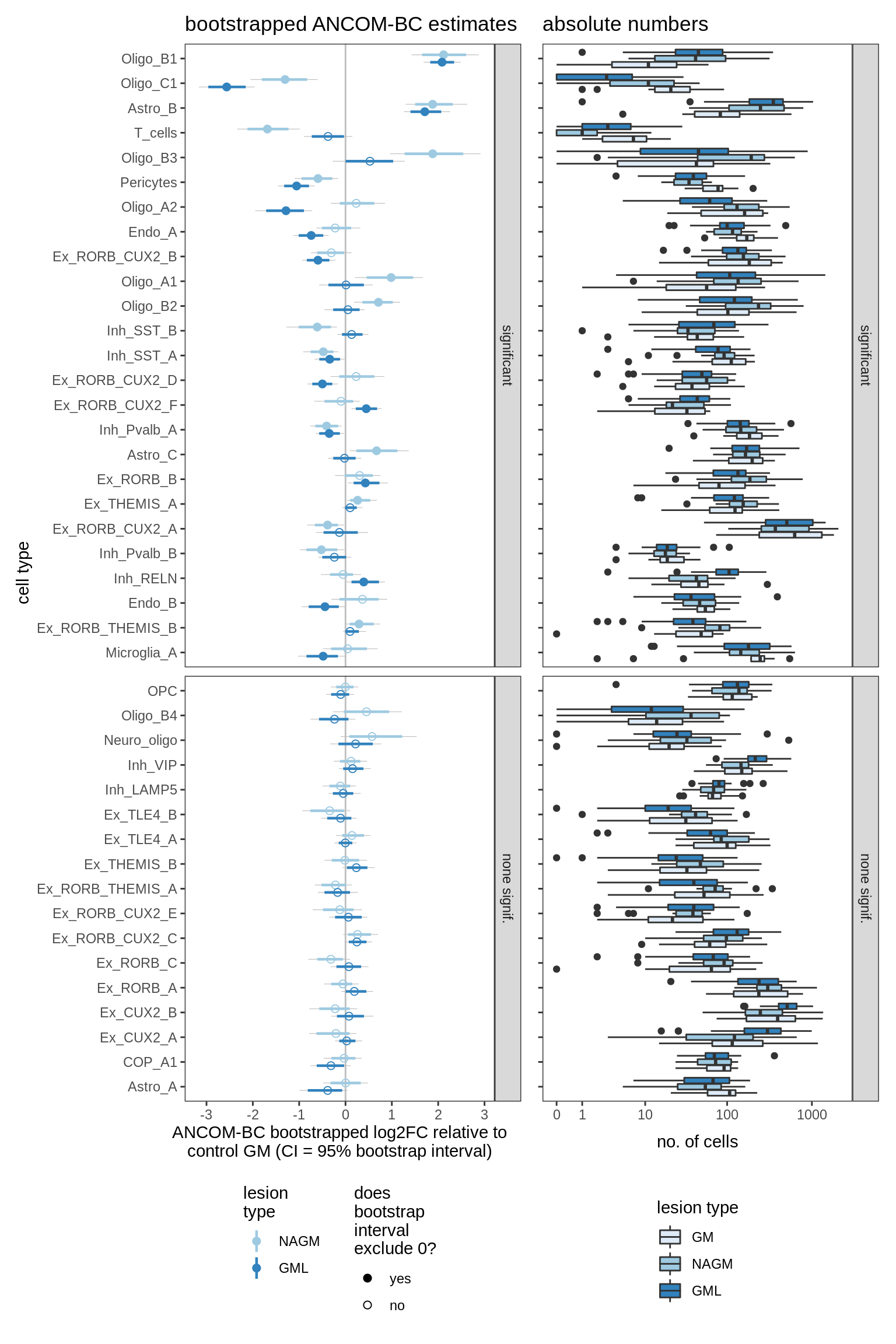
lesions_GM_4pcs
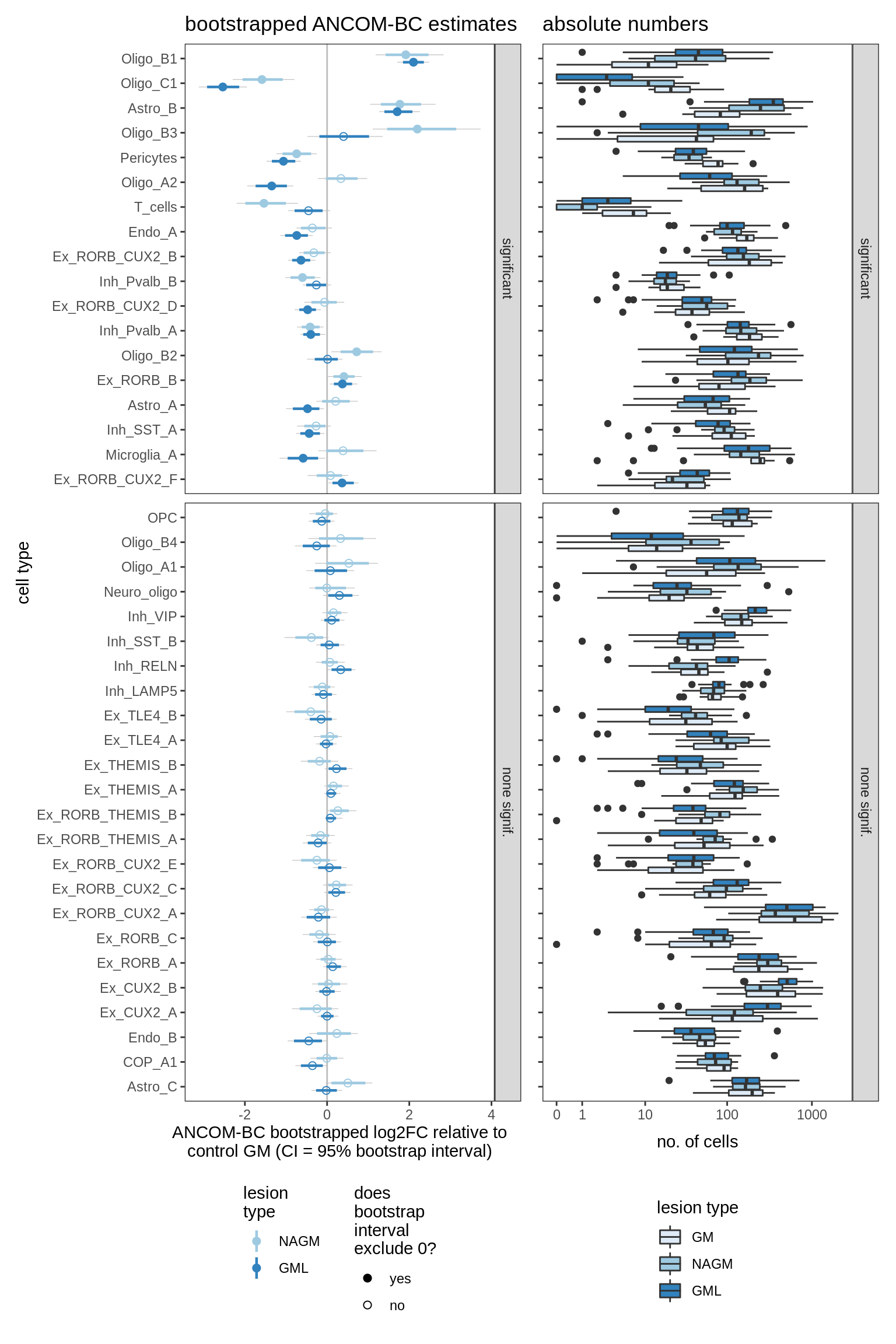
lesions_GM_5pcs
lesions_GM_6pcs
lesions_GM_7pcs
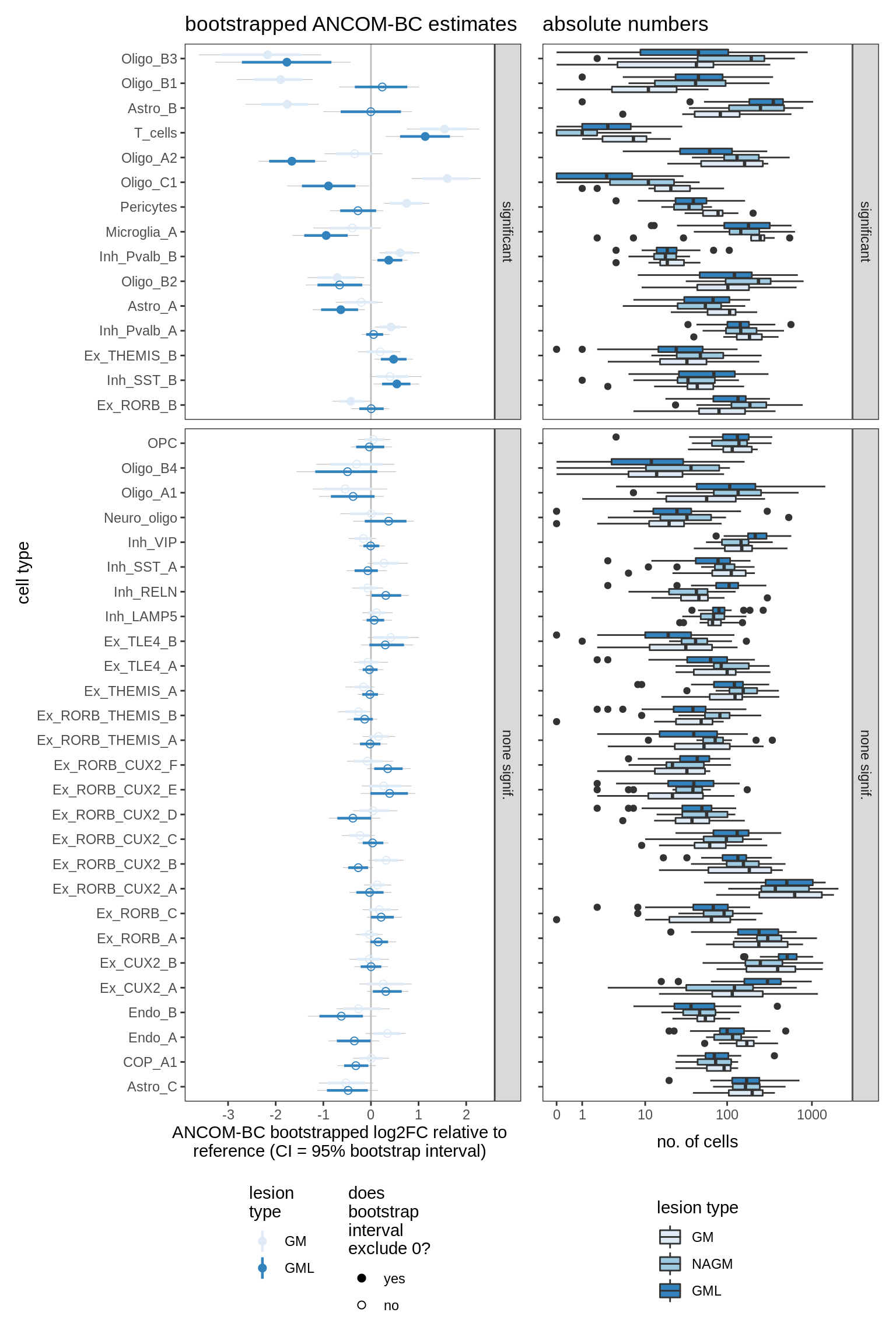
ANCOM-BC bootstrap results, lesions, significant only
for (nn in names(boots_ls)) {
cat('#### ', nn, '\n')
print(plot_boots_dt(boots_ls[[nn]],
coef_filter = "lesion_type", signif_only = TRUE))
cat('\n\n')
}lesions_WM
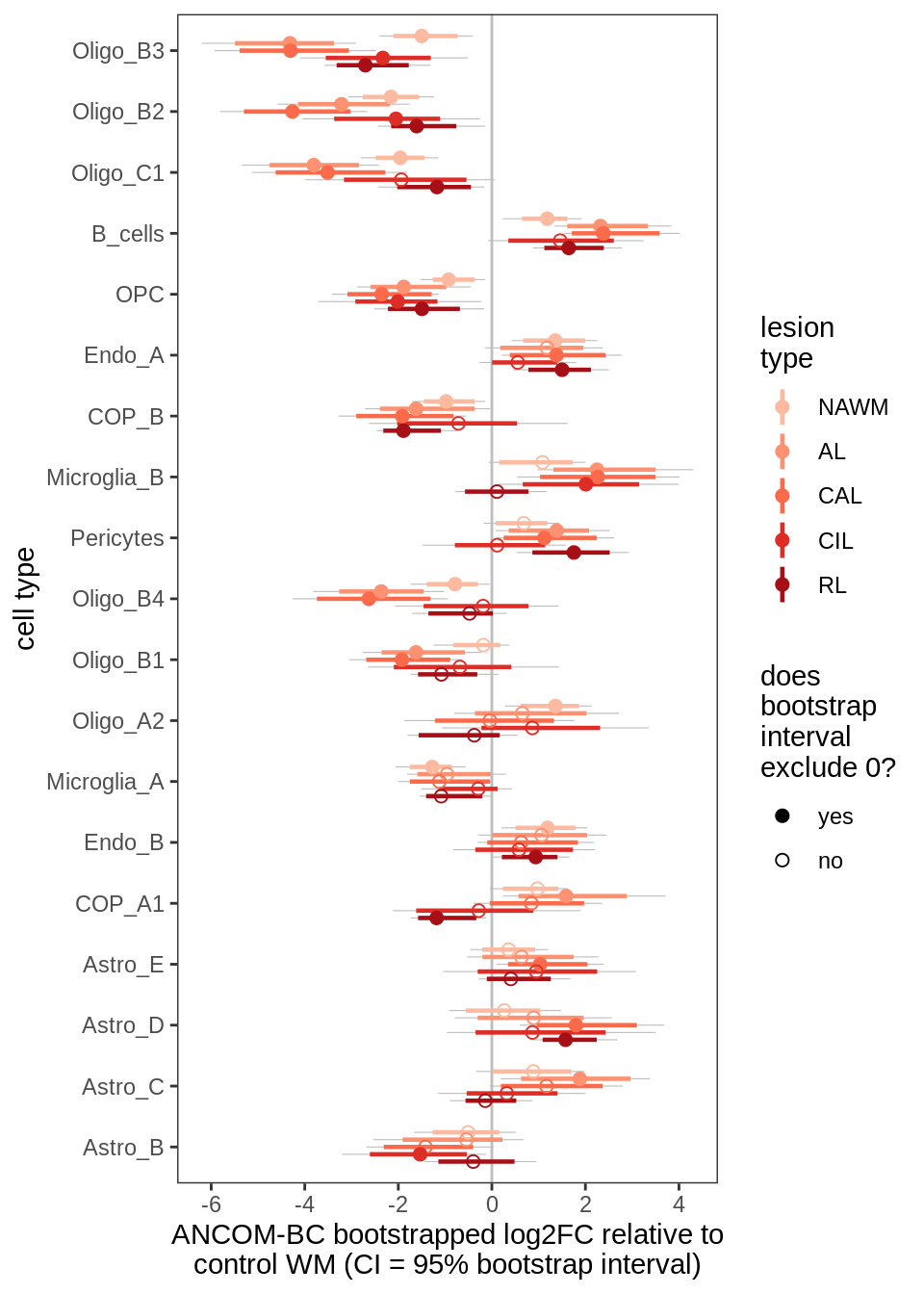
lesions_GM

lesions_GM_1pcs
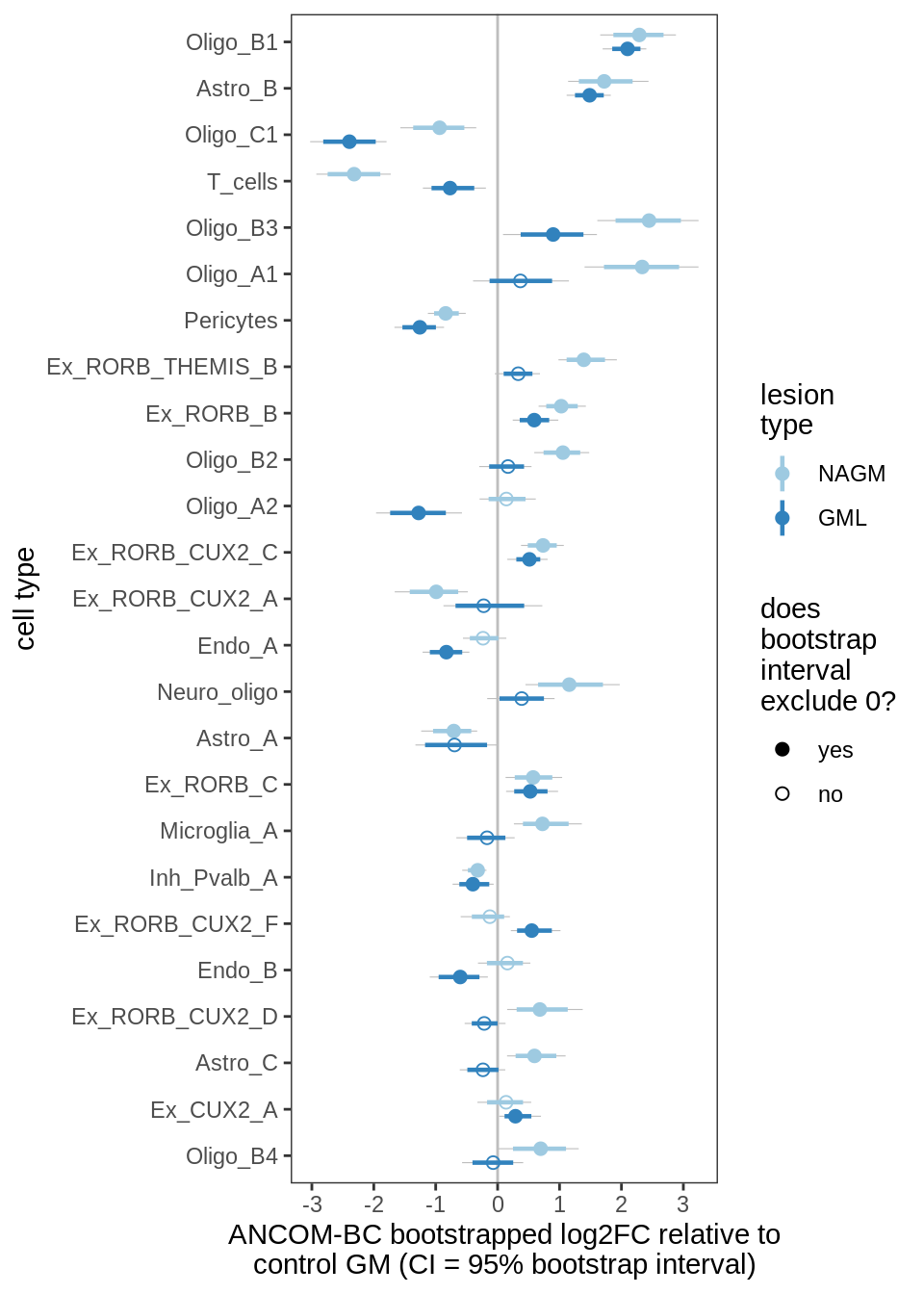
lesions_GM_2pcs

lesions_GM_3pcs
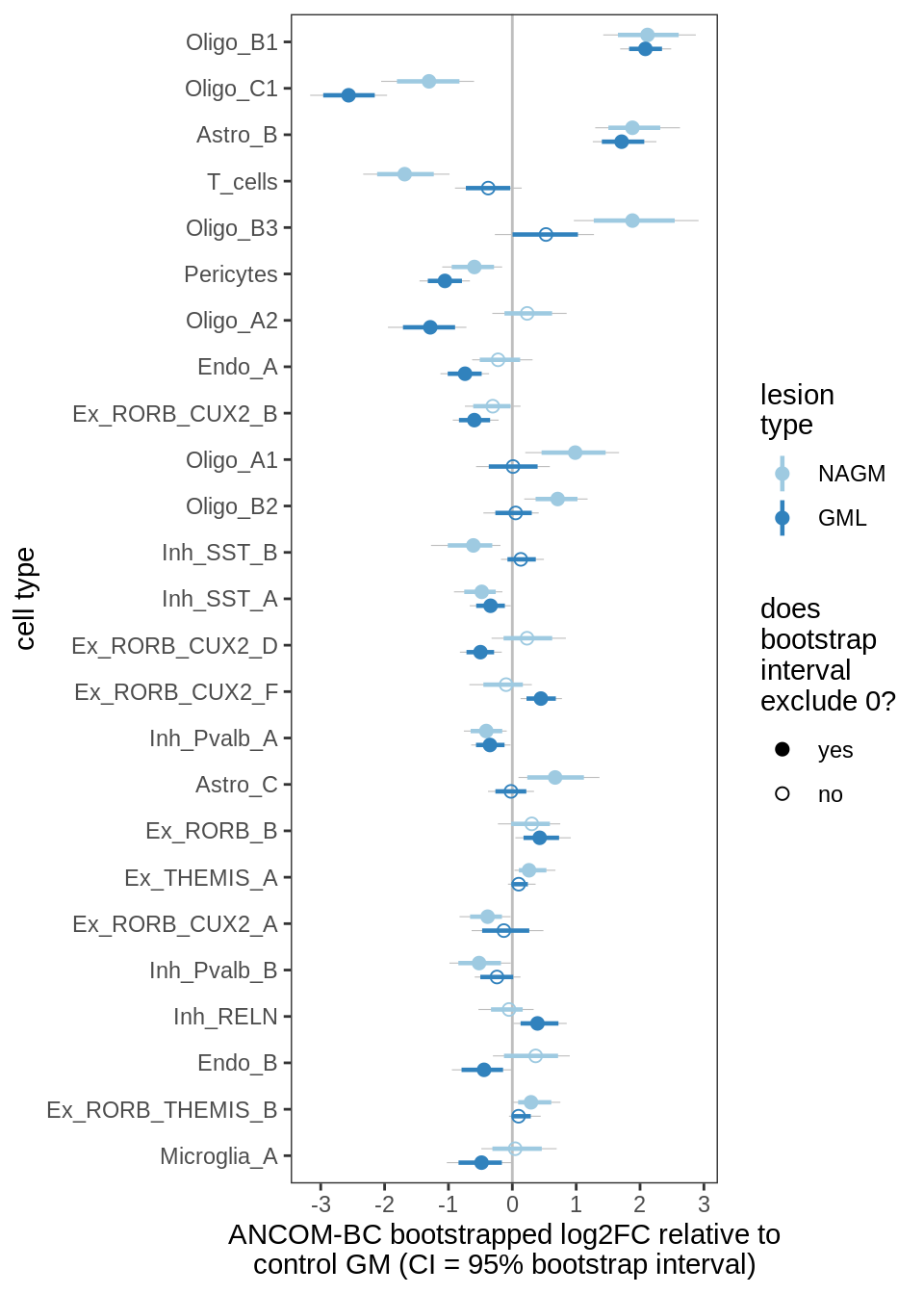
lesions_GM_4pcs
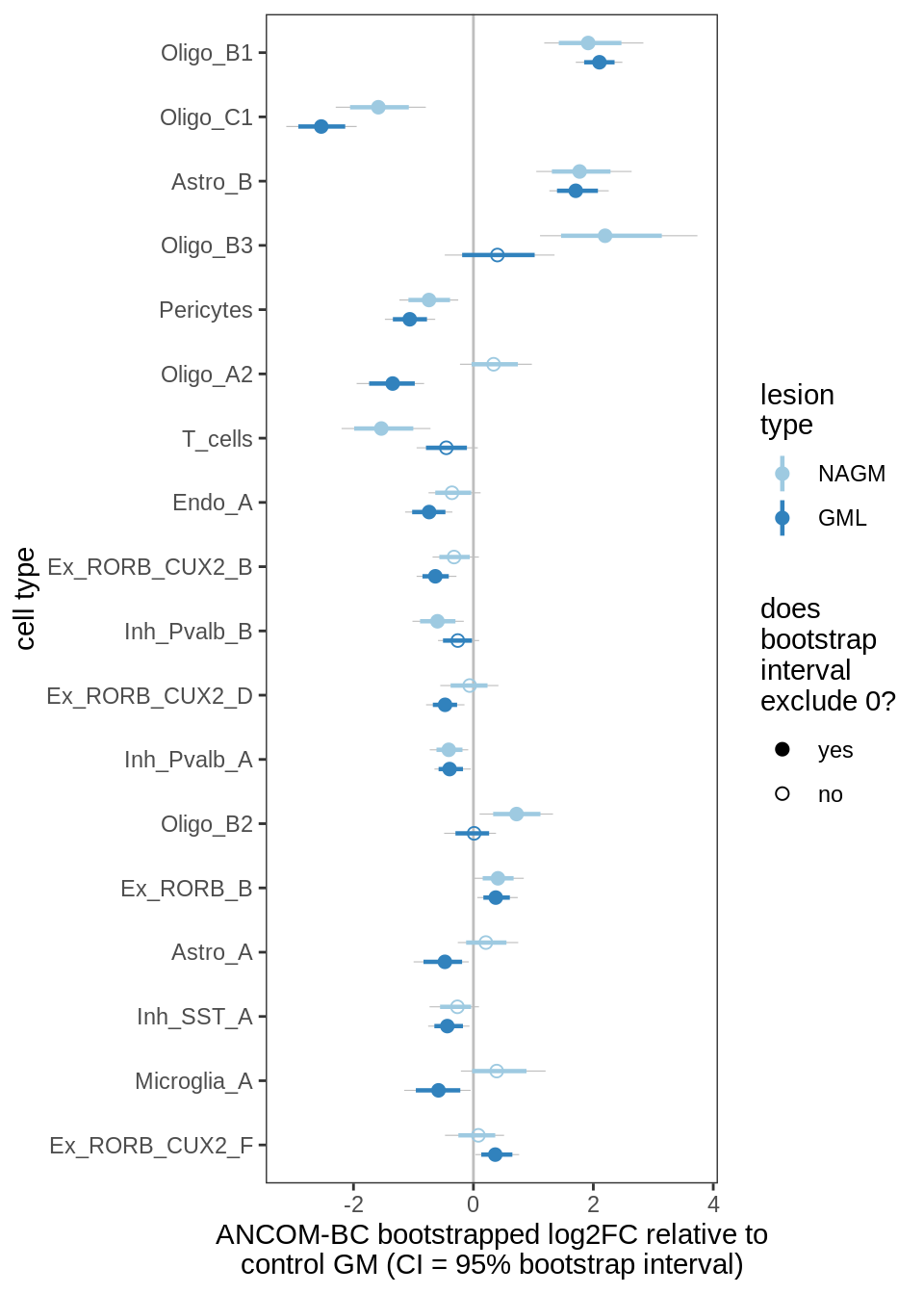
lesions_GM_5pcs
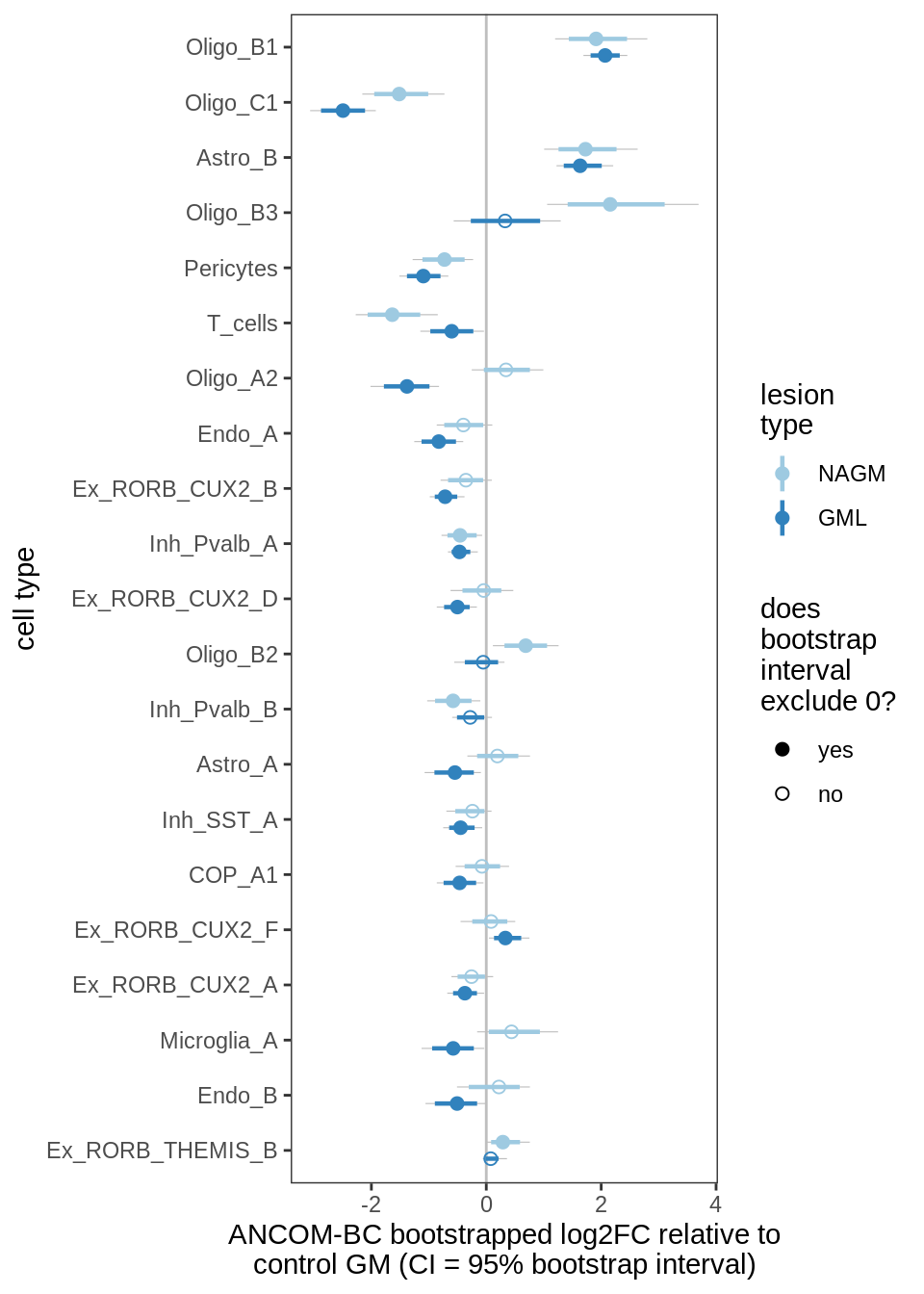
lesions_GM_6pcs
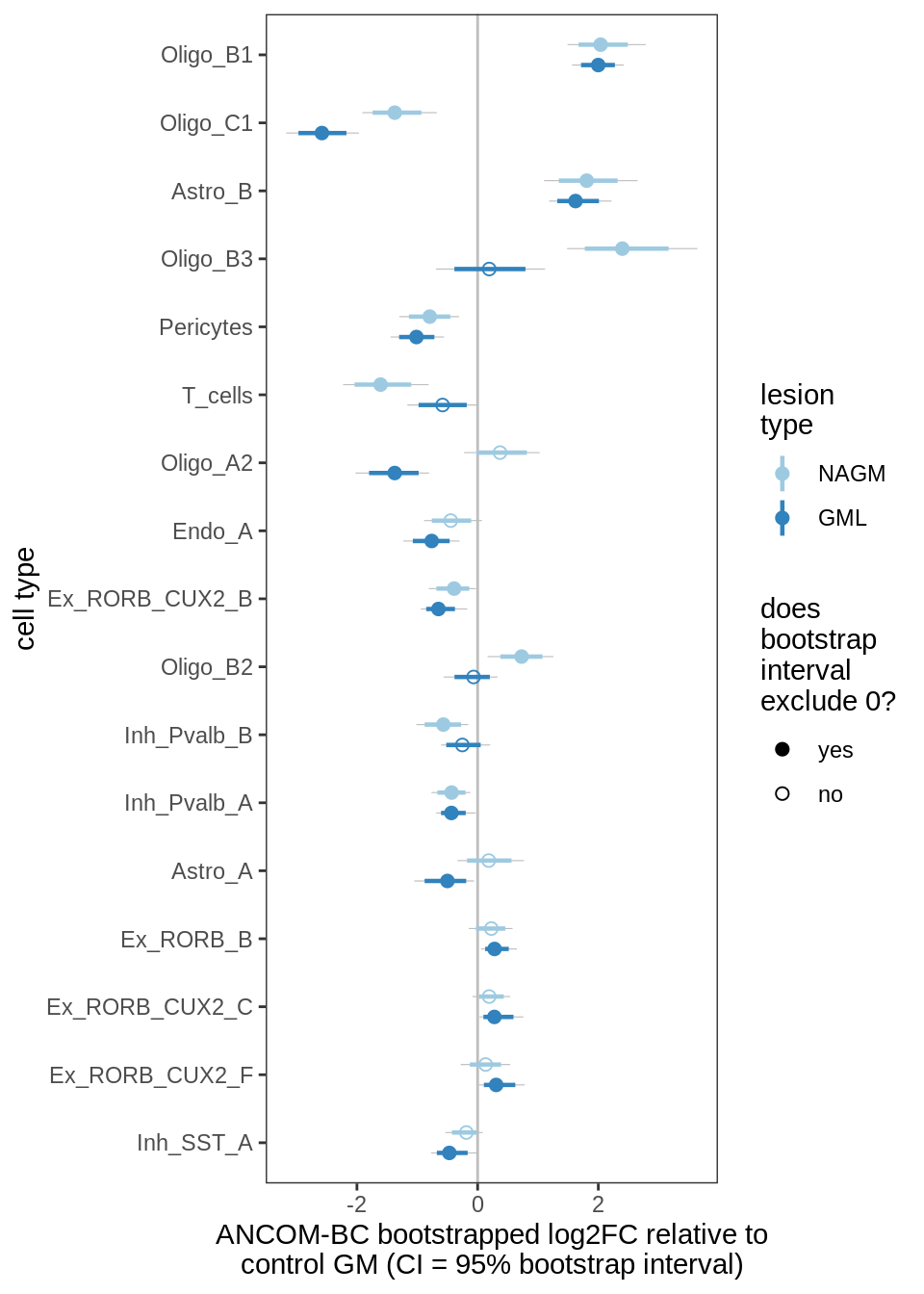
lesions_GM_7pcs

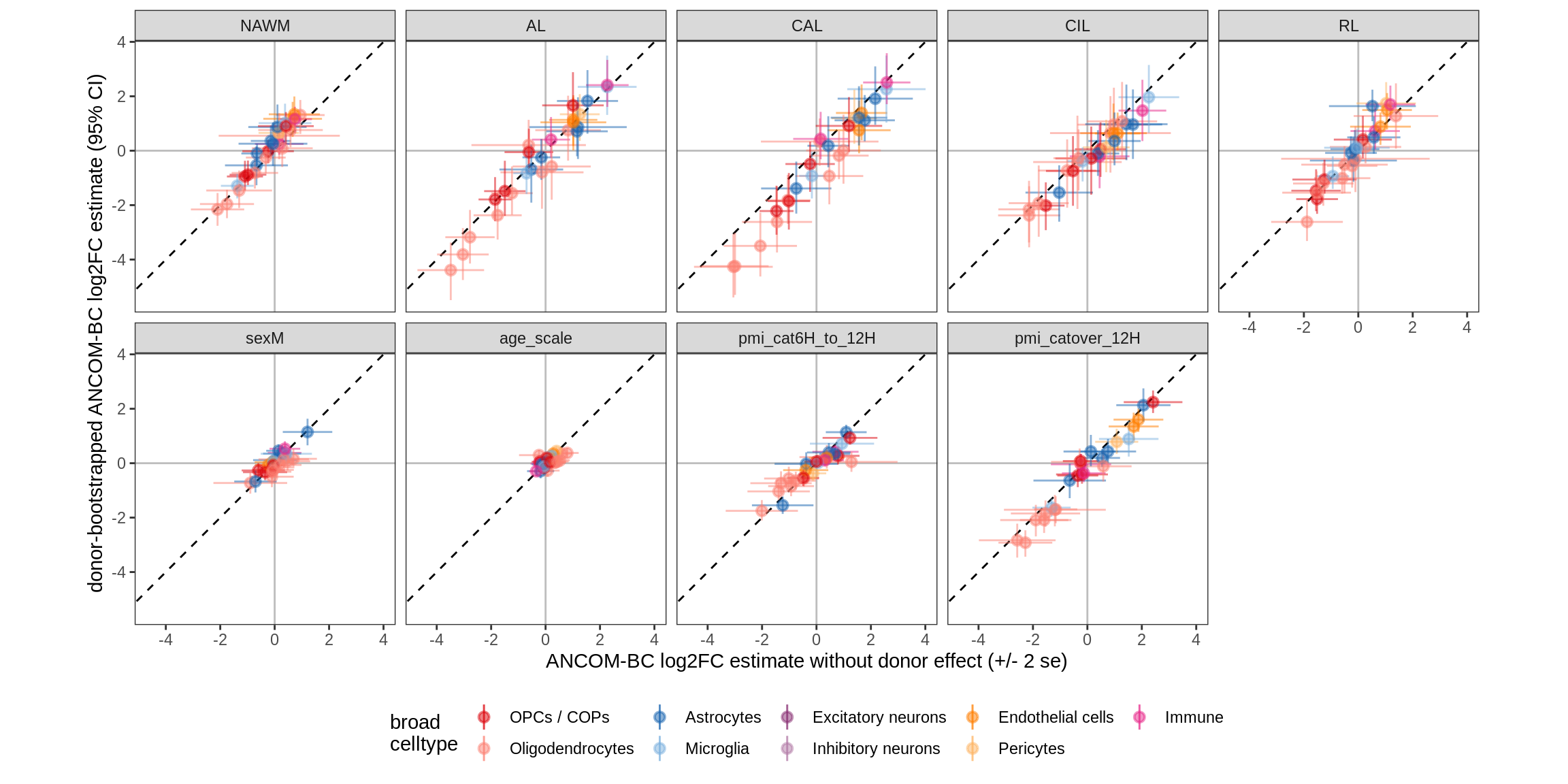
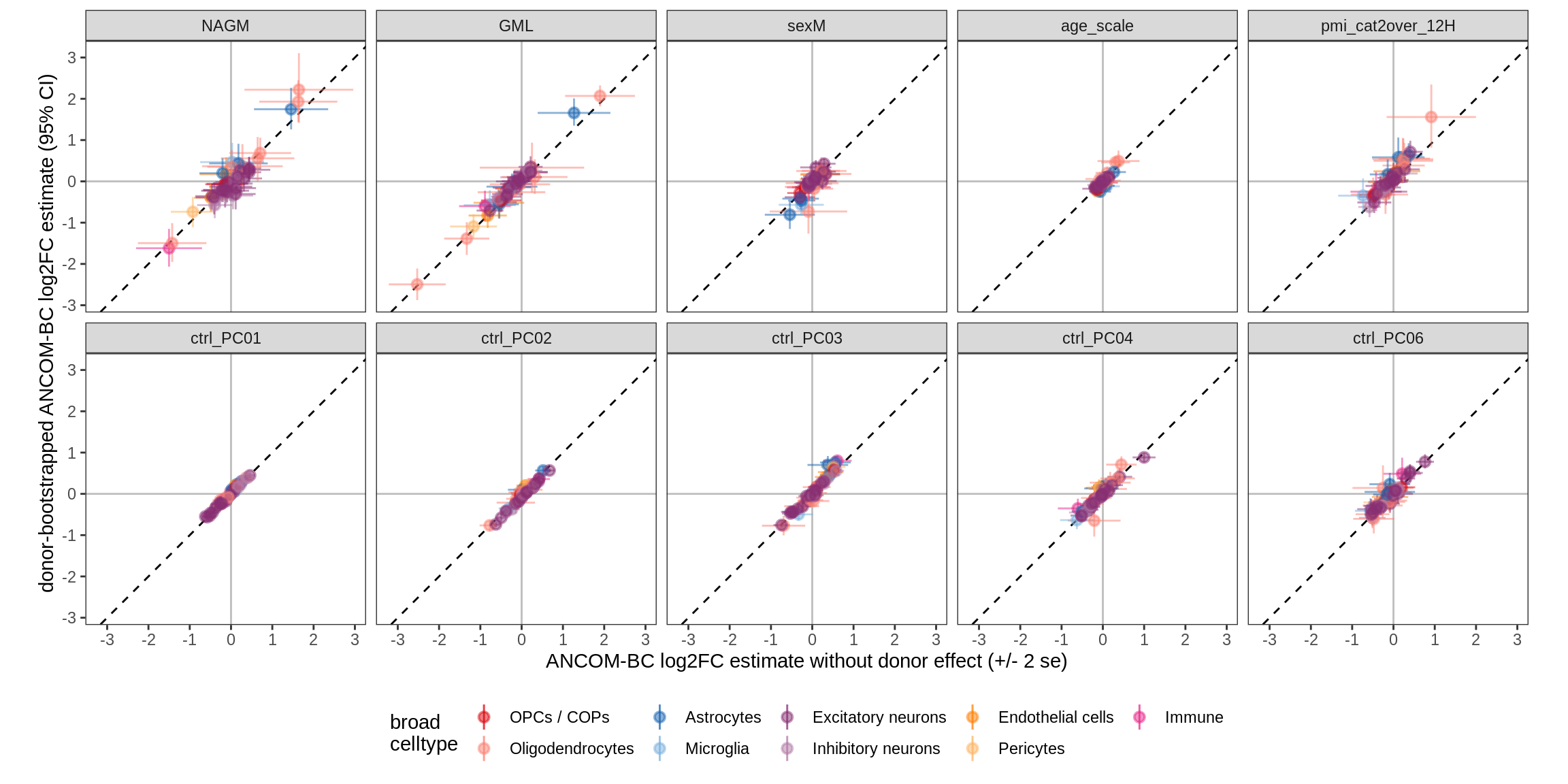
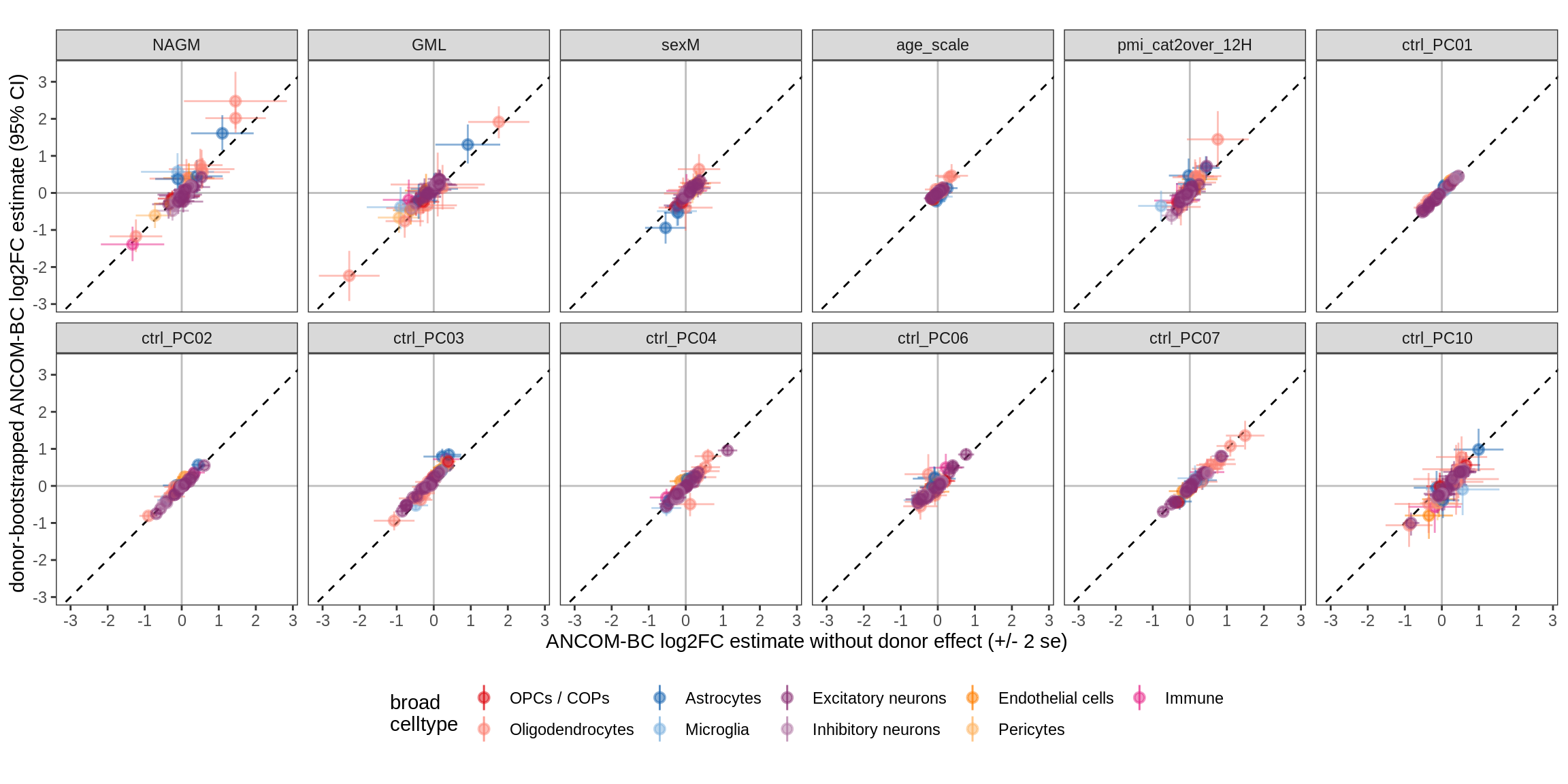
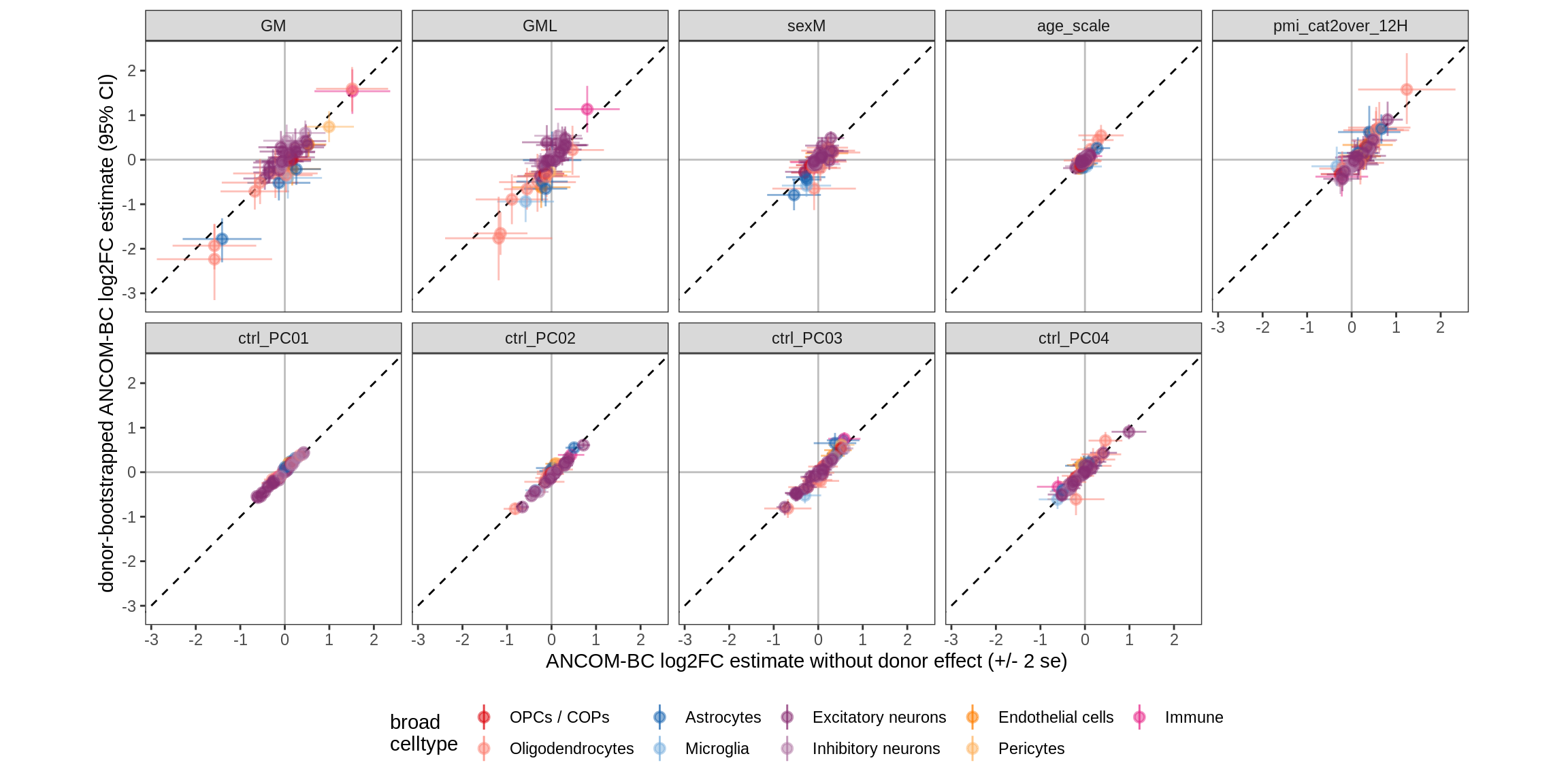
Bootstrap vs standard
for (nn in names(boots_ls)) {
cat('#### ', nn, '\n')
print(plot_boots_vs_standard(boots_ls[[nn]], ancom_ls[[nn]], labels_dt,
q_cut = 0.05))
cat('\n\n')
}lesions_WM
lesions_GM
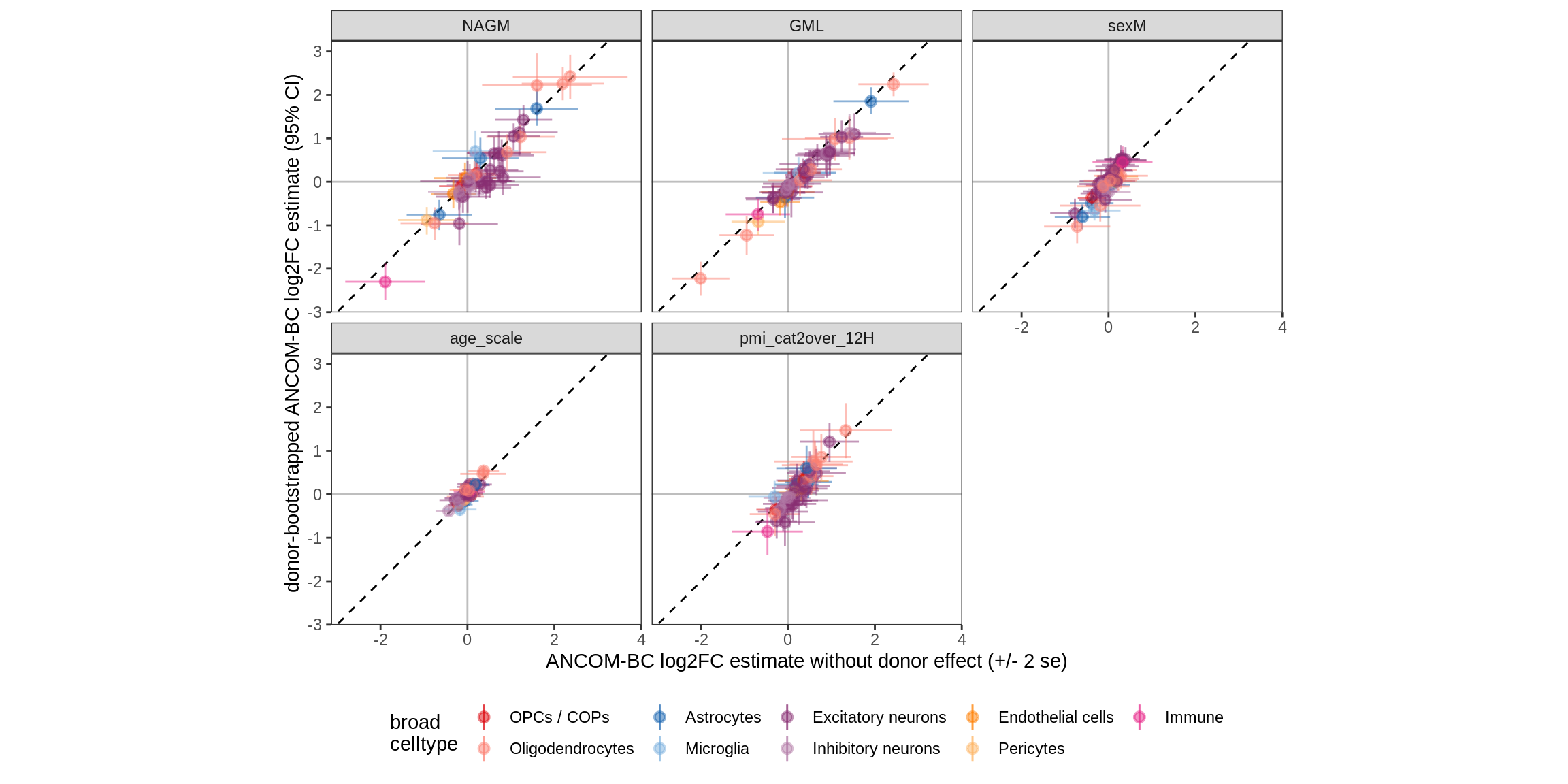
lesions_GM_1pcs
lesions_GM_2pcs

lesions_GM_3pcs
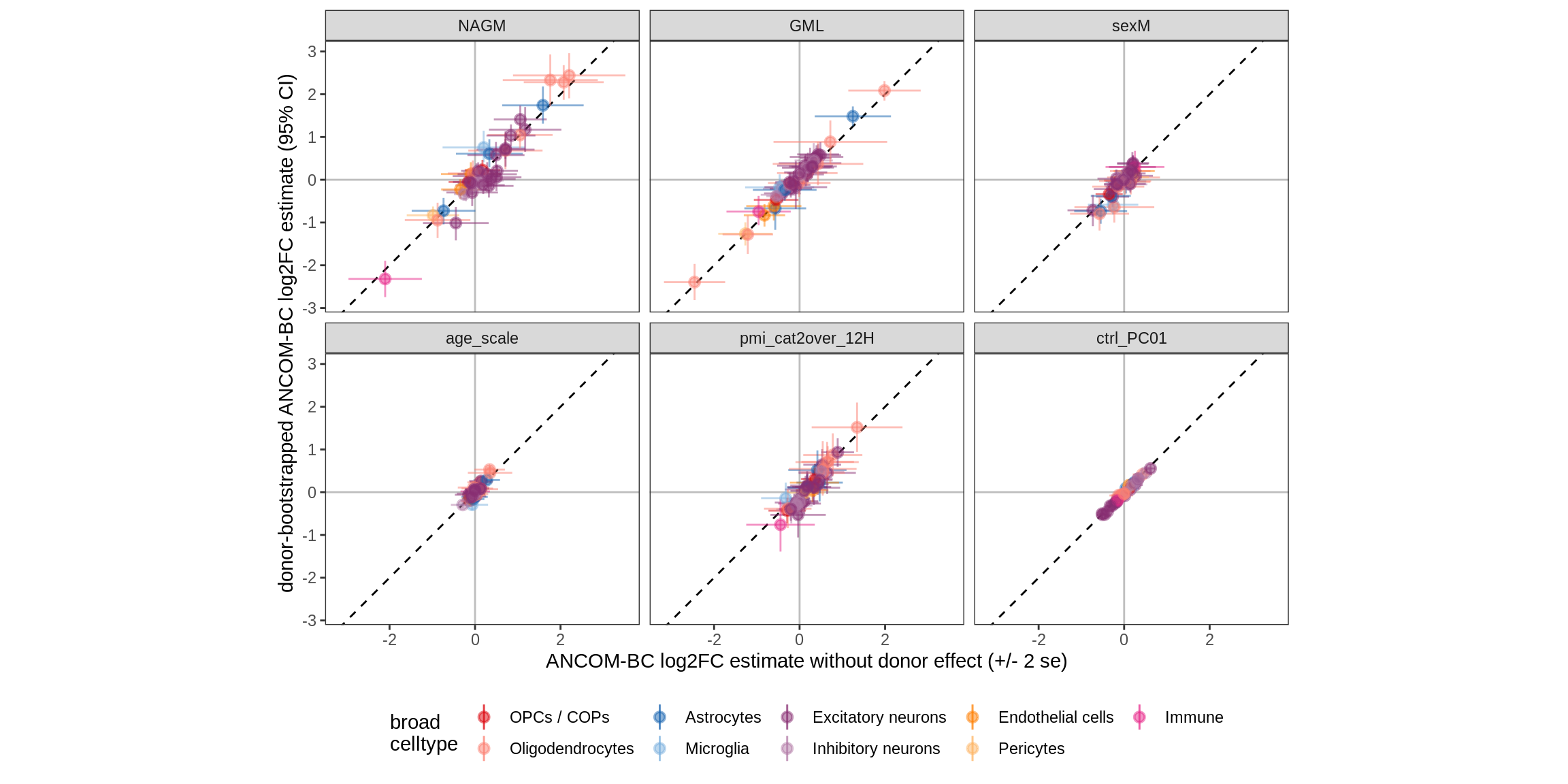
lesions_GM_4pcs

lesions_GM_5pcs
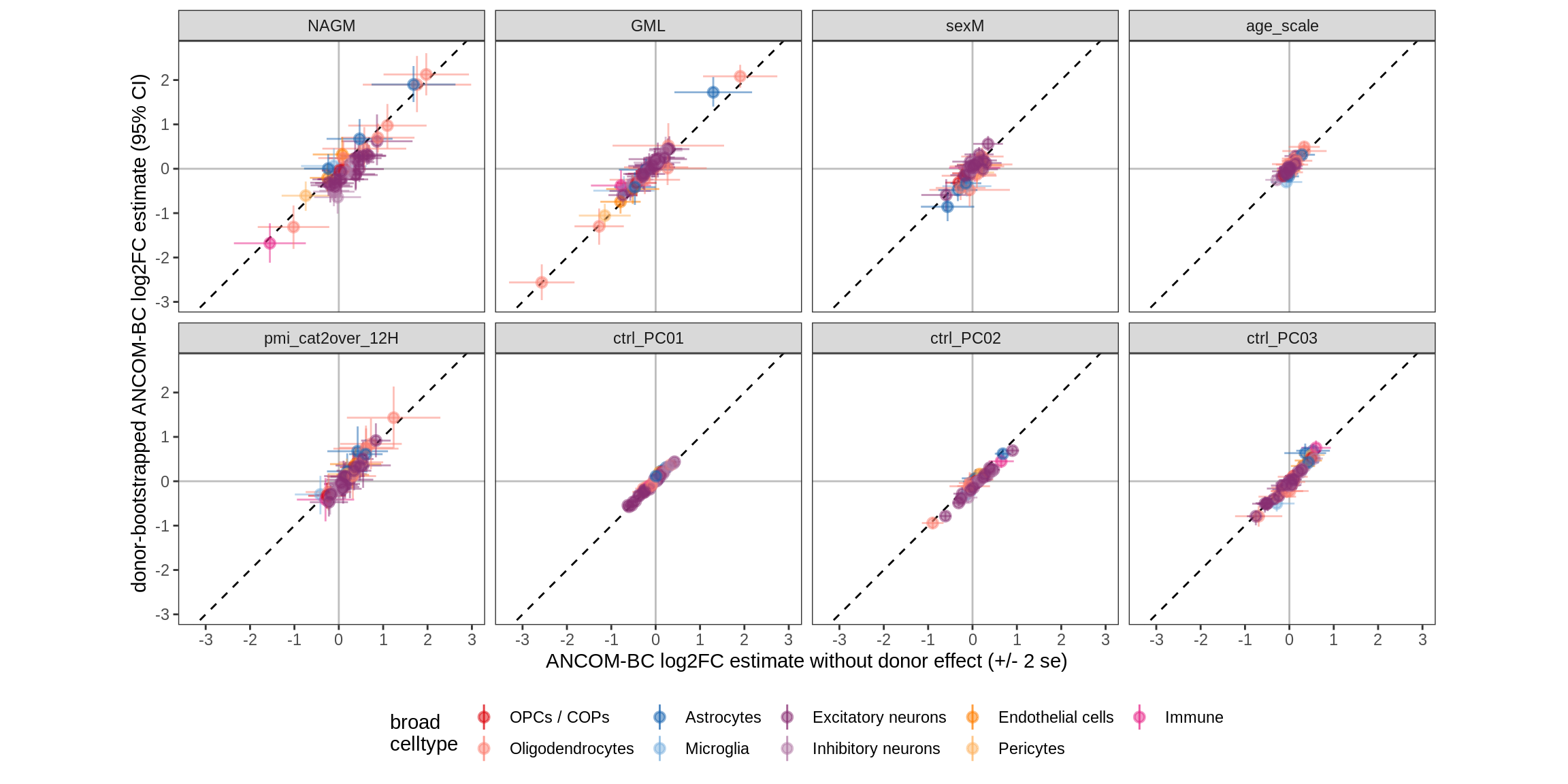
lesions_GM_6pcs
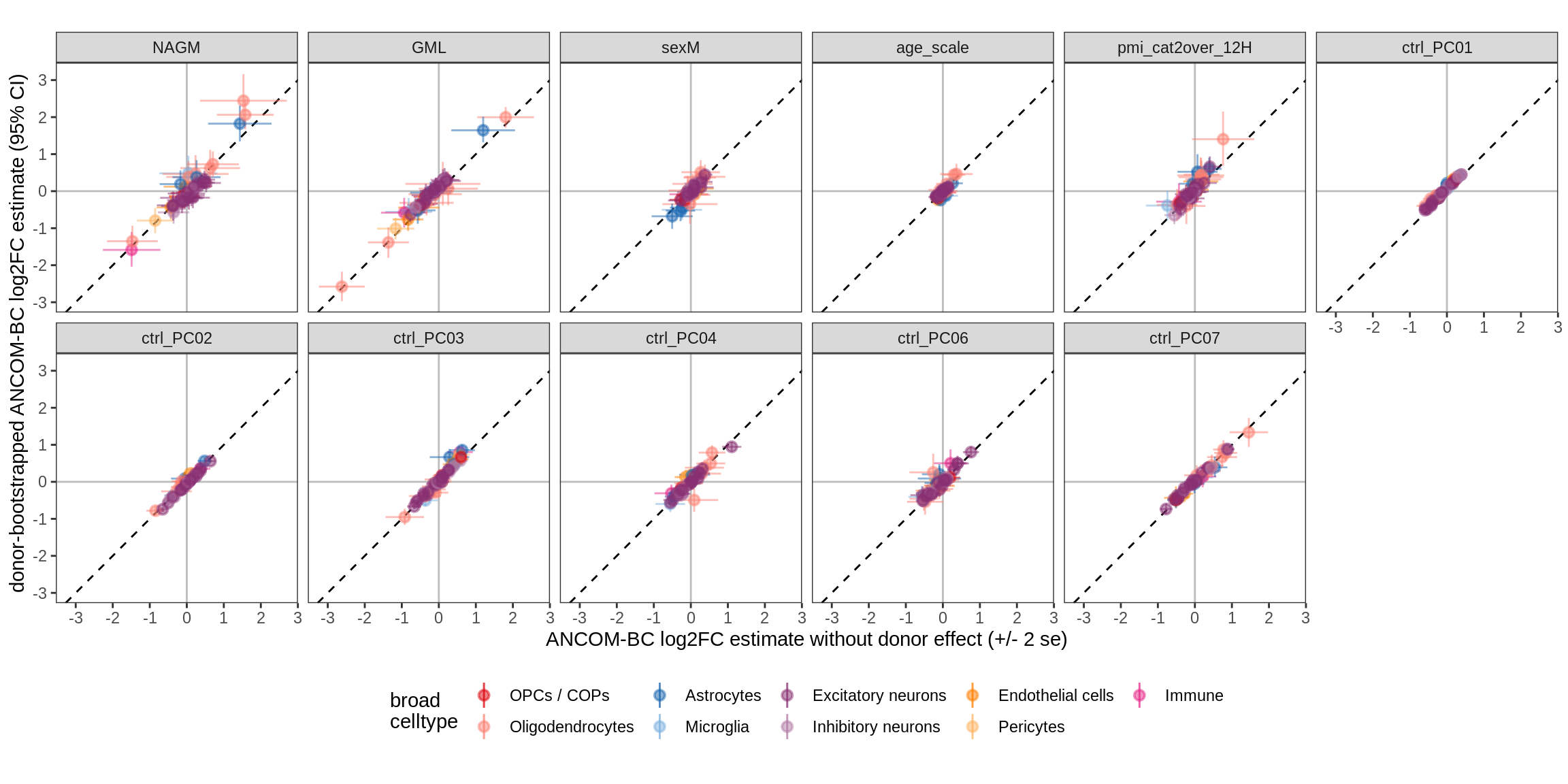
lesions_GM_7pcs
Effect of including PCs, neurons only
for (sel_coef in unique(pcs_coefs_dt$coef)) {
cat('#### ', sel_coef, '\n')
print(plot_effect_of_pcs(sel_coef,
pcs_coefs_dt[ type_broad %in% gm_pc_spec$broad_sel ] ))
cat('\n\n')
}NAGM

GML
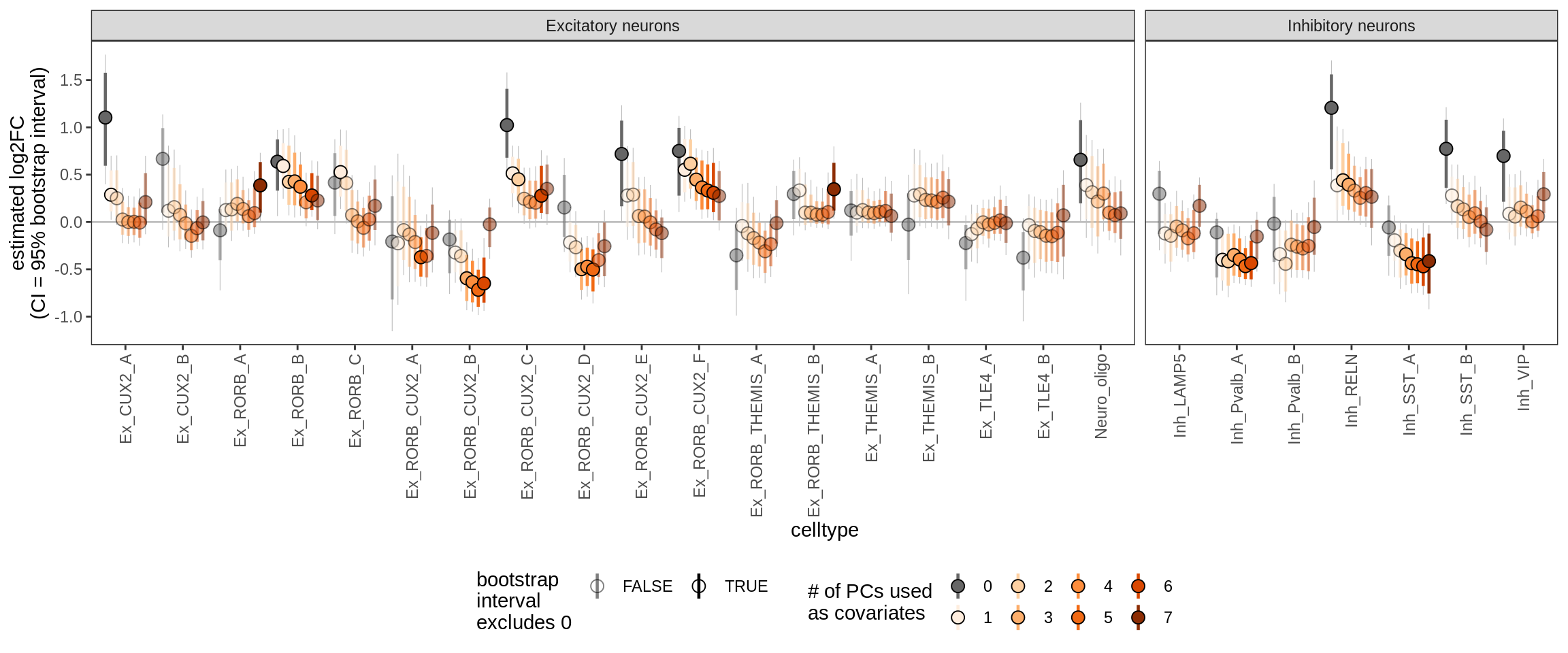
sexM
age_scale

pmi_cat2over_12H
ctrl_PC01
ctrl_PC02

ctrl_PC03

ctrl_PC04
ctrl_PC06
ctrl_PC07

Effect of including PCs, other celltypes
for (sel_coef in unique(pcs_coefs_dt$coef)) {
cat('#### ', sel_coef, '\n')
print(plot_effect_of_pcs(sel_coef,
pcs_coefs_dt[ !(type_broad %in% gm_pc_spec$broad_sel) ]))
cat('\n\n')
}NAGM
GML

sexM
age_scale
pmi_cat2over_12H

ctrl_PC01

ctrl_PC02
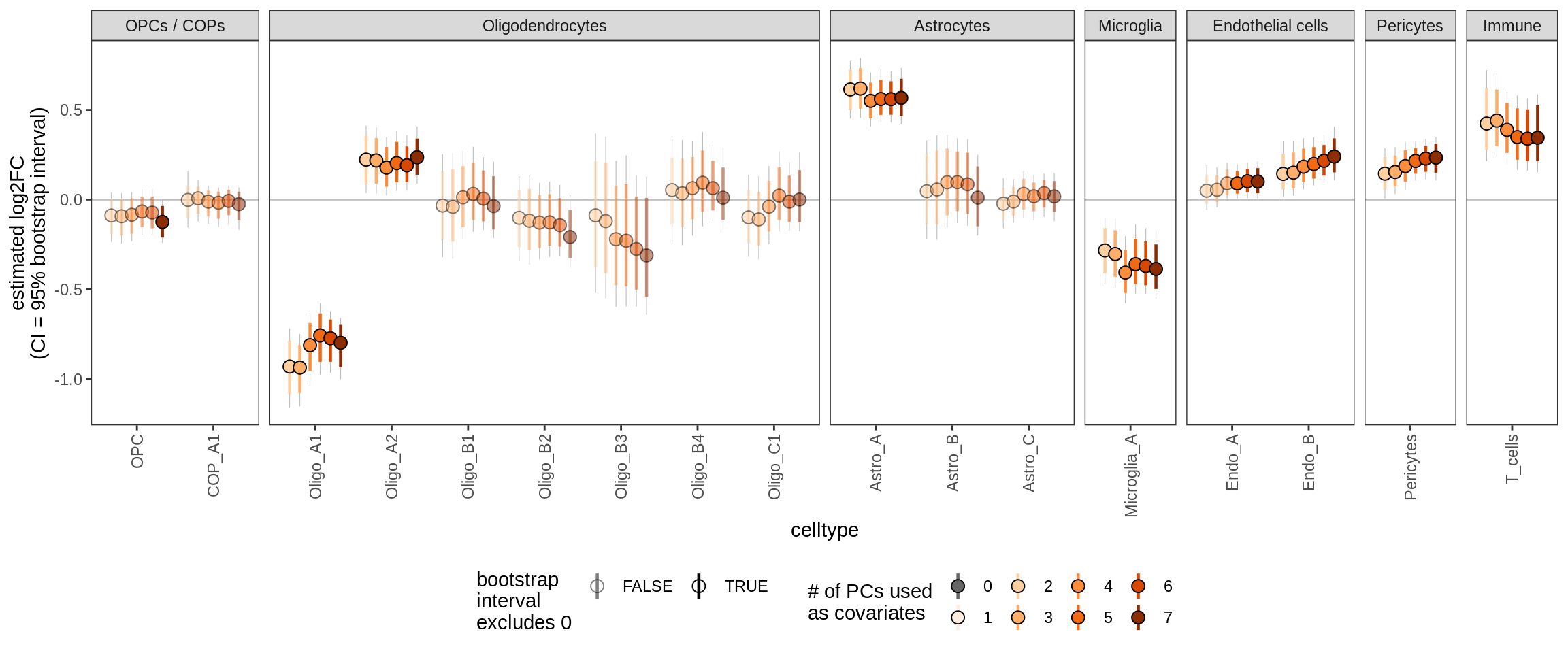
ctrl_PC03
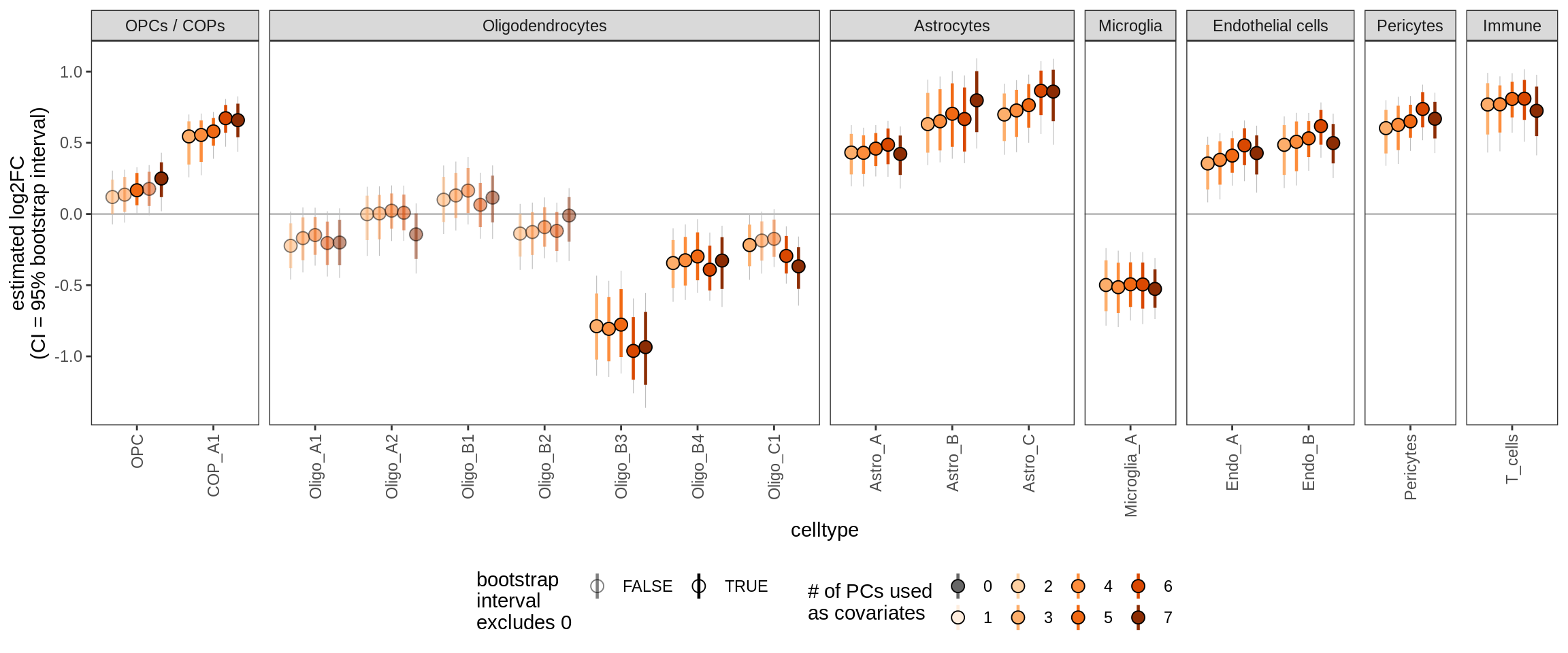
ctrl_PC04
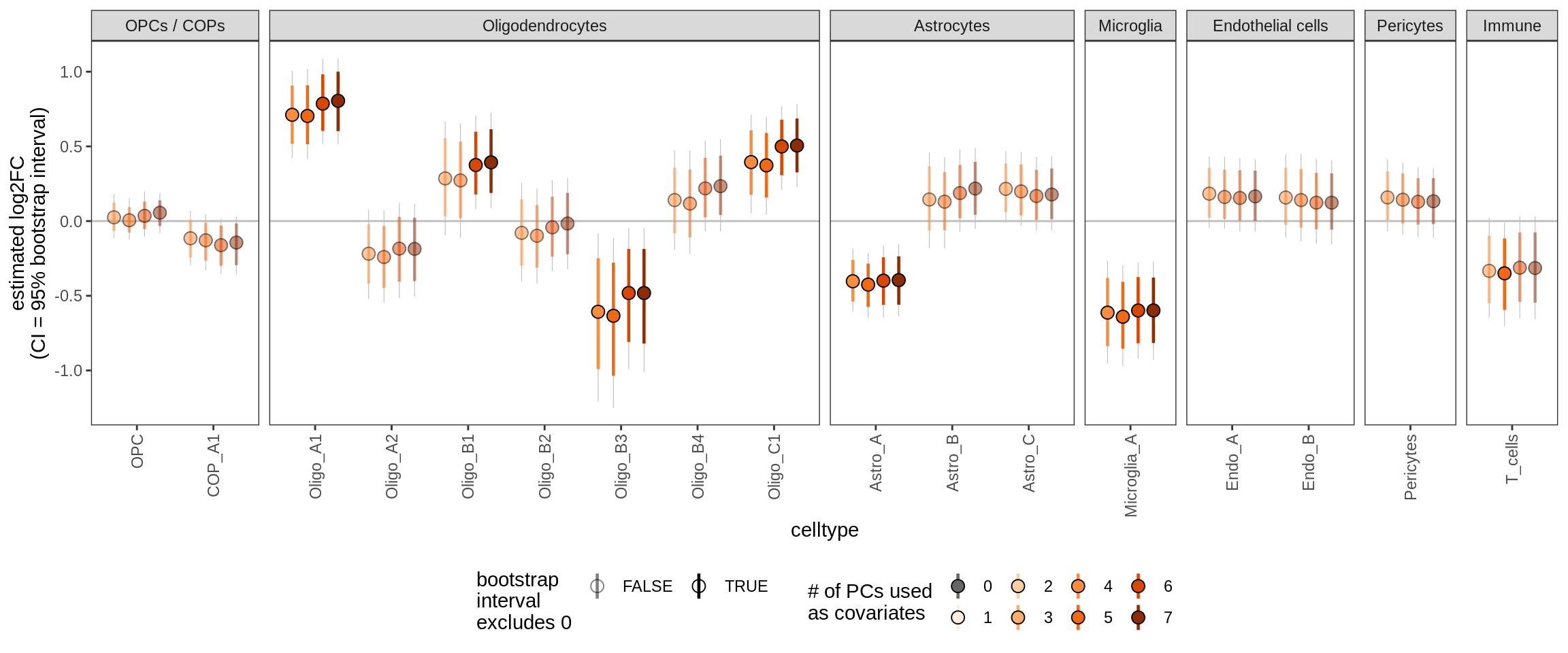
ctrl_PC06

ctrl_PC07

Effect of including PCs, lesions only
sel_coefs = c("NAGM", "GML")
cat('#### ', 'neurons only', '\n')neurons only
g = plot_effect_of_pcs(sel_coefs,
pcs_coefs_dt[ type_broad %in% gm_pc_spec$broad_sel ] ) +
labs(title = "Neurons only")
print(g)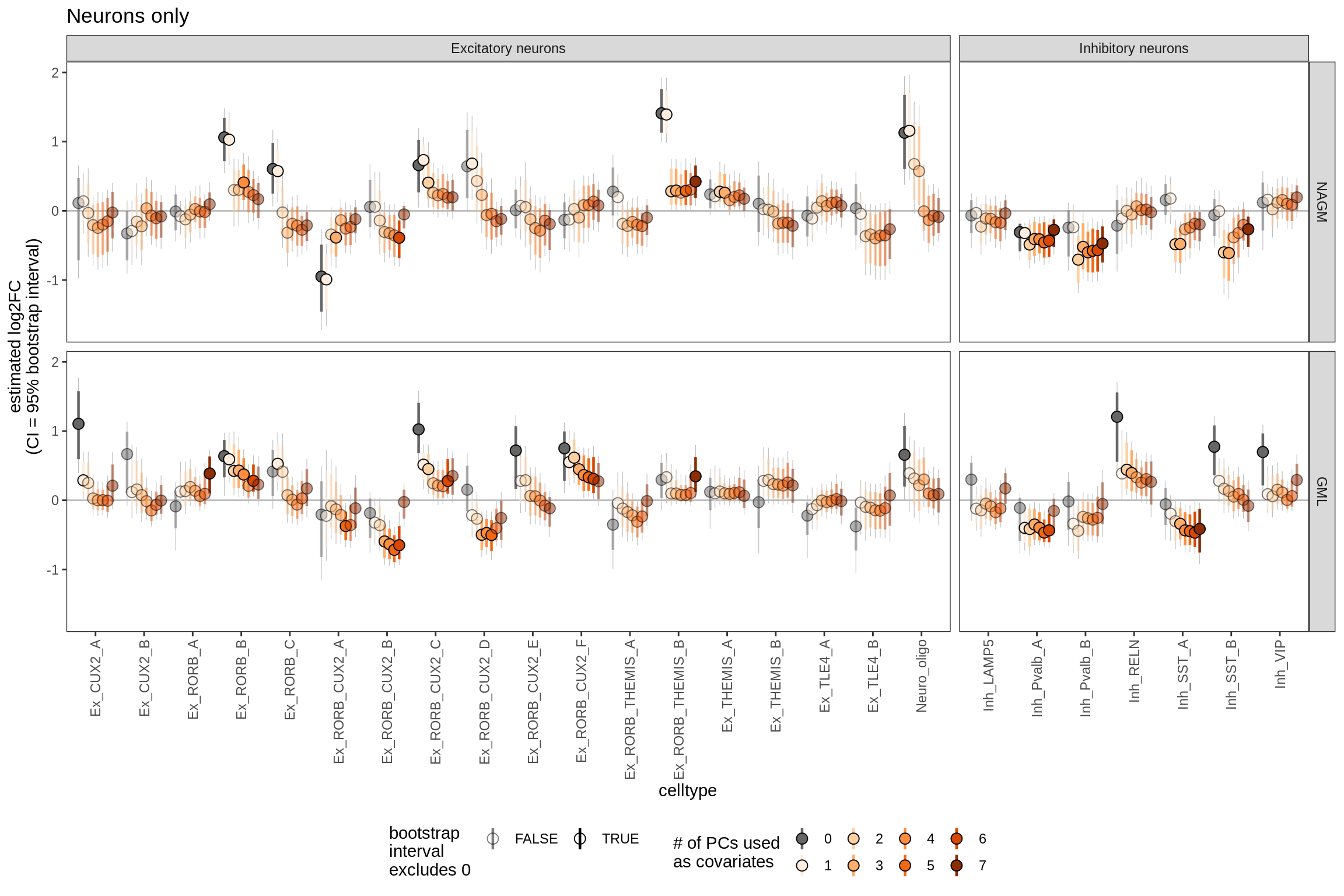
cat('\n\n')cat('#### ', 'other celltypes', '\n')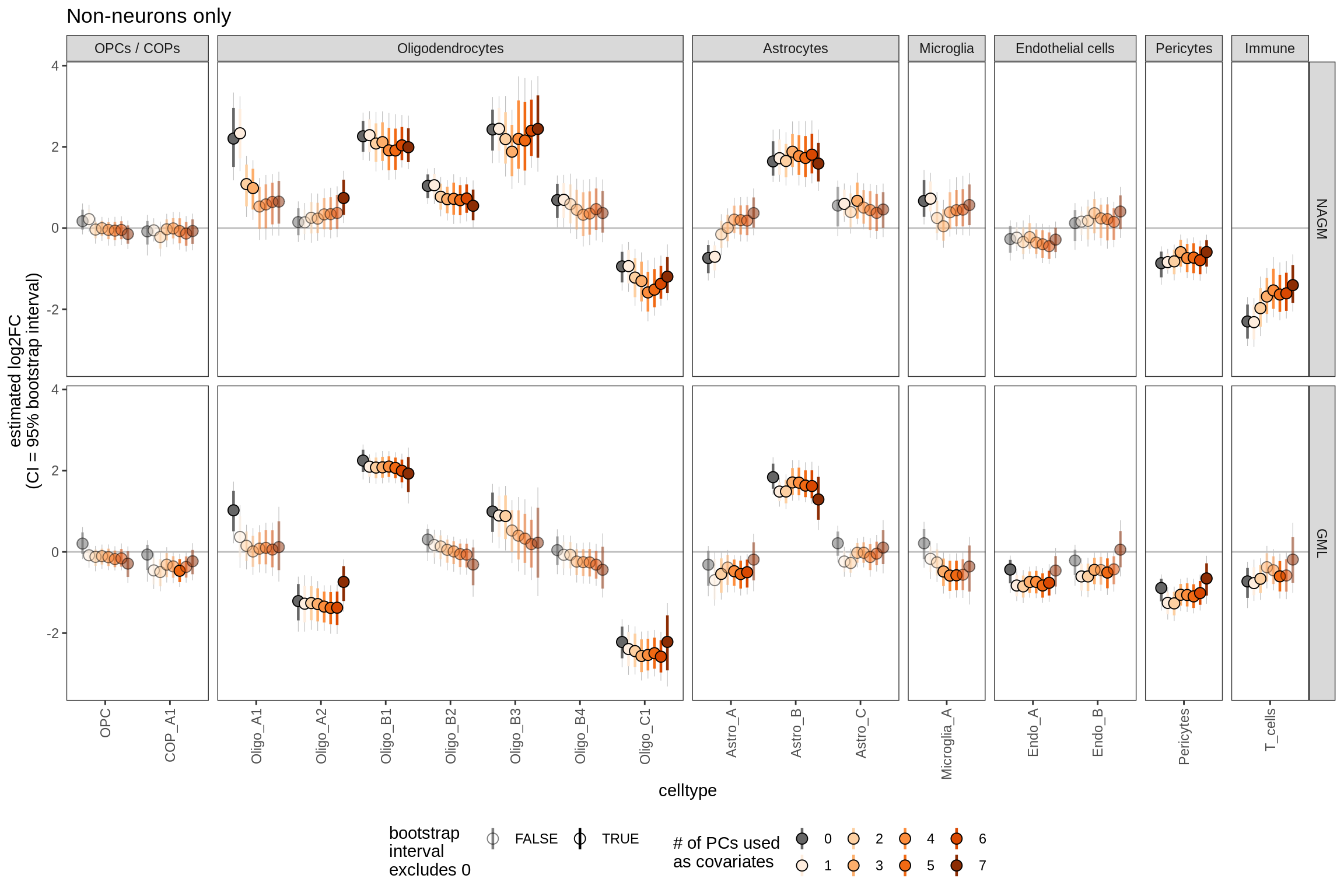
Outputs
if ( !all(file.exists(pb_pcs_ls)) ) {
for (nn in names(pb_f_ls)) {
# which files?
pb_f = pb_f_ls[[ nn ]]
pb_pcs_f = pb_pcs_ls[[ nn ]]
# load full pseudobulk, restrict to just GM
pb_all = pb_f %>% readRDS
pb_pcs = pb_all[, all_pcs_dt$sample_id]
for (v in str_subset(names(all_pcs_dt), "ctrl_PC"))
colData(pb_pcs)[[v]] = all_pcs_dt[[v]] %>% scale %>% `/`(2)
# save
saveRDS(pb_pcs, file = pb_pcs_f)
}
}devtools::session_info()- Session info ---------------------------------------------------------------
setting value
version R version 4.1.2 (2021-11-01)
os Red Hat Enterprise Linux 8.2 (Ootpa)
system x86_64, linux-gnu
ui X11
language (EN)
collate en_US.UTF-8
ctype C
tz Europe/Amsterdam
date 2022-03-23
pandoc 2.5 @ /apps/rocs/pRED/2020.08/cascadelake/software/Pandoc/2.5/bin/ (via rmarkdown)
- Packages -------------------------------------------------------------------
package * version date (UTC) lib source
abind 1.4-5 2016-07-21 [5] CRAN (R 4.1.2)
ade4 1.7-18 2021-09-16 [5] CRAN (R 4.1.2)
ANCOMBC * 1.4.0 2021-10-26 [3] Bioconductor
annotate 1.72.0 2021-10-26 [3] Bioconductor
AnnotationDbi 1.56.2 2021-11-09 [3] Bioconductor
ape 5.5 2021-04-25 [5] CRAN (R 4.1.2)
assertthat * 0.2.1 2019-03-21 [5] CRAN (R 4.1.2)
backports 1.4.0 2021-11-23 [5] CRAN (R 4.1.2)
beachmat 2.10.0 2021-10-26 [3] Bioconductor
beeswarm 0.4.0 2021-06-01 [3] CRAN (R 4.1.2)
Biobase * 2.54.0 2021-10-26 [3] Bioconductor
BiocGenerics * 0.40.0 2021-10-26 [3] Bioconductor
BiocManager 1.30.16 2021-06-15 [3] CRAN (R 4.1.2)
BiocNeighbors 1.12.0 2021-10-26 [3] Bioconductor
BiocParallel * 1.28.3 2021-12-09 [1] Bioconductor
BiocSingular 1.10.0 2021-10-26 [3] Bioconductor
BiocStyle * 2.22.0 2021-10-26 [3] Bioconductor
biomformat 1.22.0 2021-10-26 [3] Bioconductor
Biostrings 2.62.0 2021-10-26 [3] Bioconductor
bit 4.0.4 2020-08-04 [5] CRAN (R 4.1.2)
bit64 4.0.5 2020-08-30 [5] CRAN (R 4.1.2)
bitops 1.0-7 2021-04-24 [5] CRAN (R 4.1.2)
blme 1.0-5 2021-01-05 [3] CRAN (R 4.1.2)
blob 1.2.2 2021-07-23 [5] CRAN (R 4.1.2)
bluster 1.4.0 2021-10-26 [3] Bioconductor
boot 1.3-28 2021-05-03 [5] CRAN (R 4.1.2)
broom 0.7.10 2021-10-31 [5] CRAN (R 4.1.2)
bslib 0.3.1 2021-10-06 [5] CRAN (R 4.1.2)
cachem 1.0.6 2021-08-19 [5] CRAN (R 4.1.2)
callr 3.7.0 2021-04-20 [5] CRAN (R 4.1.2)
caTools 1.18.2 2021-03-28 [5] CRAN (R 4.1.2)
cellranger 1.1.0 2016-07-27 [5] CRAN (R 4.1.2)
circlize * 0.4.13 2021-06-09 [3] CRAN (R 4.1.2)
cli 3.2.0 2022-02-14 [1] CRAN (R 4.1.2)
clue 0.3-60 2021-10-11 [5] CRAN (R 4.1.2)
cluster 2.1.2 2021-04-17 [5] CRAN (R 4.1.2)
coda 0.19-4 2020-09-30 [5] CRAN (R 4.1.2)
codetools 0.2-18 2020-11-04 [5] CRAN (R 4.1.2)
colorspace 2.0-3 2022-02-21 [1] CRAN (R 4.1.2)
ComplexHeatmap * 2.10.0 2021-10-26 [3] Bioconductor
conos * 1.4.5 2022-01-21 [1] CRAN (R 4.1.2)
cowplot 1.1.1 2020-12-30 [5] CRAN (R 4.1.2)
crayon 1.5.0 2022-02-14 [1] CRAN (R 4.1.2)
data.table * 1.14.2 2021-09-27 [5] CRAN (R 4.1.2)
DBI 1.1.1 2021-01-15 [5] CRAN (R 4.1.2)
DelayedArray 0.20.0 2021-10-26 [3] Bioconductor
DelayedMatrixStats 1.16.0 2021-10-26 [3] Bioconductor
deldir 1.0-6 2021-10-23 [5] CRAN (R 4.1.2)
desc 1.4.0 2021-09-28 [5] CRAN (R 4.1.2)
DESeq2 1.34.0 2021-10-26 [3] Bioconductor
devtools 2.4.3 2021-11-30 [5] CRAN (R 4.1.2)
digest 0.6.29 2021-12-01 [5] CRAN (R 4.1.2)
doParallel 1.0.16 2020-10-16 [5] CRAN (R 4.1.2)
dplyr 1.0.7 2021-06-18 [5] CRAN (R 4.1.2)
dqrng 0.3.0 2021-05-01 [5] CRAN (R 4.1.2)
DropletUtils * 1.14.1 2021-11-08 [3] Bioconductor
edgeR * 3.36.0 2021-10-26 [3] Bioconductor
ellipsis 0.3.2 2021-04-29 [5] CRAN (R 4.1.2)
emmeans 1.7.1-1 2021-11-29 [3] CRAN (R 4.1.2)
estimability 1.3 2018-02-11 [3] CRAN (R 4.1.2)
evaluate 0.15 2022-02-18 [1] CRAN (R 4.1.2)
fansi 1.0.2 2022-01-14 [1] CRAN (R 4.1.2)
farver 2.1.0 2021-02-28 [5] CRAN (R 4.1.2)
fastmap 1.1.0 2021-01-25 [5] CRAN (R 4.1.2)
fitdistrplus 1.1-6 2021-09-28 [5] CRAN (R 4.1.2)
forcats * 0.5.1 2021-01-27 [5] CRAN (R 4.1.2)
foreach 1.5.1 2020-10-15 [5] CRAN (R 4.1.2)
fs 1.5.1 2021-11-30 [5] CRAN (R 4.1.2)
future 1.23.0 2021-10-31 [5] CRAN (R 4.1.2)
future.apply 1.8.1 2021-08-10 [5] CRAN (R 4.1.2)
gargle 1.2.0 2021-07-02 [5] CRAN (R 4.1.2)
genefilter 1.76.0 2021-10-26 [3] Bioconductor
geneplotter 1.72.0 2021-10-26 [3] Bioconductor
generics 0.1.1 2021-10-25 [5] CRAN (R 4.1.2)
GenomeInfoDb * 1.30.1 2022-01-30 [1] Bioconductor
GenomeInfoDbData 1.2.7 2022-03-15 [3] Bioconductor
GenomicRanges * 1.46.1 2021-11-18 [3] Bioconductor
GetoptLong 1.0.5 2020-12-15 [3] CRAN (R 4.1.2)
ggbeeswarm * 0.6.0 2017-08-07 [3] CRAN (R 4.1.2)
ggplot.multistats * 1.0.0 2019-10-28 [1] CRAN (R 4.1.2)
ggplot2 * 3.3.5 2021-06-25 [5] CRAN (R 4.1.2)
ggrepel * 0.9.1 2021-01-15 [5] CRAN (R 4.1.2)
ggridges 0.5.3 2021-01-08 [5] CRAN (R 4.1.2)
git2r 0.29.0 2021-11-22 [5] CRAN (R 4.1.2)
glmmTMB 1.1.2.3 2021-09-20 [3] CRAN (R 4.1.2)
GlobalOptions 0.1.2 2020-06-10 [3] CRAN (R 4.1.2)
globals 0.14.0 2020-11-22 [5] CRAN (R 4.1.2)
glue 1.6.2 2022-02-24 [1] CRAN (R 4.1.2)
goftest 1.2-3 2021-10-07 [5] CRAN (R 4.1.2)
googledrive 2.0.0 2021-07-08 [5] CRAN (R 4.1.2)
googlesheets4 * 1.0.0 2021-07-21 [5] CRAN (R 4.1.2)
gplots 3.1.1 2020-11-28 [5] CRAN (R 4.1.2)
gridExtra 2.3 2017-09-09 [5] CRAN (R 4.1.2)
grr 0.9.5 2016-08-26 [1] CRAN (R 4.1.2)
gtable 0.3.0 2019-03-25 [5] CRAN (R 4.1.2)
gtools 3.9.2 2021-06-06 [5] CRAN (R 4.1.2)
HDF5Array 1.22.1 2021-11-14 [3] Bioconductor
hexbin 1.28.2 2021-01-08 [5] CRAN (R 4.1.2)
highr 0.9 2021-04-16 [5] CRAN (R 4.1.2)
hms 1.1.1 2021-09-26 [5] CRAN (R 4.1.2)
htmltools 0.5.2 2021-08-25 [5] CRAN (R 4.1.2)
htmlwidgets 1.5.4 2021-09-08 [5] CRAN (R 4.1.2)
httpuv 1.6.3 2021-09-09 [5] CRAN (R 4.1.2)
httr 1.4.2 2020-07-20 [5] CRAN (R 4.1.2)
ica * 1.0-2 2018-05-24 [5] CRAN (R 4.1.2)
igraph * 1.2.11 2022-01-04 [1] CRAN (R 4.1.2)
IRanges * 2.28.0 2021-10-26 [3] Bioconductor
irlba 2.3.5 2021-12-06 [5] CRAN (R 4.1.2)
iterators 1.0.13 2020-10-15 [5] CRAN (R 4.1.2)
janitor 2.1.0 2021-01-05 [2] CRAN (R 4.1.2)
jquerylib 0.1.4 2021-04-26 [5] CRAN (R 4.1.2)
jsonlite 1.8.0 2022-02-22 [1] CRAN (R 4.1.2)
KEGGREST 1.34.0 2021-10-26 [3] Bioconductor
KernSmooth 2.23-20 2021-05-03 [5] CRAN (R 4.1.2)
knitr 1.37 2021-12-16 [1] CRAN (R 4.1.2)
later 1.3.0 2021-08-18 [5] CRAN (R 4.1.2)
lattice 0.20-45 2021-09-22 [5] CRAN (R 4.1.2)
lazyeval 0.2.2 2019-03-15 [5] CRAN (R 4.1.2)
leiden 0.3.9 2021-07-27 [5] CRAN (R 4.1.2)
leidenAlg 1.0.2 2022-03-03 [1] CRAN (R 4.1.2)
lifecycle 1.0.1 2021-09-24 [5] CRAN (R 4.1.2)
limma * 3.50.0 2021-10-26 [3] Bioconductor
listenv 0.8.0 2019-12-05 [5] CRAN (R 4.1.2)
lme4 1.1-27.1 2021-06-22 [5] CRAN (R 4.1.2)
lmerTest 3.1-3 2020-10-23 [3] CRAN (R 4.1.2)
lmtest 0.9-39 2021-11-07 [5] CRAN (R 4.1.2)
locfit 1.5-9.4 2020-03-25 [5] CRAN (R 4.1.2)
lubridate 1.8.0 2021-10-07 [5] CRAN (R 4.1.2)
magrittr * 2.0.2 2022-01-26 [1] CRAN (R 4.1.2)
MASS * 7.3-54 2021-05-03 [5] CRAN (R 4.1.2)
Matrix * 1.3-4 2021-06-01 [5] CRAN (R 4.1.2)
Matrix.utils 0.9.8 2020-02-26 [1] CRAN (R 4.1.2)
MatrixGenerics * 1.6.0 2021-10-26 [3] Bioconductor
matrixStats * 0.61.0 2021-09-17 [5] CRAN (R 4.1.2)
mclust * 5.4.9 2021-12-17 [1] CRAN (R 4.1.2)
memoise 2.0.1 2021-11-26 [5] CRAN (R 4.1.2)
metapod 1.2.0 2021-10-26 [3] Bioconductor
mgcv 1.8-38 2021-10-06 [5] CRAN (R 4.1.2)
microbiome 1.16.0 2021-10-26 [3] Bioconductor
mime 0.12 2021-09-28 [5] CRAN (R 4.1.2)
miniUI 0.1.1.1 2018-05-18 [5] CRAN (R 4.1.2)
minqa 1.2.4 2014-10-09 [5] CRAN (R 4.1.2)
multcomp 1.4-17 2021-04-29 [5] CRAN (R 4.1.2)
multtest 2.50.0 2021-10-26 [3] Bioconductor
munsell 0.5.0 2018-06-12 [5] CRAN (R 4.1.2)
muscat * 1.8.0 2021-10-26 [3] Bioconductor
mvtnorm 1.1-3 2021-10-08 [5] CRAN (R 4.1.2)
nlme 3.1-153 2021-09-07 [5] CRAN (R 4.1.2)
nloptr 1.2.2.3 2021-11-02 [5] CRAN (R 4.1.2)
nnls * 1.4 2012-03-19 [2] CRAN (R 4.1.2)
numDeriv 2016.8-1.1 2019-06-06 [5] CRAN (R 4.1.2)
parallelly 1.29.0 2021-11-21 [5] CRAN (R 4.1.2)
patchwork * 1.1.0.9000 2022-03-23 [1] Github (thomasp85/patchwork@79223d3)
pbapply 1.5-0 2021-09-16 [5] CRAN (R 4.1.2)
pbkrtest 0.5.1 2021-03-09 [5] CRAN (R 4.1.2)
permute 0.9-5 2019-03-12 [3] CRAN (R 4.1.2)
phyloseq * 1.38.0 2021-10-26 [3] Bioconductor
pillar 1.7.0 2022-02-01 [1] CRAN (R 4.1.2)
pkgbuild 1.2.1 2021-11-30 [5] CRAN (R 4.1.2)
pkgconfig 2.0.3 2019-09-22 [5] CRAN (R 4.1.2)
pkgload 1.2.4 2021-11-30 [5] CRAN (R 4.1.2)
plotly 4.10.0 2021-10-09 [5] CRAN (R 4.1.2)
plyr 1.8.6 2020-03-03 [5] CRAN (R 4.1.2)
png 0.1-7 2013-12-03 [5] CRAN (R 4.1.2)
polyclip 1.10-0 2019-03-14 [5] CRAN (R 4.1.2)
prettyunits 1.1.1 2020-01-24 [5] CRAN (R 4.1.2)
processx 3.5.2 2021-04-30 [5] CRAN (R 4.1.2)
progress 1.2.2 2019-05-16 [5] CRAN (R 4.1.2)
promises 1.2.0.1 2021-02-11 [5] CRAN (R 4.1.2)
ps 1.6.0 2021-02-28 [5] CRAN (R 4.1.2)
purrr * 0.3.4 2020-04-17 [5] CRAN (R 4.1.2)
R.methodsS3 1.8.1 2020-08-26 [5] CRAN (R 4.1.2)
R.oo 1.24.0 2020-08-26 [5] CRAN (R 4.1.2)
R.utils 2.11.0 2021-09-26 [5] CRAN (R 4.1.2)
R6 2.5.1 2021-08-19 [5] CRAN (R 4.1.2)
RANN 2.6.1 2019-01-08 [5] CRAN (R 4.1.2)
rbibutils 2.2.7 2021-12-07 [5] CRAN (R 4.1.2)
RColorBrewer * 1.1-2 2014-12-07 [5] CRAN (R 4.1.2)
Rcpp 1.0.8.3 2022-03-17 [1] CRAN (R 4.1.2)
RcppAnnoy 0.0.19 2021-07-30 [5] CRAN (R 4.1.2)
RCurl 1.98-1.6 2022-02-08 [1] CRAN (R 4.1.2)
Rdpack 2.1.3 2021-12-08 [5] CRAN (R 4.1.2)
readxl * 1.3.1 2019-03-13 [5] CRAN (R 4.1.2)
registry 0.5-1 2019-03-05 [5] CRAN (R 4.1.2)
remotes 2.4.2 2021-11-30 [5] CRAN (R 4.1.2)
reshape2 1.4.4 2020-04-09 [5] CRAN (R 4.1.2)
reticulate * 1.22 2021-09-17 [5] CRAN (R 4.1.2)
rhdf5 2.38.0 2021-10-26 [3] Bioconductor
rhdf5filters 1.6.0 2021-10-26 [3] Bioconductor
Rhdf5lib 1.16.0 2021-10-26 [3] Bioconductor
rjson 0.2.20 2018-06-08 [5] CRAN (R 4.1.2)
rlang 1.0.2 2022-03-04 [1] CRAN (R 4.1.2)
rmarkdown 2.13 2022-03-10 [1] CRAN (R 4.1.2)
ROCR 1.0-11 2020-05-02 [5] CRAN (R 4.1.2)
rpart 4.1-15 2019-04-12 [5] CRAN (R 4.1.2)
rprojroot 2.0.2 2020-11-15 [5] CRAN (R 4.1.2)
RSQLite 2.2.9 2021-12-06 [5] CRAN (R 4.1.2)
rsvd 1.0.5 2021-04-16 [5] CRAN (R 4.1.2)
Rtsne 0.15 2018-11-10 [5] CRAN (R 4.1.2)
S4Vectors * 0.32.3 2021-11-21 [3] Bioconductor
sandwich 3.0-1 2021-05-18 [5] CRAN (R 4.1.2)
sass 0.4.0 2021-05-12 [5] CRAN (R 4.1.2)
ScaledMatrix 1.2.0 2021-10-26 [3] Bioconductor
scales * 1.1.1 2020-05-11 [5] CRAN (R 4.1.2)
scater * 1.22.0 2021-10-26 [3] Bioconductor
scattermore 0.7 2020-11-24 [5] CRAN (R 4.1.2)
sccore 1.0.1 2021-12-12 [1] CRAN (R 4.1.2)
scran * 1.22.1 2021-11-14 [3] Bioconductor
sctransform 0.3.2 2020-12-16 [5] CRAN (R 4.1.2)
scuttle * 1.4.0 2021-10-26 [3] Bioconductor
seriation * 1.3.1 2021-10-16 [3] CRAN (R 4.1.2)
sessioninfo 1.2.2 2021-12-06 [5] CRAN (R 4.1.2)
Seurat * 4.0.5 2021-10-17 [5] CRAN (R 4.1.2)
SeuratObject * 4.0.4 2021-11-23 [5] CRAN (R 4.1.2)
shape 1.4.6 2021-05-19 [3] CRAN (R 4.1.2)
shiny 1.7.1 2021-10-02 [5] CRAN (R 4.1.2)
SingleCellExperiment * 1.16.0 2021-10-26 [3] Bioconductor
snakecase 0.11.0 2019-05-25 [2] CRAN (R 4.1.2)
sparseMatrixStats 1.6.0 2021-10-26 [3] Bioconductor
spatstat.core 2.3-2 2021-11-26 [5] CRAN (R 4.1.2)
spatstat.data 2.1-0 2021-03-21 [5] CRAN (R 4.1.2)
spatstat.geom 2.3-0 2021-10-09 [5] CRAN (R 4.1.2)
spatstat.sparse 2.0-0 2021-03-16 [5] CRAN (R 4.1.2)
spatstat.utils 2.2-0 2021-06-14 [5] CRAN (R 4.1.2)
statmod 1.4.36 2021-05-10 [5] CRAN (R 4.1.2)
stringi 1.7.6 2021-11-29 [5] CRAN (R 4.1.2)
stringr * 1.4.0 2019-02-10 [5] CRAN (R 4.1.2)
SummarizedExperiment * 1.24.0 2021-10-26 [3] Bioconductor
survival 3.2-13 2021-08-24 [5] CRAN (R 4.1.2)
tensor 1.5 2012-05-05 [5] CRAN (R 4.1.2)
testthat 3.1.1 2021-12-03 [5] CRAN (R 4.1.2)
TH.data 1.1-0 2021-09-27 [5] CRAN (R 4.1.2)
tibble 3.1.6 2021-11-07 [5] CRAN (R 4.1.2)
tidyr 1.1.4 2021-09-27 [5] CRAN (R 4.1.2)
tidyselect 1.1.1 2021-04-30 [5] CRAN (R 4.1.2)
TMB 1.7.22 2021-09-28 [3] CRAN (R 4.1.2)
TSP 1.1-11 2021-10-06 [3] CRAN (R 4.1.2)
usethis 2.1.3 2021-10-27 [5] CRAN (R 4.1.2)
utf8 1.2.2 2021-07-24 [5] CRAN (R 4.1.2)
uwot 0.1.11 2021-12-02 [5] CRAN (R 4.1.2)
variancePartition 1.24.0 2021-10-26 [3] Bioconductor
vctrs 0.3.8 2021-04-29 [5] CRAN (R 4.1.2)
vegan 2.5-7 2020-11-28 [3] CRAN (R 4.1.2)
vipor 0.4.5 2017-03-22 [3] CRAN (R 4.1.2)
viridis * 0.6.2 2021-10-13 [5] CRAN (R 4.1.2)
viridisLite * 0.4.0 2021-04-13 [5] CRAN (R 4.1.2)
whisker 0.4 2019-08-28 [5] CRAN (R 4.1.2)
withr 2.5.0 2022-03-03 [1] CRAN (R 4.1.2)
workflowr 1.7.0 2021-12-21 [1] CRAN (R 4.1.2)
xfun 0.30 2022-03-02 [1] CRAN (R 4.1.2)
XML 3.99-0.8 2021-09-17 [5] CRAN (R 4.1.2)
xtable 1.8-4 2019-04-21 [5] CRAN (R 4.1.2)
XVector 0.34.0 2021-10-26 [3] Bioconductor
yaml 2.3.5 2022-02-21 [1] CRAN (R 4.1.2)
zlibbioc 1.40.0 2021-10-26 [3] Bioconductor
zoo 1.8-9 2021-03-09 [5] CRAN (R 4.1.2)
[1] /gpfs/homefs/global/home/macnairw/R/x86_64-pc-linux-gnu-library/4.1.2-foss
[2] /apps/rocs/2020.08/cascadelake/software/R-Roche-bundle/2021.12-foss-2020a-R-4.1.2
[3] /apps/rocs/2020.08/cascadelake/software/R-bundle-Bioconductor/3.14-foss-2020a-R-4.1.2
[4] /apps/rocs/2020.08/cascadelake/software/ncdf4/1.18-foss-2020a-R-4.1.2
[5] /apps/rocs/2020.08/cascadelake/software/R/4.1.2-foss-2020a/lib64/R/library
------------------------------------------------------------------------------
sessionInfo()R version 4.1.2 (2021-11-01)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Red Hat Enterprise Linux 8.2 (Ootpa)
Matrix products: default
BLAS/LAPACK: /apps/rocs/2020.08/cascadelake/software/OpenBLAS/0.3.9-GCC-9.3.0/lib/libopenblas_skylakexp-r0.3.9.so
locale:
[1] LC_CTYPE=C LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats4 grid stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] ggbeeswarm_0.6.0 ggrepel_0.9.1
[3] reticulate_1.22 mclust_5.4.9
[5] MASS_7.3-54 phyloseq_1.38.0
[7] ANCOMBC_1.4.0 ica_1.0-2
[9] purrr_0.3.4 nnls_1.4
[11] muscat_1.8.0 DropletUtils_1.14.1
[13] edgeR_3.36.0 limma_3.50.0
[15] googlesheets4_1.0.0 scran_1.22.1
[17] scater_1.22.0 scuttle_1.4.0
[19] SingleCellExperiment_1.16.0 SummarizedExperiment_1.24.0
[21] Biobase_2.54.0 GenomicRanges_1.46.1
[23] GenomeInfoDb_1.30.1 IRanges_2.28.0
[25] S4Vectors_0.32.3 BiocGenerics_0.40.0
[27] MatrixGenerics_1.6.0 matrixStats_0.61.0
[29] BiocParallel_1.28.3 ggplot.multistats_1.0.0
[31] patchwork_1.1.0.9000 seriation_1.3.1
[33] ComplexHeatmap_2.10.0 SeuratObject_4.0.4
[35] Seurat_4.0.5 conos_1.4.5
[37] igraph_1.2.11 Matrix_1.3-4
[39] readxl_1.3.1 forcats_0.5.1
[41] ggplot2_3.3.5 scales_1.1.1
[43] viridis_0.6.2 viridisLite_0.4.0
[45] assertthat_0.2.1 stringr_1.4.0
[47] data.table_1.14.2 magrittr_2.0.2
[49] circlize_0.4.13 RColorBrewer_1.1-2
[51] BiocStyle_2.22.0
loaded via a namespace (and not attached):
[1] rsvd_1.0.5 ps_1.6.0
[3] foreach_1.5.1 lmtest_0.9-39
[5] rprojroot_2.0.2 crayon_1.5.0
[7] rbibutils_2.2.7 spatstat.core_2.3-2
[9] rhdf5filters_1.6.0 Matrix.utils_0.9.8
[11] nlme_3.1-153 backports_1.4.0
[13] rlang_1.0.2 XVector_0.34.0
[15] ROCR_1.0-11 microbiome_1.16.0
[17] irlba_2.3.5 callr_3.7.0
[19] nloptr_1.2.2.3 rjson_0.2.20
[21] bit64_4.0.5 glue_1.6.2
[23] sctransform_0.3.2 processx_3.5.2
[25] pbkrtest_0.5.1 parallel_4.1.2
[27] vipor_0.4.5 spatstat.sparse_2.0-0
[29] AnnotationDbi_1.56.2 spatstat.geom_2.3-0
[31] tidyselect_1.1.1 usethis_2.1.3
[33] fitdistrplus_1.1-6 variancePartition_1.24.0
[35] XML_3.99-0.8 tidyr_1.1.4
[37] zoo_1.8-9 xtable_1.8-4
[39] evaluate_0.15 Rdpack_2.1.3
[41] cli_3.2.0 zlibbioc_1.40.0
[43] miniUI_0.1.1.1 whisker_0.4
[45] bslib_0.3.1 rpart_4.1-15
[47] shiny_1.7.1 BiocSingular_1.10.0
[49] xfun_0.30 clue_0.3-60
[51] pkgbuild_1.2.1 multtest_2.50.0
[53] cluster_2.1.2 caTools_1.18.2
[55] TSP_1.1-11 biomformat_1.22.0
[57] KEGGREST_1.34.0 tibble_3.1.6
[59] ape_5.5 listenv_0.8.0
[61] permute_0.9-5 Biostrings_2.62.0
[63] png_0.1-7 future_1.23.0
[65] withr_2.5.0 bitops_1.0-7
[67] plyr_1.8.6 cellranger_1.1.0
[69] dqrng_0.3.0 coda_0.19-4
[71] pillar_1.7.0 gplots_3.1.1
[73] GlobalOptions_0.1.2 cachem_1.0.6
[75] multcomp_1.4-17 fs_1.5.1
[77] GetoptLong_1.0.5 DelayedMatrixStats_1.16.0
[79] vctrs_0.3.8 ellipsis_0.3.2
[81] generics_0.1.1 devtools_2.4.3
[83] tools_4.1.2 beeswarm_0.4.0
[85] munsell_0.5.0 emmeans_1.7.1-1
[87] DelayedArray_0.20.0 pkgload_1.2.4
[89] fastmap_1.1.0 compiler_4.1.2
[91] abind_1.4-5 httpuv_1.6.3
[93] sessioninfo_1.2.2 plotly_4.10.0
[95] GenomeInfoDbData_1.2.7 gridExtra_2.3
[97] glmmTMB_1.1.2.3 workflowr_1.7.0
[99] lattice_0.20-45 deldir_1.0-6
[101] utf8_1.2.2 later_1.3.0
[103] dplyr_1.0.7 jsonlite_1.8.0
[105] ScaledMatrix_1.2.0 pbapply_1.5-0
[107] sparseMatrixStats_1.6.0 estimability_1.3
[109] genefilter_1.76.0 lazyeval_0.2.2
[111] promises_1.2.0.1 doParallel_1.0.16
[113] R.utils_2.11.0 goftest_1.2-3
[115] spatstat.utils_2.2-0 rmarkdown_2.13
[117] sandwich_3.0-1 cowplot_1.1.1
[119] blme_1.0-5 statmod_1.4.36
[121] Rtsne_0.15 uwot_0.1.11
[123] HDF5Array_1.22.1 survival_3.2-13
[125] numDeriv_2016.8-1.1 yaml_2.3.5
[127] htmltools_0.5.2 memoise_2.0.1
[129] locfit_1.5-9.4 digest_0.6.29
[131] mime_0.12 registry_0.5-1
[133] RSQLite_2.2.9 future.apply_1.8.1
[135] remotes_2.4.2 vegan_2.5-7
[137] blob_1.2.2 R.oo_1.24.0
[139] splines_4.1.2 Rhdf5lib_1.16.0
[141] googledrive_2.0.0 RCurl_1.98-1.6
[143] broom_0.7.10 hms_1.1.1
[145] rhdf5_2.38.0 colorspace_2.0-3
[147] BiocManager_1.30.16 shape_1.4.6
[149] sass_0.4.0 Rcpp_1.0.8.3
[151] RANN_2.6.1 mvtnorm_1.1-3
[153] fansi_1.0.2 parallelly_1.29.0
[155] R6_2.5.1 ggridges_0.5.3
[157] lifecycle_1.0.1 bluster_1.4.0
[159] minqa_1.2.4 testthat_3.1.1
[161] leiden_0.3.9 jquerylib_0.1.4
[163] snakecase_0.11.0 desc_1.4.0
[165] RcppAnnoy_0.0.19 TH.data_1.1-0
[167] iterators_1.0.13 TMB_1.7.22
[169] htmlwidgets_1.5.4 beachmat_2.10.0
[171] polyclip_1.10-0 mgcv_1.8-38
[173] globals_0.14.0 leidenAlg_1.0.2
[175] lubridate_1.8.0 codetools_0.2-18
[177] metapod_1.2.0 gtools_3.9.2
[179] prettyunits_1.1.1 R.methodsS3_1.8.1
[181] gtable_0.3.0 DBI_1.1.1
[183] git2r_0.29.0 tensor_1.5
[185] httr_1.4.2 highr_0.9
[187] KernSmooth_2.23-20 stringi_1.7.6
[189] progress_1.2.2 farver_2.1.0
[191] reshape2_1.4.4 annotate_1.72.0
[193] hexbin_1.28.2 sccore_1.0.1
[195] boot_1.3-28 grr_0.9.5
[197] BiocNeighbors_1.12.0 lme4_1.1-27.1
[199] ade4_1.7-18 geneplotter_1.72.0
[201] scattermore_0.7 DESeq2_1.34.0
[203] bit_4.0.4 spatstat.data_2.1-0
[205] janitor_2.1.0 pkgconfig_2.0.3
[207] gargle_1.2.0 lmerTest_3.1-3
[209] knitr_1.37